The human umbilical cord must reflect some critical evolutionary compromise; otherwise, it
is hard to see the advantages of having the fetal
lifeline to its placental respiratory and nutritional source be merely
two arteries and a single vein. These vessels are packaged in a cord
up to 70-plus cm long that floats in a fluid space that becomes progressively
smaller throughout gestation. Finally, this fluid space may contain
substances potentially noxious to fetoplacental hemodynamics, such
as cytokines and possibly meconium. That cord pathology is comparatively
infrequent speaks to the cord's remarkable resilience. The umbilical cord epithelium is continuous with amniotic epithelium and
tightly adherent to the underlying Wharton's jelly. The cord has
no vasa vasorum, lymphatics, or nerves. Mast cells in the compressible
Wharton's jelly may protect against umbilical thrombosis. The
Wharton's jelly cell population includes many histiocytes, cells
that are facultative macrophages. After meconium exposure or cord trauma (e.g., cordocentesis), the cells may phagocytose debris and become
pigment-laden. The vascular muscle is a decussating helix of smooth muscle fibers. The
umbilical vein has a well-developed internal elastic lamina; the arteries
have elastic tissue only in the media. The umbilical arteries have
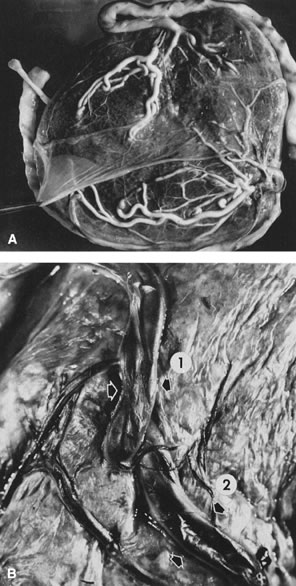
been described to anastomose within the first 2 to 3 cm of cord proximal
to the chorionic plate.24 Such an anastomosis would facilitate uniform placental perfusion. This
Hyrtl's anastomosis is usually at the site of cord attachment to
the chorionic disk, but it may be on the chorionic plate between smaller
branches of the chorionic arteries. We dissected umbilical arteries
in 69 consecutive term deliveries and found a variety of anastomotic
patterns. In 9 of the 69 (13%), the umbilical arteries completely fused
to form a common channel. In 28 (41%), a transverse anastomosis was
found within 2 cm of the chorionic insertion. Five cases (7%) showed
an anastomosis on the chorionic plate distal to an umbilical arterial
branch point. In six cases (9%), a visible anastomosis was not probe-patent.25 Any communication that allowed the potential for pressure and flow equilibration
between the umbilical arteries would allow greater capacity
for ventilation-perfusion matching, for the placenta to redistribute
blood away from focal placental lesions to better-perfused areas. Variance
in anastomotic patterns may explain the variability of fetal compromise
when focal placental injury such as infarct and abruption occur. However, measurable
differences between umbilical arterial Doppler waveforms
are both uncommon and not clearly associated with perinatal compromise.26 Recently, the Doppler resistance index before and after this anastomosis
was studied at a median gestational age of 33.1 weeks (range 25.5 to 40.3 weeks).26 Anastomosis blood flow was always unidirectional toward the umbilical
artery with a lower resistance index. When the cord insertion was marginal
or velamentous, umbilical arteries were more likely to be fused, but
in all cases, the anastomoses served to equalize pressures between
the umbilical arteries. The authors suggest that this provides a “safety
valve” against arterial compression that permits persistent
perfusion of the complete placental disk despite cord compression.27 The majority of vascular resistance lies in the placenta itself;28 maintenance of low umbilical arterial resistance is important for normal
fetoplacental hemodynamics. After transit through the placental capillary
bed, blood pressure has continued to drop so that by the time blood
reaches the common umbilical vein, its flow is nonpulsatile.28 Pulsatile umbilical venous flow has been associated with poor pregnancy
outcome,29 especially in the growth-restricted fetus.30 Umbilical blood flow tends to increase in proportion to fetal weight, about 100 to 130 mL/kg
per minute between 14 and 28 weeks of gestation.28 Cord length also increases with increasing fetal weight.31 Other factors associated with increased cord length include parity,32 the size of the uterine environment,33 increased fetal movement,33 male gender,33 and possibly genetics, as there may be an increased risk of recurrent
long umbilical cords in women with a past pregnancy with a long umbilical
cord.31 Decreased umbilical cord length is associated with decreased fetal movements
from any cause dating from early in gestation, including Down syndrome, skeletal
dysplasias, central nervous system lesions that impair
fetal movement, amnion bands (in which fetal movement may be constrained
after amnion rupture), and uterine structural malformations and
multifetal gestations, in which the uterine environment is constrained
or crowded.31 The association of oligohydramnios with a shorter cord is inconsistent,34 likely reflecting different times of onset and severity of oligohydramnios. Umbilical
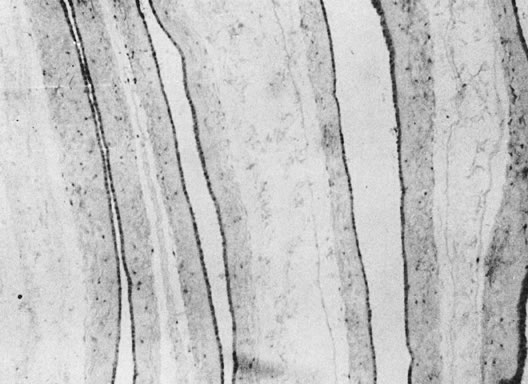
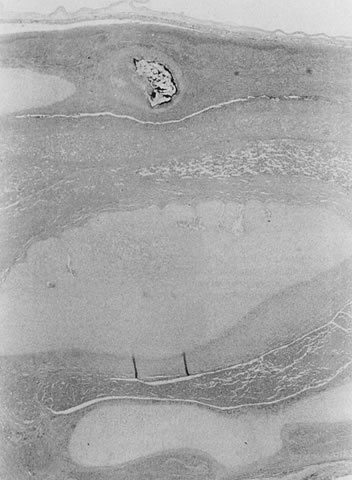
coiling (Fig. 3) may serve to enhance cord hemodynamics, as arterial pulsations transmitted
to the vein may help pump blood back up the cord from the placental
capillary bed. Both abnormally straight (uncoiled) cords and excessively
coiled cords are associated with an increased risk of adverse perinatal
outcome.35 Risk factors for abnormal vascular coiling were extremes of maternal age
for hypercoiling, and obesity, gestational diabetes mellitus, and preeclampsia.35 An excessively long or coiled cord may be at increased risk for torsion. Passive
rotation of the fetus around the flaccid cord after death may
lead to cord twisting and, in the absence of an umbilical cord pulse, severe
stricture. Careful gross and microscopic examination and detailed
clinical correlation are necessary to rule out other causes of cord
vulnerability to torsion during life (reduced intrauterine volume/oligohydramnios, abnormal
cord insertion). To some degree, this diagnosis
may be a diagnosis of exclusion in the stillborn fetus. This has been
considered a sporadic cause of fetal compromise, but a genetic predisposition
has been recently suggested, based on a familial cluster of
three subsequent pregnancies with recurrent cord torsion and recurrent
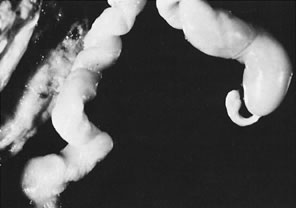
fetal death.36 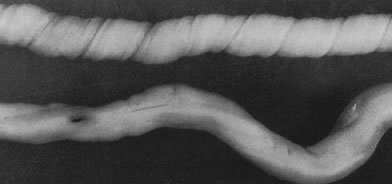 Fig 3. Normal spiraling of umbilical cord ( top) compared with one with decreased spiraling ( bottom ). Note the needle mark on the left from umbilical cord blood sampling. Fig 3. Normal spiraling of umbilical cord ( top) compared with one with decreased spiraling ( bottom ). Note the needle mark on the left from umbilical cord blood sampling.
|
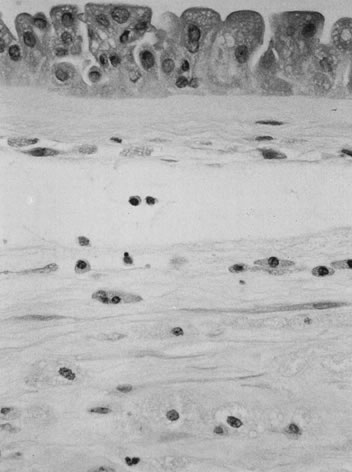
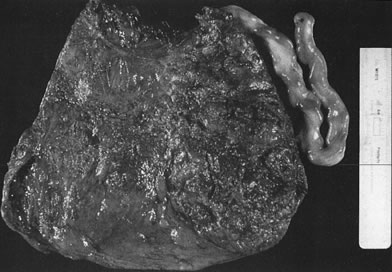
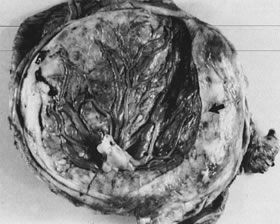
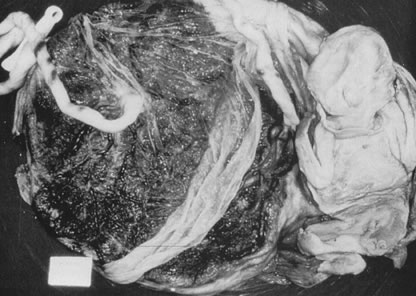
Velamentous and marginal insertions of the umbilical cord (Fig. 4) also carry potential fetal risk. When the cord is inserted on the membranes (velamentous
insertion), the chorionic arteries and vein must travel
in the chorion laeve to reach the definitive placental disk (or
an accessory lobe). Membranous vessels must accompany velamentous cord
insertion but may be found with marginally inserted cords, with accessory
lobes, or with completely normal chorionic placentation. Vasa previa
describes the case when these membranous blood vessels lie over the
cervix, between the baby and the cervix. Even dilatation of the lower
uterine segment may put these vessels under mechanical stress, and membrane
rupture can be an immediately lethal event to the fetus if the
vasa previa are large-caliber vessels. A recent series of 18 suspected
cases of vasa previa included 10 velamentous cords and 3 cases each
of bilobed placentas, accessory lobes, and marginal cord insertion. Two
of the 18 cases ended with fetal death; the remaining infants did well
with minor complications of prematurity.37 With rupture of a large-caliber vessel, fetal exsanguination may occur
within 3 minutes. Velamentous vessels not seen over the os may still
rupture, although this is far less common. Independent of the risks of
vessel rupture, malposition of the umbilical cord insertion may compromise
fetal well-being. The placental disk is approximately twice as thick
when the maternal intervillous blood space is fully distended by
normal maternal intervillous blood flow than after delivery. Even after
delivery, the chorionic plate is a “springy” flexible structure, cushioning
the vessels coursing in it. By contrast, velamentous
vessels are adjacent to the rigid myometrial wall, a structure without
a great deal of “give.” Velamentous vessels are much more
susceptible to compression and other types of mechanical trauma. In
twin pregnancies, the velamentous cord insertion is more common in the
smaller of the siblings, and the donor of the pair in cases of twin-transfusion.38 Sections through traumatized but unruptured velamentous vessels may show
necrotic smooth muscle cells, mural thrombi, and mixed vasculitis. A
marginally inserted umbilical cord may also be at risk because of the
mechanics of the angle at which the extraplacental membranes join the
chorionic disk. Intra-amniotic pressure maintains this angle open. Once
the membranes rupture, this angle may buckle, destabilizing the umbilical
cord. We have observed cases of acute fetal decompensation at
spontaneous or artificial membrane rupture, or after internal cephalic
version with a marginal umbilical cord insertion, especially when the
umbilical cord is short. Mechanical stress can compromise umbilical cord
vessels when there is segmental absence of Wharton's jelly, focal
deficiencies of muscular wall,39 or furcate insertion. In furcate insertion, the vessels split before their
insertion into the chorionic plate and may have some investment with
Wharton's jelly. However, vessels in the furcate insertion are
more susceptible to compression, trauma, or laceration. Fetal structural
defects associated with velamentous cord insertions are typically
due to deformation of a normally formed part, probably as a result of
intrauterine molding.40 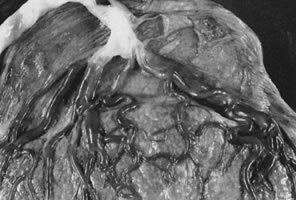 Fig 4. Placenta with velamentous insertion of cord. Membranous vessels that run
in extraplacental membranes to chorionic disk are susceptible to compression
or rupture. Fig 4. Placenta with velamentous insertion of cord. Membranous vessels that run
in extraplacental membranes to chorionic disk are susceptible to compression
or rupture.
|
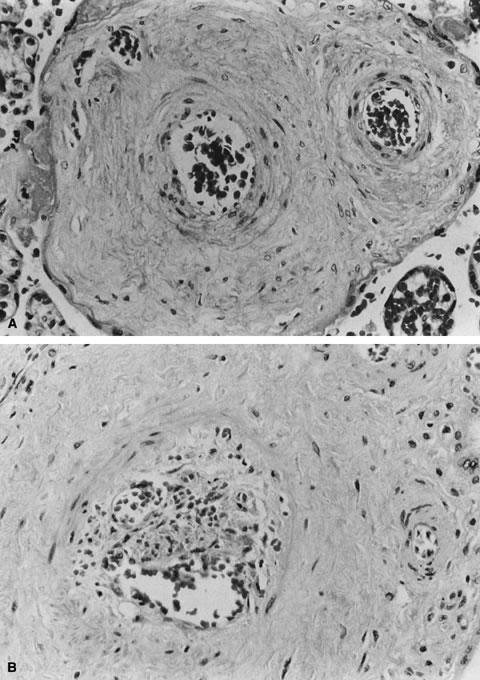
One of the risks of compression and vascular injury is thrombosis. Thanks
to the cushioning of Wharton's jelly, potentially the protective, anticoagulant
effect of cord mast cells, and the normal helical arrangement
of the vessels, cord thrombosis due to compression or mechanical
trauma is uncommon, even with extensive velamentous vessels. Spontaneous
cord thrombosis is very rare. Two recent case reports described
umbilical venous thrombosis with intrauterine fetal death, one after
abrupt onset of fetal heart rate decelerations with delivery 14 minutes
later of a nonresuscitable infant.41 Predisposing factors to cord thrombosis are unclear, but cord coiling
may be an important protective mechanism. In a recent study,42 37% of cases with overcoiled cords and 29% of undercoiled cords were cases
of fetal demise. The authors raised the possibility that abnormal
cord coiling might carry risks of neurologic sequelae to survivors. Obviously, further
study of this possibility is needed. One of us (C.M.S.) has
seen two cases in recent years of spontaneous cord thrombosis
associated with neonatal stroke, and both were in infants of mothers who
were at least heterozygous for the factor V Leiden mutation. The recommendation, based
on pathology, was that the infant and/or father be
genotyped for this mutation. Vern and colleagues43 have described avascular villi, an obliteration of the fetoplacental vasculature
in the placental microcirculation, in association with fetoplacental
heterozygosity for the factor V Leiden mutation. It may be reasonable
to obtain careful cardiovascular pedigrees from mother and father
in cases of umbilical cord thrombosis, because the potential for
hereditable thrombophilia would bear significantly on recurrence risk
assessment. Umbilical cord edema develops as a result of any forces that promote movement
of water out of the umbilical vessels and into Wharton's jelly. In
any vascular system, an increase in intravascular pressure would
lead to fluid extravasation. Therefore, it is not surprising to find
umbilical edema in the vicinity of umbilical cord knots. Umbilical
edema is also commonly seen in association with intra-amniotic infection. We
speculate that intra-amniotic inflammatory cytokines affect umbilical
epithelium permeability and/or umbilical vascular pressure to promote
umbilical cord edema. In support of the latter hypothesis, we have
observed an increase in the umbilical systolic-diastolic ratio in
cases with histologic umbilical vasculitis.44 Umbilical edema is also common in infants of a diabetic mother and may
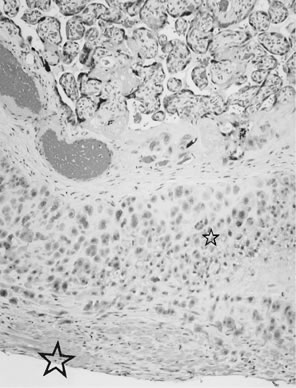
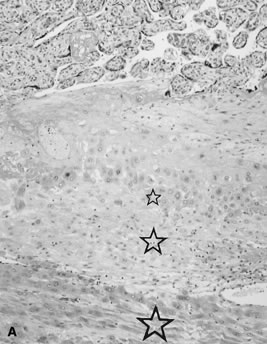
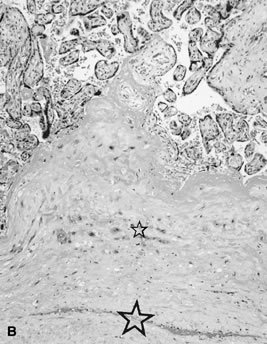
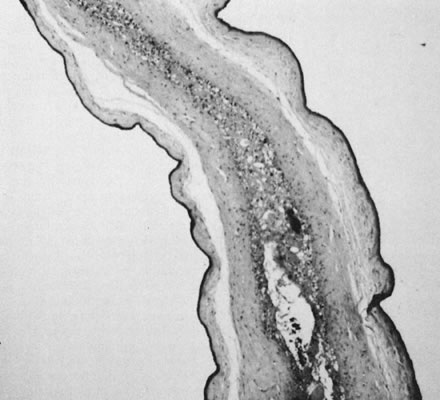
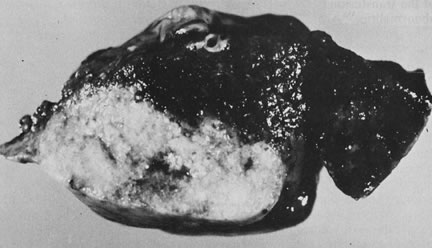
reflect the hyperdynamic state of these overgrown, plethoric infants. Absence of one artery (Fig. 5) may be due to failure of formation or to regression with a residual calcified
remnant.45,46 Embryonic remnants (most common near the fetal end of the cord) are of
little clinical significance.47 Umbilical cord length in part reflects fetal growth and in utero fetal activity.31 Umbilical cord pathology and some guides to clinical correlation are presented
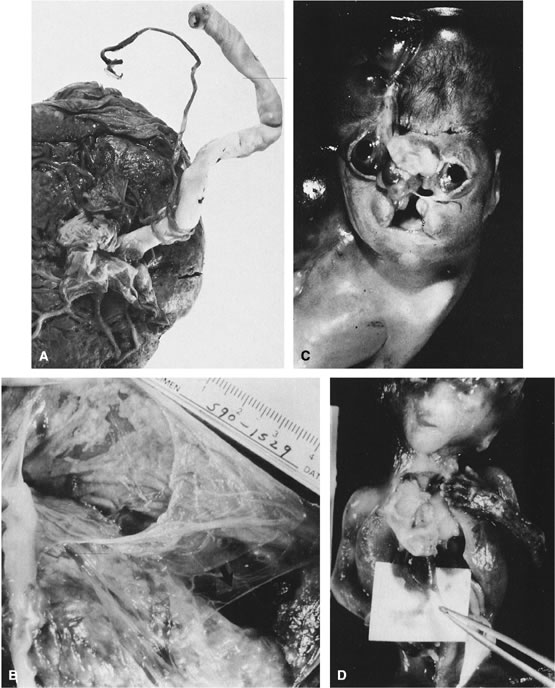
in Table 1.  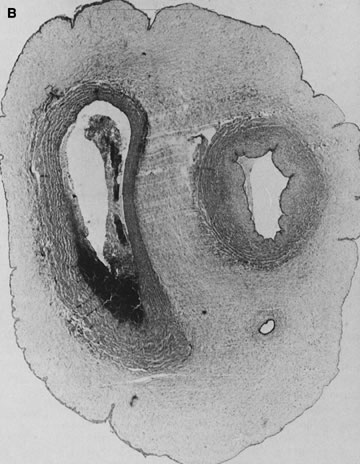 Fig 5. A. Cross-section of cord with single umbilical artery. B. On microscopic section, in addition to the single artery and vein, a small
remnant of the omphalomesenteric duct is present below the artery. This
is lined by columnar epithelium and should not be confused with
a third vessel.(Courtesy of Cynthia Kaplan, MD, State University of New York, Stony Brook) Fig 5. A. Cross-section of cord with single umbilical artery. B. On microscopic section, in addition to the single artery and vein, a small
remnant of the omphalomesenteric duct is present below the artery. This
is lined by columnar epithelium and should not be confused with
a third vessel.(Courtesy of Cynthia Kaplan, MD, State University of New York, Stony Brook)
|
Table 1. Umbilical Cord Abnormalities
Abnormality | Incidence | Significance |
Single umbilical artery | 1% | 20% congenital malformations, IUGR, small placenta,twins, trisomy 18, maternal
diabetes |
| 2.5% stillborn | |
Supernumerary vessels | Rare | Section taken within 3 cm of placental insertion, conjoined twins, remnant (vitelline
vessel) |
Short cords | ? | <32 cm, decreased fetal movement and neurodevelopmental abn, musculoskeletal
abn, difficult delivery, congenital malformations, twins, breech
presentation |
Long cords | ? | >100 cm, increased fetal movement, thrombosis, entanglements, torsions |
Edema | 2.7% | Hydrops fetalis, preeclampsia, acute chorioamnionitis |
Thin cord | 0.2% | IUGR, fetal malformations including prolapse and compression |
Omphalomesenteric duct | 1.5% | Near surface of cord, muscle in wall, intestinal-type mucosa, rarely ulceration
of gastric mucosa, ? association with Meckel's diverticulum
or intestinal atresia |
Allantoic duct | 15% | Located between the arteries, connective tissue wall,transitional-type
epithelium, ? patent urachus or cyst |
Vitelline vessels | 7% | Usually central, usually capillary-like, rarely with muscular wall, origin
of hematoma or hemangioma |
Teratoma | Rare | Benign |
Hematoma | 2% | Rupture of umbilical vein, iatrogenic most common, short cords |
True knots | 0.5–1% | Rarely tighten during labor, rarely cause of in utero hypoxia |
False knots | Frequent | Vascular redundancies, varicosities, rare thrombosis |
Thrombosis | 0.7% | Associated with perinatal death, more common in vein, reported to be more
significant in artery(ies), congenital hypercoagulable states, acute
chorioamnionitis, funisitis, vigorous fetal motor activity, commonly
associated with true knot |
Strictures | ? | Focal deficiency of Wharton's jelly, rarely due to “amniotic
bands” |
Torsion | ? | Exaggerated spiraling, including fetal movement; most occur after fetal
demise |
Decreased spiraling | ? | Single uterine artery, decreased fetal movement |
Cord rupture | 2–4% | Short cord, velamentous or marginal insertion |
Marginal insertion | 5–7% | Compression of vessels, thrombosis, rupture, twins |
Velamentous insertion | 0.3–2% | Compression of vessels, thrombosis,avulsion of cord, rupture of vasa previa, twins, diabetes, congenital malformation |
Cord prolapse | 2–4% | Long cords, multiple gestations |
Funisitis | ? | Fetal reaction to infection or cord compression, margination, angiitis, funisitis (neutrophils
in Wharton's jelly), rare before 26 weeks, candidiasis, plaques
on surface |
Necrotizing funisitis | rare | Syphilis, herpes, toxoplasmosis, often associated with villitis and chorioamnionitis |
Calcification | Rare | Necrotic inflammatory debris muscle fibers, etiology known |
Muscle degeneration | Rare (livebirths) | <1% of meconium-stained placentas, autolysis, begins at fetal end of
cord |
| Common | After fetal demise |
abn, abnormalities; IUGR, intrauterine growth retardation.
|