The union of a spermatozoon and ovum marks the beginning of a new individual and the continuation of a “life”that has existed for an incredible length of time. The fertilization of an ovum is an amazingly complex act, although it appears simple at first glance. Egg formation (oogenesis) and sperm formation (spermatogenesis) have many similarities, although they differ with regard to sex determination. There are 44 somatic chromosomes and two sex chromosomes in the normal human cell 7.
Oogenesis begins with the formation of a primary oocyte which contains 44 + XX chromosomes. This process occurs before birth and it is believed that no primary oocytes form after birth. After puberty and just before ovulation, one or more of these primary oocytes undergoes two maturation divisions. The first division is a meiotic (G. meiosis, a lessening) division in which the normal or diploid number of chromosomes found in the body cells is reduced to the haploid (G. haplous, single) number so that the normal chromosomal number of 44 somatic chromosomes and two sex chromosomes will be restored after fertilization. During this first maturation division, the secondary oocyte (22 + X) retains most of the cytoplasm, while the other half of the nucleus remains as a small first polar body. A second maturation division results in the formation of the mature ovum (22 + X) plus a second polar body.
Spermatogenesis involves important differences. The spermatogonium is formed in the testis, with a chromosome complement of 44 + XY. This Y sex chromosome determines male development. The spermatogonium develops into a relatively large spermatocyte (44 + XY). This in turn undergoes a first maturation (meiotic) division to form secondary spermatocytes, half of which are 22 + X and half of which are 22 + Y. During a second maturation division, the secondary spermatocytes again divide into spermatids, half of which are 22 + X and half of which are 22 + Y. After 1 or 2 weeks the spermatids become mature spermatozoa. These spermatozoa must undergo further changes before they can fertilize an ovum. The first change, known as “capacitation,”is a physiologic change probably associated with removal of a protective coating 9. Following this, an “acrosome reaction”occurs at the anterior extremity of the spermatozoon, where small perforations develop in the wall. Enzymes passing through these openings digest a path for the sperm through the corona and the zona pellucida.
Ovum transport is the mechanism by which the nonmotile ovum is carried by a stream of peritoneal fluid into the infundibulum of the tube. This stream is produced by sweeping movements of the fimbriae. The ovum is carried into the tubal ampulla partially by muscular contractions of the tube but mostly as a result of ciliary action. At any given normal intercourse, over 300 million sperm are deposited in the vagina near the cervical os Only a few thousand sperm reach the oviducts and only a few hundred reach the ampulla, where fertilization usually occurs. As the sperm head advances, it reaches the surface of the ovum and attaches so that its nucleus is within the membrane of the ovum. As a result, the zona pellucida changes and the entrance of other spermatozoa is inhibited. At the same time, the secondary oocyte is completing its second meiotic division and expelling the second polar body. The male and female pronuclei come into contact near the center of the ovum and mingle their chromosomes. During the process of fertilization, the chromosomes of father and mother mingle, the diploid number (46) of chromosomes is restored, and sex is determined by the presence or absence of the Y, or male, chromosome.
Cleavage
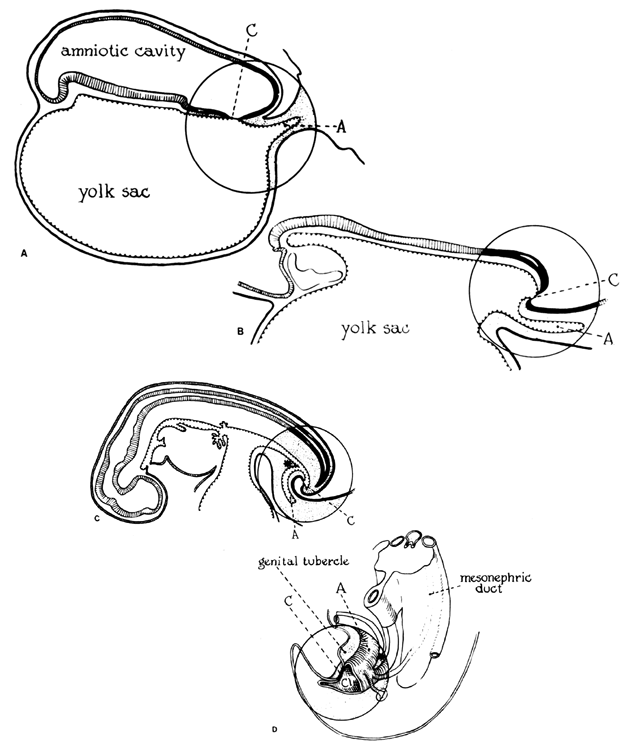
The fertilized egg undergoes a series of rapid mitotic divisions while passing down the tube. After several days, a ball of cells called the “morula”(L. morus, mulberry) is formed. After 5 days, a fluid-filled space appears in the morula, which is then called the “blastocyst”(G. blastos, germ). An inner cell mass appears on one side of the blastocyst cavity at the site of embryo. By 6 days the blastocyst has implanted and the trophoblast cells invade the succulent endometrium or decidua. During the second week, the embryo becomes a bilaminar disk composed of ectoderm and entoderrn (Fig. 1).
|
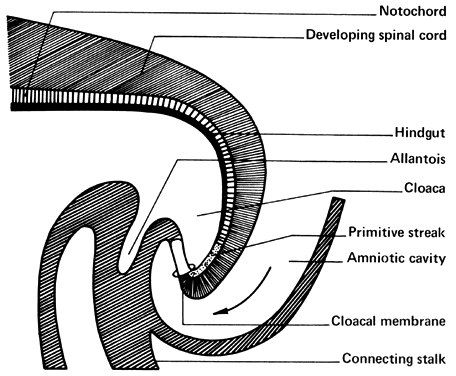
The amniotic cavity appears above the embryonic ectoderm while the blastocyst cavity develops into the yolk sac. In humans the yolk sac never develops yolk as such but makes important contributions to organogenesis. Around amnion, yolk sac, and embryo lies the extraembryonic mesoderm in which isolated lacunas appear. These coelomic spaces fuse rapidly and form large areas of extraembryonic coelom (G. koiloma, hollow). During the third week, a three-layered embryo develops, consisting of ectoderm, endoderm, and mesoderm. A notochord and the notochordal canal develop. The flat embryonic disk folds into a roughly cylindric shape, and the rapid growth of the embryo results in folding at both ends of the embryo. At the caudal end, a tail fold, which includes part of the yolk sac, develops and becomes a hindgut. Part of the hindgut becomes the cloaca, separated from the amniotic cavity by the cloacal membrane, while a ventral outpouching develops as the allantois (G. alias, sausage).
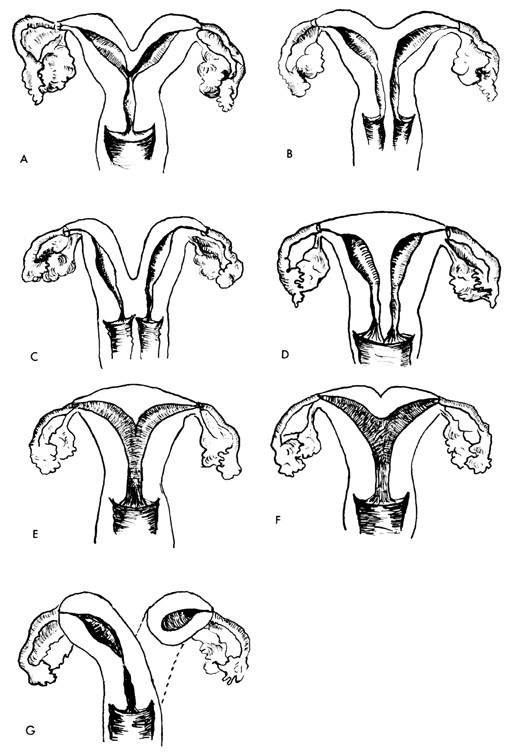
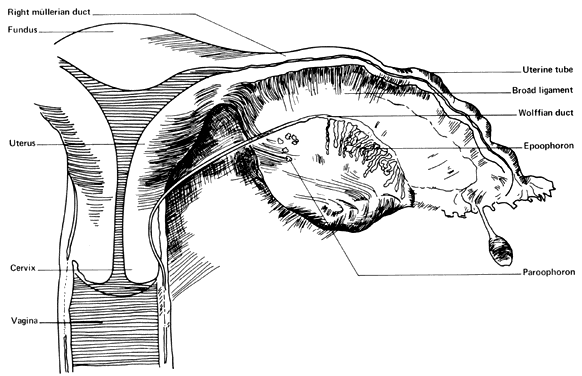
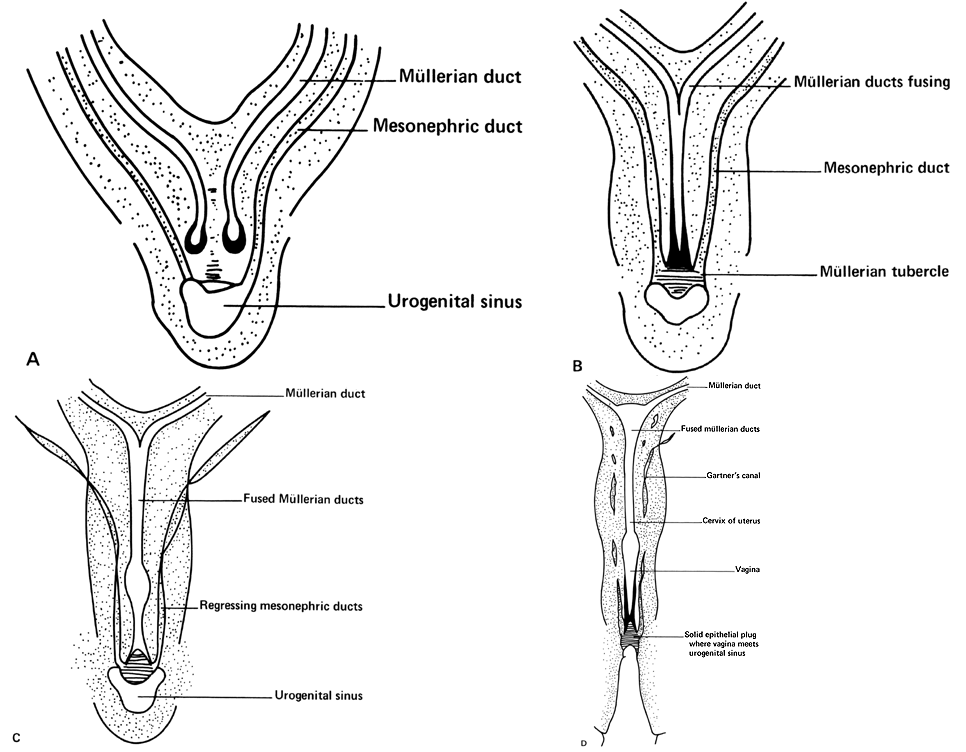
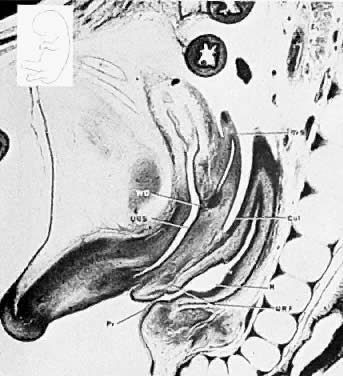
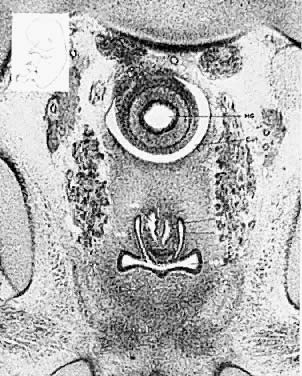
Very early in embryonic development, the primitive body cavity (an intraembryonic coelom) appears. Running longitudinally along the sides of the cavity, the anlage of the genital ridges develops. Primordial cells aggregate along these ridges to form the earliest elements of the future gonads. Two sets of future duct systems develop. The mesonephric urinary ducts are formed and connect at the caudal end with the urogenital sinus and the cloaca. The paramesonephric or müllerian ducts develop lateral and parallel to the mesonephric ducts 10. These urogenital ridges become more prominent and fuse caudally to form the genital cord. During their development, the paramesonephric ( müllerian) ducts migrate until they lie side by side in the midline. These paramesonephric ducts develop initially at the cranial ends where funnel-shaped openings communicate with the future peritoneal cavity. As these ducts develop caudally, they cross the ventral aspect of the mesonephric ducts to fuse in the midline, resulting in a Y-shaped tubular structure. The caudal portion of this fused paramesonephric duct forms an elevation known as the müllerian tubercle, which protrudes into the urogenital sinus. The caudal ends of the mesonephric ducts enter the urogenital sinus on either side of the müllerian tubercle. Under the influence of the ovarian hormones (and due to the lack of male hormones in females) the paramesonephric ducts continue their development to form the genital tract, while the mesonephric system regresses except for certain vestigial structures. The anterior or cranial portions of the paramesonephric duct system develop into the oviducts or fallopian tubes; the middle portion develops into the body of the uterus, while the caudal fused portions become the primordium of the cervix and the upper vagina. With further development they fuse to form a double-barreled organ which terminates in the müllerian tubercle located in the dorsal portion of the urogenital sinus. In the early embryo, sexual differentiation is not apparent until the tenth week of development. After this time the mesonephric or wolffian ducts regress in the female, leaving a few vestigial fragments, chiefly in the broad ligament. The paramesonephric or müllerian ducts continue their developmenl to form vagina, uterus, and tubes (Table 1).
TABLE 1. Timetable of Development of the Female Genital Tract
Size (mm) | Age (wk)* | |
0.08 | 7.5 days | Bilaminar disk embryo |
0.21 | 13 days | Yolk sac forming, hindgut, foregut |
0.36 | 20 days | Allantois formed |
1.5 | 2+ | Gonocytes are located in yolk sac entoderm |
Urogenital ridge begins to form | ||
2 | 3 | Wolffian duct appears as coalescence of pronephric ducts |
Allantois well formed, joins hindgut at cloaca | ||
Cloacal membrane forms as cloaca contacts hind-end ectoderm | ||
4 | 4 | Wolffian ducts open into urogenital sinus (cloaca) |
5 | Primordium of gonad appears | |
6 | 4+ | Primordial germ cells in mesenchyme |
7 | 1 mo + | Indifferent gonad developing |
5 | Wolffian ducts open to urogenital sinus | |
Ureteric buds | ||
External genital tubercle forming | ||
8 | Sex cords budding from coelomic epithelium | |
Anlage of adrenal cortex forms | ||
Urorectal septum starts partition of cloaca | ||
10 | Free ends of sex cords form rete testis | |
11 | 5.5 | Müllerian funnel and duct begin to form |
Primary bifurcation of ureteric bud | ||
Sex difference in external genitalia: urethral groove shorter in | ||
female | ||
Urethral folds develop lateral to central groove | ||
15 | 5 ¾ | Differentiation of gonad into testis in male |
Ureter opens separately from wolffian duct into urogenital sinus | ||
16 | 6 | Cloaca subdividing; urorectal septum completed |
Phallus forming from genital tubercle | ||
18 | Secondary bifurcation of ureteric bud | |
Urethral membrane (and cloacal membrane) perforate | ||
20 | 7 | Gonad recognizable as ovary; cortex forming |
Labioscrotal folds developed (lateral genital swellings) | ||
23 | 8 | |
25 | Coronary sulcus of phallus appears | |
Female phallus shows caudal curvature | ||
30 | Interstitial cells recognizable | |
32 | 9 | Müllerian ducts reach urogenital sinus |
Müllerian tubercle formed | ||
42 | Lower Müllerian ducts fused | |
45 | 10 | Wolffian ducts begin to regress in female |
Coronary sulcus of phallus forms groove (easily recognized) | ||
50 | Posterior commissure formed | |
Great increase of Leydig cells from mesenchyme | ||
Prostatic buds first appear in male | ||
56 | 11 | Sex differences in external genitals first noticeable |
Uterovaginal canal formed, fusion of Müllerian ducts with | ||
sinovaginal | ||
bulbs | ||
65 | Wolffian ducts begin to disappear | |
Longitudinal folds in urogenital sinus | ||
Sinovaginal bulbs formed, obliterate Müllerian tubercle | ||
70 | 12 | Beginning solidification of uterovaginal canal by “proliferation of |
both Müllerian and sinovaginal” epithelium | ||
94 | Sinus upgrowth along vaginal plate | |
100 | 16 | Wolffian ducts have disappeared by this stage |
External genitalia of male fully formed | ||
125 | Fetal ovarian stroma differentiates | |
150 | Primordial follicles in central part of ovary | |
Vagina canalizes from urogenital sinus forward | ||
Maximum development of Leydig cells | ||
175 | 20 | Beginning shrinkage and degeneration of Leydig cells |
180 | Vagina fully canalized; glycogenated epithelium | |
187 | Seminiferous tubules coiled; lumina | |
200 | 24 | |
240 | 7 mo (early) | Testes descend into scrotum |
250 | 28 | During cortical differentiation, medullary cords crowded centrally; |
some medullary tubules provided with germ cells | ||
persist in hilum | ||
286 | 32 | Reduction in Leydig cells (male) |
Disappearance of medullary cords in ovary (female) |
*Except as otherwise noted.
From Dougherty CM, Spencer R: Female Sex Anomalies. Hagerstown, MD: Harper & Row, 1972, p
The cumbersome terms “mesonephric”(wolffian) and “paramesonephric”( müllerian) have always been difficult to keep clearly differentiated. Mesonephric means “middle-kidney”(pronephric elements are not a factor in human development). The eponym “ müllerian duct”is deeply ingrained in the literature as relating to uterus, tubes, and vagina, while the term “paramesonephric,”meaning “‘alongside’ the middle kidney system,”is less difficult to remember. The concept of the paramesonephric ( müllerian) duct system as a double-barreled organ (suggesting a shotgun) is useful in understanding developmental anomalies, since various growth arrests and failures of development at this stage explain many of the anomalies encountered.