Disturbance in Maturation: Hemoglobin Synthesis IRON DEFICIENCY ANEMIA. IDA accounts for 75% of all the anemias diagnosed during pregnancy.4 Iron depletion without signs of anemia is very common during pregnancy. In
one study, iron stores were found to be exhausted in 25% of young, apparently
healthy women on their first prenatal visit. Another study
demonstrated that 80% of normal pregnant, iron-sufficient women if not
given iron supplementation were anemic by term. Finally, it has been
estimated that 20 million persons in the United States are iron depleted, particularly
in the socioeconomically disadvantaged population. Any
discussion of iron deficiency necessarily includes an understanding
of iron absorption, metabolism, and loss.5 Iron is necessary for RBC formation but is also intimately involved with
proteins important in intermediary metabolism. Nearly half the enzymes
and cofactors in the Krebs cycle either contain iron or require its
presence. Iron Compartments. The basic compartments of iron distribution include hemoglobin iron, storage
iron, myoglobin iron, labile pool iron, other tissue iron, and
transport iron. Table 2 illustrates the significant differences in the total body iron distribution
between the adult male and the female during the reproductive years.6 Pregnancy normally increases the amount of iron but not the percentage
distribution to each compartment; however, socioeconomic status, nutritional
status, and concurrent disease processes can modify the dispersion
of iron in each compartment. TABLE 2. Iron Compartments*
Iron Content (mg) |
Compartment | Male | Female† | Total Body Iron (%) |
Hemoglobin iron | 2500 | 1700 | 67 |
Storage iron | 1000 | 700 | 27 |
Myoglobin iron | 130 | 130 | 3.5 |
Labile pool | 80 | 80 | 2.2 |
Other tissue iron | 8 | 8 | 0.2 |
Transport iron | 3 | 3 | 0.08 |
* These values represent estimates for an iron-sufficient person.
† Nonpregnant.
Hemoglobin Iron. Assessments with radioactive tracers have shown that hemoglobin iron makes
up 65% to 70% of total body iron and averages 1700 mg in a normal
adult woman. During pregnancy, hemoglobin iron is increased by 20%. Hemoglobin
contains 0.34% of iron by weight; thus, 1 ml of packed red cells
contain approximately 1 mg of iron. Therefore, iron loss or gain
can be calculated if the change in RBC mass is known. It is principally
the hemoglobin iron that is reflected in the laboratory test such as
PCV or hemoglobin concentration. Initially, the storage iron compartment
must be depleted before clinical signs or a change in laboratory assessment
reveals actual iron deficiency in hemoglobin content. Storage Iron. Storage iron exists in two forms: ferritin and hemosiderin. In healthy, iron-sufficient
women, it totals approximately 600 to 800 mg and makes
up approximately 27% to 30% of total body iron. Depletion of this storage
compartment occurs when iron loss or use exceeds iron absorption; it
is usually decreased during pregnancy even in iron sufficient women. Ferritin contains approximately half the storage iron and is found in plasma
as well as in most of the cells in the body; several varieties of
ferritin have been demonstrated by electrophoresis and isoelectric focusing. Ferritins
indigenous to many organs and to reticuloendothelial
cells have been identified, supporting the hypothesis that these iron
proteins are organ specific and are products of different genotypes. Ferritin
has been identified by electron microscopy as a water-soluble
complex with a molecular weight of approximately 460,000, which is 20% iron. Stereochemically, it exists as a complex of ferric hydroxide
and the protein carrier apoferritin. Each apoferritin shell houses a core
of ferric hydroxide and phosphate ions, which are dispersed in a rigid
latticelike arrangement. The role of ferritin in iron absorption
is direct; a low concentration of this compound in the intestinal mucosal
cell enhances the biosynthesis of additional apoferritin, which results
in more iron absorption. The life span of apoferritin is only a
few days; the degeneration and resynthesis provide an available intracellular
iron pool. The measurement of serum ferritin has become one method
to delineate iron stores without having to resort to bone marrow
sampling. Hemosiderin is found only in cells of the reticuloendothelial system, such
as those of bone marrow, liver, and spleen (about one third in each
organ). Immunologic studies have shown that the protein components of
both ferritin and hemosiderin are identical; hemosiderin may represent
a partially denatured ferritin. Hemosiderin is approximately 25% to 30% iron
by weight, is water soluble, and, in contrast to ferritin, can
be seen with the light microscope. It represents about 50% of the storage
iron and about 12% to 15% of the total body iron. Hemosiderin represents
that quantity of iron demonstrated by Prussian blue stains of
bone marrow aspirates. This storage iron is attached to a substrate
called apohemosiderin, which is amorphous, thus lacking the firm, crystal
like molecular arrangement of ferritin. The mechanism of mobilization, use, and
contribution of storage iron in an anemia process is discussed
in the sections on iron absorption and homeostasis. While the measurement
of ferritin is involved with an assessment of iron absorption
capabilities, the assessment of hemosiderin is a measure of iron balance; until
hemosiderin becomes depleted, no signs of iron deficiency
develop (Fig. 5). Therefore, as long as storage iron is present and released normally, no
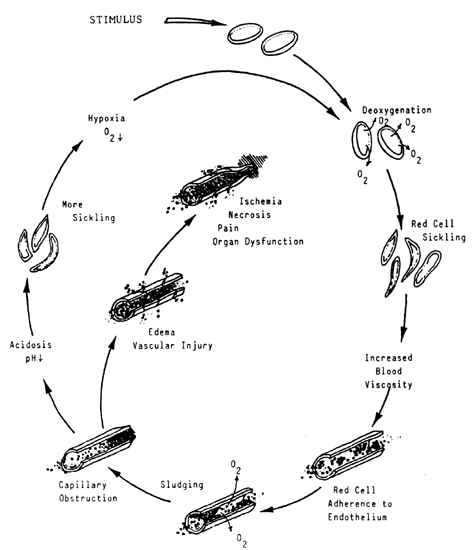
change in the peripheral blood smear is seen. 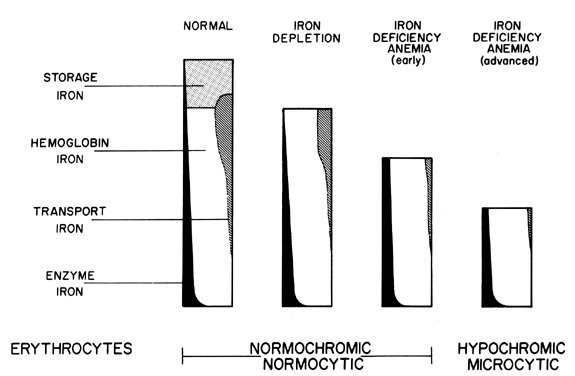 Fig. 5. Relationships of known iron compartments to various stages of clinical
iron depletions as depicted in the cellular appearance of the RBC. Fig. 5. Relationships of known iron compartments to various stages of clinical
iron depletions as depicted in the cellular appearance of the RBC.
|
During pregnancy, the increase in RBC mass gives an indirect measurement
of iron stores. The principal cause for a failure of the expansion of
the PCV is the absence of bone marrow hemosiderin. This finding indicates
exhaustion of storage iron, its absence being the earliest sign
of iron deficiency. If IDA is clinically evident by a decreased hemoglobin
concentration; by a characteristic microcytic, hypochromic blood
film; and by altered RBC indices, the iron stores will be nonexistent. On
the other hand, bone marrow stores decrease to minimal levels during
pregnancy, even in iron sufficient women, in the absence of iron supplementation. Since
a bone marrow examination is not practical during
pregnancy unless severe hematologic disease is suspected, reductions
in serum ferritin, PCV, hemoglobin concentration, and RBC indices are
the first laboratory indications of inadequate iron stores. Iron stores
are reduced in IDA, blood loss, and nutritional anemias; however, hemolytic
processes, hereditary anemias, and ineffective iron utilization
during infection or inflammation may be associated with normal to increased
storage iron levels and concomitant reductions in hemoglobin levels, PCV, and
RBC indices. Myoglobin Iron. Myoglobin iron accounts for approximately 3% to 4% of total body iron (130 mg) and
is relatively constant between men and women, even in pregnancy. Each
myoglobin molecule consists of a heme moiety surrounded by
a helical chain of 150 amino acids. The myoglobin is less than 1% iron
and has a molecular weight of approximately 17,000. It is found in
most muscle cells and appears to serve as an oxygen reservoir when cellular
damage from hypoxia occurs. Function of this iron component peculiar
to pregnancy has not been reported. Labile Iron Pool. The labile iron pool represents approximately 80 mg of iron constantly
in exchange between the plasma, interstitial, and intracellular compartments. An
intracellular protein produced by the liver appears to be
responsible for short-term binding and release of the labile iron. This
protein has been termed acetate-extractable ferroprotein and has a molecular
weight of approximately 12,000. It has been isolated from cells
in the lung, liver, intestinal mucosa, spleen, and kidneys, as well
as from RBCs. Iron kinetic studies have shown that approximately 80% of
this transitional protein eventually is reincorporated into hemoglobin
and that the remaining 20% is associated with storage compounds. The
labile iron pool offers a method of studying the clearance rate, incorporation
data, and daily iron turnover; ferrokinetic studies indicate
that in IDA the incorporation of iron into hemoglobin and subsequently
into normal RBCs is almost 100%. In contrast, in hemolytic or megaloblastic
anemia, the incorporation of iron is rapid and complete, but
there is ineffective erythropoiesis and early destruction of the erythroblasts. Patients
with aplastic anemia or thalassemia also demonstrate
an incorporation rate that is markedly reduced in the presence of normal
erythrocytes. Enzymatic Iron. Tissue iron is constant during pregnancy and represents approximately 6 to 8 mg
of iron, or between 0.2% and 0.5% of total body iron. Although
extremely small, this compartment is very important because it includes
enzymes such as the cytochrome peroxidase catalase dehydrogenase, as
well as acyl-CoA and various other oxidases. Changes in enzymatic
iron are reflected in decreased efficiency of the mitochondrial systems; thus, severe
anemias may affect fetal growth and development by such
a mechanism. In addition, maternal-fetal steroidogenesis and host response
to various disease states may be modified by functional changes
in these critical enzyme systems. As shown in Fig. 5, the amount of parenchymal iron is altered only in severe IDA, and it
is one of the last compartments to become depleted. Transport Iron. Transport iron represents less than 0.1% of the total iron (3 mg) in both
men and women, and yet, kinetically, it is the most active compartment, being
replaced approximately every 2.5 hours. The metal in this
category represents the interchange between all compartments previously
mentioned and is not altered during pregnancy. Transport iron is loosely
bound to a specific protein (i.e., transferrin), as shown in Fig. 6. Transferrin migrates electrophoretically with the β-globulins and
has a molecular weight of approximately 80,000; it is called apotransferrin
when not bound to iron. Although the kinetic properties of each
apotransferrin are similar, cellular specificity is evident, since 19 genetically
distinct molecular variants have been described. It is synthesized
principally by the liver but also by other tissues of the reticuloendothelial
system and is the only iron form that shows diurnal
variation; the highest values are obtained in the morning. 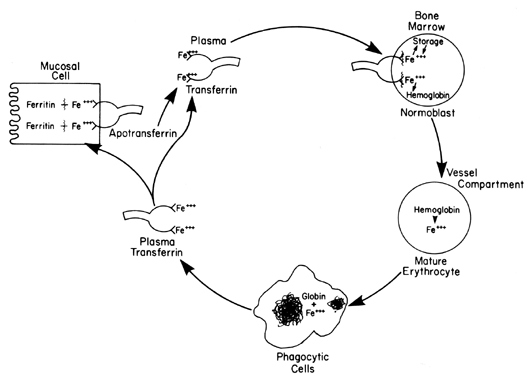 Fig. 6. Transportation sequence of iron involving intestinal absorption, marrow
hemoglobin incorporation, RBC destruction, return to labile iron pool, and
storage deposition. Fig. 6. Transportation sequence of iron involving intestinal absorption, marrow
hemoglobin incorporation, RBC destruction, return to labile iron pool, and
storage deposition.
|
A trivalent iron atom may be bound at both ends of the polypeptide chain (see Fig. 6), and binding affinity is increased when one site is occupied. Transferrin
receives absorbed iron and transports it to the immature marrow normoblasts. The
transferrin in the plasma in iron-replete subjects is
one third saturated, leaving two thirds unoccupied; this is measured as
the unsaturated iron binding capacity (UIBC). When the serum iron is
decreased, the UIBC is increased and vice versa. Iron Absorption. Iron absorption depends on many factors (Table 3), reflecting the diet, the status of the bowel lumen, and mucosal abnormalities, as
well as systemic factors.7 Iron is principally absorbed in the duodenum and proximal small intestine. The
iron presented to the gastrointestinal tract is usually in one
of three forms: the ferrous form (from elemental iron), hemoglobin (from
animal protein sources), and the trivalent or ferric form (from vegetable
complexes) (Fig. 7). The ferrous salts are best, since they need no conversion to be absorbed; the
ferric iron in vegetable protein complexes must be reduced to
the divalent state before it can pass into the mucosal cell. Hemoglobin
iron is readily absorbed after being hydrolyzed in the gut lumen into
heme and globin. The globin, although degraded by intestinal enzymes
into small peptides, remains an integral factor in absorption, since
it continues to stabilize the heme iron in the ferrous state. TABLE 3. Conditions Affecting Iron Absorption
| Increased Absorption | Decreased Absorption |
Iron content |
Form of iron | Heme iron | Enteric-coated capsules |
| Adequate ferrous salt | |
| Iron deficiency | |
Intraluminal factors |
Intestinal secretions | Hydrochloric acid | Achlorhydria |
| Bile | |
| Intrinsic factor | |
Stomach contents | Ascorbic and other acids, | Oxalates, phytates, phosphorus, |
| cysteine | carbonate |
Intestinal motility | Atropine | Cathartics |
Chelators | | EDTA, desferrioxamine |
Metallic cations | | Cobalt, nickel |
Mucosal factors |
Disease states | Intermittent outlet obstruction | Gastrectomy, lymphoma |
| | Chronic diarrhea (sprue) |
Cellular | Decreased mucosal iron | Increased mucosal iron |
Systemic factors |
Erythropoiesis | Acute blood loss | Aplastic anemia |
| Hemolytic anemia | Transfusion, chronic infection |
| Hypoxia | |
Iron requirements | Pregnancy, growth | Weight loss, thalassemia |
| Iron deficiency | Hemochromatosis | 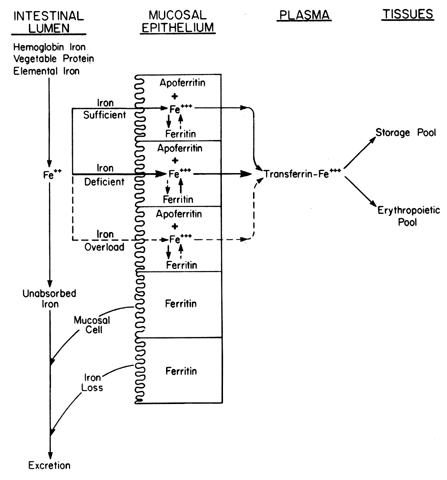 Fig. 7. Mechanism of iron absorption from the intestinal lumen through the mucosal
epithelium and into various iron compartments. Fig. 7. Mechanism of iron absorption from the intestinal lumen through the mucosal
epithelium and into various iron compartments.
|
Gastric acid degrades the organic iron complexes (vegetable protein) and
reduces the trivalent mineral to ferrous iron. Moreover, hydrochloric
acid serves to prevent alkaline dietary constituents from forming insoluble
complexes with ferrous iron, to prevent various agents from oxidizing
iron to the ferric state, and to prevent iron precipitating as
insoluble ferric hydroxide. While gastric juice may facilitate elemental
iron absorption, it does not appear to have any effect on hemoglobin
iron absorption. The alkaline milieu of the duodenum and upper small
bowel favors absorption, since the ferrous iron has greater solubility
in these surroundings; this is true for all three sources of iron, but
it is particularly necessary for hemoglobin iron. Therefore, agents
that form alkaline-soluble complexes promote iron absorption; these
include the reducing agents ascorbic acid, fructose succinic acid, lactate, pyruvate, and
sorbitol and certain amino acids such as cysteine. Certain
drugs such as hydroquinone and alcohol may also increase iron
absorption by increasing gastric acid production, while medications that
decrease intestinal motility, such as atropine or reserpine, also
enhance iron absorption. Bile may have a limited role in facilitating
iron absorption, but pancreatic secretions are thought to be noncontributory. In contrast, oxidants and certain medications reduce iron absorption by
increasing the concentration of the trivalent form. Other substances
form insoluble complexes with the ferrous iron; chelators have a similar
mode of action. Metallic cation competitors decrease absorption by
competing for binding sites, while other agents reduce absorption by reducing
gastric acidity. Cathartics and disease processes reduce iron
absorption by increasing intestinal motility, reducing the time of exposure
of mucosal cells to iron. Surgical removal of the gastric or duodenal
mucosa limits the surface area for absorption. Finally, another factor, pica, has long been associated with IDA. Pica
is the ingestion of various substances that have no dietary value; pagophagia (ice), geophagia (clay), and amylophagia (starch) are examples. In
some ethnic groups, it is a common associated finding in over 60% to 80% of
patients with proved IDA. The “cravings” these
patients experience are not always for nonfoods, but may be for vegetables (carrots
or celery) or commercial items such as “fast foods” (invariably
not a good source of iron). Formerly all forms of
pica were thought to retard absorption of iron. Now it is clear that pica
is a sign of IDA rather than a cause. A history of pica in an adult
suggests that the patient has IDA, since the abnormal craving usually
disappears in a few days after the institution of iron supplementation. It
is not proved but may be inferred that certain food fetishes during
pregnancy may represent pica due to the increased iron demands of
pregnancy. This hypothesis is supported by the observation that most
pica occurs after the 20th week, a time during which the iron demands
increase most rapidly. The relationship of pica to IDA is not completely
understood; however, it seems to be related to a decrease of iron-containing
enzymes in the oral mucosal cells of these patients. The regulatory mechanism of iron uptake is closely related to the plasma
iron turnover rate (see Fig. 6). It appears that transferrin from the labile pool is deposited into the
developing mucosal cells destined to form the basal layer of the crypts
of Lieberkühn in the duodenum. This protein controls the amount
of iron absorbed by trapping the metal entering the cell. The conversion
of the absorbed ferrous iron into ferritin is a metabolic process
that requires a chelator, such as ascorbic acid, an energy source (probably
adenosine triphosphate), and, finally, oxidation of the divalent
iron to the ferric iron, which requires ceruloplasmin (see Fig. 7). This sequence is adequate for elemental iron and vegetable protein iron
absorbed as the simple ferrous cation. However, hemoglobin or myoglobin
iron must be further degraded by a microsomal enzyme called heme
oxygenase. Once the mucosal cells are saturated with ferritin iron absorption
is halted. If the iron stores are reduced, very little ferritin
is present in the mucosal lining and iron is absorbed rapidly through
the cell, by transferrin, into the bloodstream. In contrast, during
iron sufficiency, increased amounts of cellular ferritin are present
and the iron is either stored as ferritin or is not absorbed. Ferritin
is lost as the duodenal mucosal cell matures and works its way from the
base of the crypt to the luminal border. The mucosal cell control of
absorption is not, however, absolute; it can be overcome if the concentration
of iron at the intestinal lumen increases sufficiently, as in
toxic overdoses. If large amounts of iron are presented chronically, hemochromatosis
may eventually result. The transport of iron from the mucosal cell to the utilization pool is
performed by transferrin (see Fig. 6). When iron is required by the marrow, transferrin obtains trivalent iron
from the ferritin in the mucosal cells. The ferric iron binds to one
of the glycoprotein moieties of transferrin, is transported to the
marrow, and is used for hemoglobin synthesis. The transferrin binds briefly
on the normoblast membrane, where it delivers one or both iron atoms. During
the binding process, transferrin may partially enter the
cell by a process of endomitosis and actually participate in hemoglobin
formation. The release of iron is an active process that can be abolished
by enzyme inhibitors. Although direct transfer of transferrin to
the normoblast is thought to be the major pathway, rhophenocytosis (accepting
of transferrin by reticuloendothelial cells) may represent a
minor pathway. Once the iron transferrin complex is attached to the receptor
site inside the normoblast, it is conjugated to a protein molecule
with a molecular weight of approximately 20,000 and transported to
the mitochondria, where heme synthesis occurs; this process takes 8 to 10 minutes. As
the erythrocyte matures, it retains its capacity to take
up iron and synthesize heme until after the reticulocyte stage, in
which the mitochondria are lost (see Fig. 2). After the erythroid cells mature, they are extruded from the bone marrow
into the peripheral circulation and have a normal life span of 120 days. The
life cycle is complete when the red cells are destroyed and
hemoglobin degradation takes place within the reticuloendothelial system, chiefly
the liver and the spleen (see Fig. 6). Digestion of the red cells occurs within a few hours, and the plasma
iron is redistributed, 80% into new hemoglobin and 20% as storage iron. The
mechanism by which the reticuloendothelial cells transfer the iron
to transferrin appears to involve ceruloplasmin (ferroxidase), which
is also involved in the binding of iron within the mucosal cells. Iron Homeostasis. The conservation of iron in humans is tenacious, with only 0.1% of the
total amount of body iron lost each day. This amount is easily replaced
in the nonpregnant adult if the dietary source is adequate. The average
amount of iron excreted by the adult averages 0.9 mg/day, with most
being lost in the intestinal tract as desquamated gastrointestinal
cells, blood, and bile. Additionally, epidermal cell loss and sweat produce
a dally iron loss of 0.2 mg. In areas of high temperature and humidity, an
additional 0.5 mg/day may be lost, but this loss rarely produces
IDA. Finally, a small amount (0.1 mg) of iron is excreted dally
in the urine. Both sexes lose a similar amount of iron through these mechanisms; the
basal loss is approximately 14 μg/kg/day. In women during
their reproductive years, the iron losses are compounded by menses. Although
the blood loss is relatively constant in successive periods, the
individual variation between women is large. Most normal, iron-sufficient
women lose an average of 25 to 45 ml of blood through menses
each month, which approximates 0.7 to 1.4 mg/day in terms of iron loss. The
blood loss is lower in the younger age-groups, in women taking
oral contraceptives, in women with good nutrition, and in higher socioeconomic
groups. Blood loss is exaggerated in women over 40 years old, those
with intrauterine contraceptive devices, patients after tubal
sterilization, females of high parity, women of lower socioeconomic groups, and
those with poor nutrition. A menstrual blood loss of 50 to 60 ml
seems to be the upper limit of normal, since women whose losses
have exceeded this amount eventually develop IDA. Therefore, the total
basal iron loss of a woman in the reproductive years would appear to
average 1 mg for menstrual loss in addition to the 1 mg obligatory loss
experienced by men and women alike. Many factors, both physiologic and those related to disease states, can
cause a healthy woman to become iron deficient. Pregnancy has a marked
effect on iron homeostasis (Table 4).8 In healthy menstruating women, the loss of 2 mg/day can be overcome by
a daily food intake: of 1800 to 2200 calories, which contain 11 to 13 mg
of iron. However, even in an iron-sufficient state, large amounts
of iron must be borrowed from iron stores to complete a pregnancy (Table 4). During the first half of pregnancy, iron requirements are not increased; in
the absence of menses, an intake of 11 to 13 mg/day is adequate. After
the 20th week, however, the RBC mass begins to expand and the
fetus requires more iron. Even with increased absorption, the amount
of dietary iron is not adequate to prevent a reduction in iron stores. Obviously, if
dietary iron does not meet the requirements, then the storage
iron must supply the needs. Since the average North American diet
provides about 6 mg of iron per 1000 calories and the pregnant woman
of ideal weight should consume 1800 to 2200 calories each day, the average
pregnant woman should receive between 10 and 12 mg of elemental
iron daily.9 Even though the absorption rate increases from 10% in the first trimester
to 30% during the latter half of pregnancy, the iron acquired from
the diet alone during this time ranges from 450 to 500 mg; thus, approximately 400 to 500 mg
must be supplied by storage iron during pregnancy. In
women who are deficient in storage iron prior to pregnancy, this
further requirement may lead to overt IDA. It should also be noted
that if the storage iron is insufficient at the beginning of pregnancy, the
maternal hemoglobin mass will not be expanded until the fetal demands
are met. The lack of expansion of the RBC mass thus may be an indication
of inadequate iron stores. Post partum, the amount of iron in
the expanded hemoglobin mass not lost at delivery can be returned to
the iron stores and the anemia can be partially balanced. However, in
most women this is not sufficient replacement, and almost all women will
remain deficient in storage iron unless they receive supplementation
during gestation. TABLE 4. Iron Homeostasis in Pregnancy,*Expressed in Milligrams of Elemental
Iron
Maternal Loss | Maternal Gain | Borrowed |
(Source) | (Source) | From Stores |
500 (RBC mass | 490 (diet)† | |
expansion) | | |
300 (fetal/placental) | 270 (returned to storage | |
| after delivery) | |
190 (basal loss) | | |
230 (loss at | | |
delivery) | | |
TOTAL: 1220 | TOTAL: 760 | 460 |
* Lasts 20 weeks.
† Average of 3 to 4 mg/day actual absorption.
Laboratory Assessment. The laboratory diagnosis of IDA depends on the severity of iron depletion. In
the mildest stage, iron deficiency is manifested by a decrease
in serum ferritin, but serum iron, PCV, and hemoglobin values are usually
normal (see Fig. 5). Iron deficiency without anemia is the next stage; absence of storage
iron, manifested by reduced serum ferritin, low serum iron, and decreased
transferrin saturation without anemia are characteristic. IDA is
first reflected by a reduced RBC mass, then reductions in PCV and hemoglobin
levels following hypochromasia and microcytosis (see Fig. 5). Iron assessment tests must be altered during pregnancy. A relative decrease
in serum iron occurs from approximately 12 weeks through 32 to 34 weeks
owing to the increase in PV. However, as the RBC mass approaches
the increase in PV, the serum iron rises to normal nonpregnant levels. During
the early postpartum period, serum iron again rapidly decreases
over the first 4 to 5 days before returning to normal at the end of
the first week. This is probably related to ineffective release of storage
iron owing to the change in hormonal milieu. Transferrin begins
to increase from 12 weeks through 34 to 36 weeks, but a slight decrease
occurs toward term. During the first 7 days after delivery, the transferrin
concentration increases before it returns to normal levels approximately 10 days
post partum. These changes are also thought to be
hormonally mediated, since similar changes have been observed in women
taking oral contraceptives. These observations are present only in iron
sufficient pregnant women and are not significant enough to affect
the diagnosis of IDA. The most consistent findings in the blood of a patient with IDA is a decrease
in the PCV and hemoglobin concentration, with concomitant hypochromia
and microcytosis observed in the blood film. Serum iron, serum
ferritin, UIBC, and transferrin saturation may be used to confirm IDA, although
they are not routinely obtained during antepartal screening.10 IDA is usually suspected if the serum iron is below 60 μg/dL, the serum
ferritin is below 30μ g/L, the transferrin saturation is below 20%, and
the UIBC is above 350 μg/dL; the normal values are shown
in Table 1. Values of less than 30 μg/dL, less than 10 μg/L, less than 10%, and
greater than 400 μg/dL in the serum iron, ferritin, transferrin
saturation, and UIBC, respectively, are diagnostic of IDA. Some authors
have advocated further evaluation in an attempt to detect early iron-deficient
states; however, these have yielded limited clinical information. In iron-deficient states, zinc may replace iron in the protoporphyrin ring; thus, the
measurement of RBC zinc protoporphyrin (RBC ZP) may be
an accurate predictor of IDA. This test is relatively rapid and is not
costly, although it is not specific for IDA. In addition, the measurement
of iron chelating agents such as desferrioxamine, followed by measurement
of mobilized iron stores excreted in the urine, has been of value
in some cases. The measurement of iron stores by nuclear magnetic
resonance may also be helpful in cases of early IDA. In mild cases of
IDA, the measurement of free erythrocyte protoporphyrin (FEP) is increased
fivefold. Measurement of FEP has been advocated as a screening test
for IDA, but the results are not related to the severity of the anemia, nor
is it specific for IDA. Differential Diagnosis. Although iron depletion or IDA can be easily treated in most cases, it
is important to rule out more severe hematologic or systemic diseases. Hypochromic
anemia, caused by the following conditions, may be confused
with IDA: (1) chemical toxicity related to the intake of chloramphenicol, lead, alcohol, or
isoniazid; (2) inflammatory processes; (3) malignancy; (4) pyridoxine-responsive anemia; and (5) hemoglobinopathies. Drug effects may be toxic (lead, alcohol) or idiosyncratic (isoniazid). Maternal
encephalopathy and basophilic stippling of the normal red cells
may be observed with lead poisoning. On the other hand, toxic dosages
of alcohol or reactions to drugs (chloramphenicol) usually cause a
deficiency in porphyrin synthesis, which leads to sideroblastic anemia. On
the peripheral smear, the red cells are hypochromic, but the presence
of target cells and ring sideroblasts help differentiate this process
from IDA. Characteristically, in toxic anemias the serum iron is
elevated and the transferrin saturation is increased; the opposite is
found in IDA. Examination of the bone marrow, if performed, reveals the
presence of ring sideroblasts, along with increased iron stores common
in porphyrin synthesis deficiencies. Chronic diseases, such as malignancies, and inflammatory processes, including
arthritis and the collagen-vascular diseases, may cause anemia, but
usually the blood film reveals normochromic and normocytic cells. Unfortunately, the
symptoms from these processes are protean, and they
may be difficult to differentiate from those of IDA if the peripheral
blood picture is similar. As with IDA, patients with chronic disorders
will characteristically have low serum iron levels. Elevated-to-normal
serum ferritins are seen in these disorders, while patients with IDA
exhibit decreased ferritin values. In those affected with inflammatory
processes, the transferrin saturation is decreased and UIBC is increased, as
seen in IDA. On the other hand, patients with malignancies
exhibit an increased transferrin saturation and decreased UIBC (see Fig. 3). Another screening device to differentiate IDA from acute and chronic
inflammation is the erythrocyte sedimentation rate. It almost always
is increased in neoplasia or acute and chronic inflammation over that
found during normal pregnancy, whereas in severe IDA it is normal. Erythrocyte
survival times are slightly decreased in IDA and may also be
shortened in chronic diseases. Infestation (e.g., hookworms), although rare in this country, must be included in the differential
diagnosis in developing countries, in world travelers, or in
areas of poor sanitation. Other causes of excess intestinal iron loss, such
as diverticulitis, intestinal cancer, and peptic ulcer disease, are
rare during the menstrual years and even more infrequent during pregnancy. Pyridoxine-responsive anemias are characterized by hypochromic, microcytic
erythrocytes with increased ring sideroblasts and prominent target
cells. Unlike IDA, serum iron levels, transferrin saturations, and serum
ferritins are increased in patients with pyridoxine-responsive anemia. The
UIBC is characteristically decreased. In hemoglobin synthesis abnormalities, such as heterozygous thalassemia, a
microcytic, hypochromic pattern may also be demonstrated, and these
abnormalities may be confused with IDA. RBC morphology usually shows
more basophilic stippling and target cells in thalassemia patients; these
are not common in patients with IDA. In a person of African or Mediterranean
origin, thalassemia should be ruled out by hemoglobin electrophoresis. Other
aids in diagnosis include the erythrocyte count, the
MCV, and the RC. Almost 85% of patients with heterozygous thalassemia
have an RBC mass greater than 5 million/mm3 despite a reduced hemoglobin concentration. In contrast, only 3% of adults
with IDA have RBC counts over 5 million/mm3 The MCV is reduced in heterozygous thalassemia; values of 55 to 70 cu μ per
cell are the rule, whereas values below 70 cu μ per cell are
very uncommon in IDA. Reticulocytosis of greater than 5% is much more
commonly noted in thalassemia patients than in those with IDA. In addition, serum
iron concentration and transferrin are not usually altered
in the hereditary anemias; however, serum ferritin levels are characteristically
normal to increased. In the final analysis, the response to iron therapy may prove to be diagnostic. This
method is commonly used in the low-risk patient and can
be harmful only in patients who have allergic reactions or in those who
have iron overload due to other hematologic diseases. In any case, in “therapeutic” diagnosis of IDA, the patient should be followed
carefully to detect iron-unresponsive anemia. If the patient with
IDA receives sufficient medication, an intense reticulocytosis should
occur between the seventh and tenth day after the initiation of therapy. A
significant increase in hemoglobin values should be evident in 3 to 4 weeks, and
the hemoglobin concentration should approach normal
values within 2 months. This may not occur during pregnancy, however, since
the increase in RBC mass and transfer of iron to the fetus may
continue the iron-depletion process. In this case, if other factors such
as inflammatory diseases, chronic infections, or hemoglobinopathies
are not suspected, the iron therapy should be continued until the end
of the pregnancy, since eventually there will be a response. Iron Therapy. Iron can be administered as elemental salt (orally), as an iron-carbohydrate
complex (parenterally), or as a blood transfusion. The oral route
is preferable. Table 5 lists the common iron preparations used for iron supplementation and the
cost of these compounds. The ferrous sulfate preparations are the least
expensive and the most logical choice for most patients.11 Other iron salts or forms of ferrous sulfate are no better absorbed and
are more expensive. Some iron preparations have been combined with vitamins. These
complexes are likewise no more effective than simple iron
preparations and are more expensive. This, plus the fact that they
are usually given in a one-a-day dosage, makes this therapy as a singular
form of iron supplementation an unlikely choice. One of the principal
uses of these combination preparations, however, might be in the iron-sufficient
woman who exhibits evidence of good iron stores, in whom
daily administration of iron might be sufficient. Nevertheless, hematologic
assessment of the patient's iron status during the second
and third trimester needs to be completed to determine if this amount
of iron is adequate. TABLE 5. Iron Supplementation Preparations
| Cost per 100 | Retail Cost |
Tablets or Capsules | for 9 Mo |
Ferrous sulfate | $2.89 | $21.82 |
Fer-In-Sol caps | NA | |
Feosol tablets | NA | |
Ferrous gluconate | | |
Generic | $3.99 | $30.16 |
Fergon | $6.99 | $52.84 |
Ferro-Sequels | $23.19 | $163.03 |
Slow FE | $25.47 | $195.03 |
Feosol elixir | $10.99/16 oz | $86.55 |
Ferrous sulfate elixir | $6.29/16 oz | $49.54 |
Parenteral Imferon | NA | |
Transfusion | 1 unit | $193.00 |
NA, Not available.
Sustained-release capsules were previously thought to be poorly absorbed. More
recent data using refined techniques have shown the reabsorption
to be 80% normal; therefore, consideration of their use in patients
who may be iron intolerant owing to gastrointestinal side-effects is
reasonable. In addition, slow-release iron tablets may provide a reasonable
alternative in these parturients. For other patients who experience
intestinal distress, ferrous sulfate liquid can be used. This preparation
should be well diluted in juice or water so that staining of the
teeth does not occur. Most of the oral agents contain 200 to 300 mg of iron complex, yielding
approximately 40 to 60 mg of elemental iron.11 Likewise, the ferrous sulfate syrup (40 mg/ml) or the pediatric drops (120 mg/ml) yield 20% of
this dose in elemental iron. The ideal dosage
is 60 mg of elemental iron administered three to four times daily. The
value of the spaced dosage is related to the absorption rate, which
is maximal about 4 to 6 hours after each dose. Most commonly, the iron
is administered with meals during pregnancy, since this seems to reduce
the incidence of gastric irritation. Other side-effects, such as diarrhea
and constipation, occur in approximately 5% and 15% of pregnant
women, respectively. However, diarrhea has been reported in about 3% and
constipation in as many as 10% to 12% when patients are given placebos. Therefore
gastrointestinal problems may be related to pregnancy
and not to iron therapy. It is recommended by some that oral iron be used in pregnancy regardless
of the hemoglobin status because iron loss due to pregnancy will certainly
exceed the amount obtained in the diet.12 Support for iron supplementation in the nonanemic pregnant woman for the
prevention of iron depletion comes from nutritional surveys demonstrating
that diets in the United States usually contain an average of 5 to 10 mg
of iron daily. Unfortunately, iron is better absorbed from meat
and dairy products than from cereals and most vegetables. Since much
of our population, including the impoverished, subsist on a high percentage
of processed, gluten-positive food, it is doubtful that a woman
receives adequate iron during pregnancy. A food fortification program
for the prevention of IDA has been suggested in which iron would be
added to bread, flour, and cereals. However, some of these sources may
not be well absorbed. An increase in hemochromatosis in iron-sufficient
persons (nonpregnant) receiving iron supplementation in their food
has been reported in studies from Scandinavia; this finding has not been
reported in populations in which IDA is common. Finally, when one
views the cumulative effects of a diet chronically low in iron, adolescence, previously
excessive menstrual loss, and early successive pregnancies, almost
every pregnant woman in our society appears in need of
supplemental iron in therapeutic doses. Certainly all pregnant women with
PCV less than 35% to 37% and hemoglobin levels below 12 g/dL should
receive iron, particularly if other laboratory tests support the diagnosis
of iron deficiency or depletion. The therapy usually is continued
throughout pregnancy and during the postpartum period. For the patient
with IDA, progress should be monitored, and if the hemoglobin level
is unchanged after 6 weeks of therapy, other hematologic problems should
be considered. Treatment with parenteral iron is rarely necessary unless the patient with
IDA cannot or will not take the iron by the oral route, has an absorption
disorder, or must undergo a surgical procedure within 1 to 2 weeks. Parenteral
iron is available as a complex with either dextran or
sorbitol and can be given intramuscularly or intravenously.13 Minor side-effects such as skin staining, discomfort at the site of injection, pruritus, malaise, and a metallic taste occur in 8% to 10% of
patients. Moderate reactions such as lymphadenopathy, phlebitis, severe
headaches, hyperpyrexia, and leukemoid reactions occur in 1% to 2%. Severe
systemic adverse reactions such as anaphylaxis, renal toxicity, and
bronchial spasm occur in approximately 0.5% of patients. The incidence
of these reactions can be reduced by eliminating those patients
with a history of eczema, asthma, or other allergic phenomena. When parenteral agents are administered, a test dose of 0.5 ml should be
given and the patient observed for 15 to 30 minutes before the remainder
of the dose is given.13 Dosages of 5 mg/kg or 250 mg per each gram of hemoglobin less than 14 g/dL
are standard. The rate of infusion for the intravenous administration
should not exceed 50 mg/minute. There have been no adverse experiences
associated with the human fetus or newborn. Infants of mothers who
were treated with parenteral iron during the course of gestation, just
prior to delivery, or during lactation showed no signs of hemochromatosis. Severe anemia, particularly in the patient with acute hemorrhage, is probably
best treated by transfusion. Transfusion obviously provides an
immediate improvement in the anemia and is a ready source of iron for
the production of new RBCs. Transfusion is not often used in the treatment
of IDA owing to its expense and to the potential for serious adverse
effects (i.e., hepatitis, human immunodeficiency virus (HIV), and transfusion reaction). It
is useful in patients with IDA who are undergoing major surgery
and who require immediate replacement of RBCs. In the normal postpartum
patient, transfusions are rarely needed unless the PCV is less than 20% to 25% or
the subject is symptomatic, since most patients can be
adequately treated with oral iron therapy. In summary, patients with iron depletion or deficiency will rarely be detected
prior to changes in the hemoglobin or PCV level. Supplemental
iron therapy is recommended in most pregnant women because (1) iron depletion
is so common, (2) therapy is safe and inexpensive, (3) lack of
response to supplements is an indication for further testing, (4) other
testing is avoided, in most cases, and (5) the effects of an undetected
common disorder like IDA on the mother and fetus are not fully known. Therefore, unless
more severe hematologic disease is suspected, a
therapeutic trial of iron should be given, at least to those at risk
for iron depletion, if not to all pregnant women.14 Finally, diagnosis of almost any anemia may be handled on an ambulatory
basis. However, there should be active hematologic consultation when
the diagnosis is unsure, severe disease is suspected, or the perinatologist
is unfamiliar with the diagnostic requirements. Thalassemia. Another disorder in hemoglobin production is thalassemia, which is caused
by an alteration in the rate of synthesis of the α- or β-chain. The
hemoglobinopathy is classified based on which chain is affected. Clinical
symptoms may also be used to classify thalassemia as thalassemia
major, intermedia, and minims. In general, only the homozygous
forms are in the major category, while the heterozygotes (α, β, α-β , δ )demonstrate variable degrees of symptomatology.15 Homozygous Forms. α-Thalassemia appears to be most severe, with fetal death occurring
in most cases. The poor outcome is due to the increased production of α-chains, which
combine to form Bart's hemoglobin, a high-oxygen-affinity
hemoglobin that functions poorly for oxygen transport. Electrophoresis
on fetal blood shows 85% to 100% Bart's hemoglobin, with
only small amounts of hemoglobin A and hemoglobin F. The infant
develops severe anemia and hydrops similar to that noted in those with
Rh isoimmunization. Homozygous α-thalassemia is the most common
form of hydrops fetalis in Southeast Asia, but is rare in this country. Homozygous β-thalassemia (Cooley's anemia) is characterized
by severe anemia, usually leading to death in early childhood. On electrophoresis, the
hemoglobin is usually 40% to 70%; hemoglobin A2 is mildly increased, while hemoglobin A is reduced and varies with the
severity of the anemia. Treatment involves repeated transfusions, and
pregnancy is infrequent since the patient usually dies in childhood. Heterozygous Forms. Patients heterozygous for thalassemia rarely manifest signs or symptoms
of the disease. Heterozygous β-thalassemia is the most common form; patients
usually exhibit only mild hypochromic, microcytic anemia, with
elevation of the hemoglobin A2 (above 3.5%) and a mildly increased hemoglobin F percentage (2% to 5%). Heterozygous α-thalassemia is difficult to detect by laboratory
means because the affected globin chain is common to all types of hemoglobin. Therefore, there
is a reduction in the amount of hemoglobin A, F, and
A2, while the percentage of these compounds remains the same as in normal
persons. This finding, combined with the fact that anemia is usually
mild, also complicates the detection process. Although family pedigrees
or intricate hematologic studies are usually required for a firm diagnosis
in adults, α-thalassemia is easier to delineate in the neonate, since
Bart's hemoglobin and small amounts of hemoglobin H (four β-chains) may
be present. Concomitant pregnancy reveals few adverse
maternal or fetal effects, except a slight increase in spontaneous
abortions. In general, the maternal outcome is related to the severity
of the anemia; those more severely affected have an increased maternal
morbidity, although fertility does not appear to be affected. The
double heterozygote (α-β-thalassemia) is also difficult to
diagnose and may be more common than previously suspected. Pregnancy statistics
are sparse, although most authors suggest that the anemia and
other symptoms are more severe than with α-thalassemia or β-thalassemia
trait alone. Prenatal Diagnosis. The prenatal diagnosis of thalassemia syndromes should be offered to any
couple with a prior affected child or those at risk because of ethnic
origin or pedigree. Prospective parents not having an affected child
previously need verification of carrier status based on MCV, zinc protoporphyrin, hemoglobin
electrophoresis, and pedigree analysis before
fetal assessment is undertaken. A sample of fetal deoxyribonucleic acid (DNA) can
be obtained by either chorionic villus sampling, amniocentesis, or
percutaneous umbilical (cord) blood sampling. DNA analysis of
that portion of the α-globin or β-globin gene containing the
deletion or mutation can be identified using restriction fragment length
polymorphisms and Southern blotting techniques. More recently, direct
DNA sequence analysis has become available by performing polymerase
chain reaction amplification of fetal DNA. Using this method of detection, prenatal
diagnosis can usually be confirmed within 7 days of fetal
sampling.16 Management. Management of the thalassemia traits during pregnancy is much the same
as for the patient with normal hemoglobin. Proper diagnosis is difficult, since
the mild microcytic, hypochromic anemia can be easily confused
with IDA, a condition that frequently coexists. Folic acid supplementation
is recommended because of the increased utilization and high
erythrocyte turnover. Blood transfusion or other invasive therapy should
be avoided, if possible, unless severe anemia or other problems intercede. Disturbance in Maturation: DNA Synthesis (Megaloblastic Anemia) Although not specific, the term megaloblastic anemia is used by most hematologists to describe a group of hypoproliferative
disorders that have a characteristic morphologic appearance, ineffective
erythropoiesis, and a moderate hemolysis of the circulating RBCs. Pernicious
anemia (lack of vitamin B12) and folate deficiencies are the prototypes of this disorder. The underlying
biochemical defect is impaired thymidylate formation, an essential
rate-limiting initial step in the DNA synthesis of the body's
cells, which require tetrahydrofolic acid as a coenzyme. The megaloblastic changes occur as a result of a maturation defect in the
marrow affecting erythrocytic, leukocytic, and thrombocytic cell lines. The
lack of vitamin B12 or folate slows DNA synthesis and delays the nuclear maturation of chromatin
of the immature cell into the dense cyanotic figure associated
with the normoblast during normal erythropoiesis (see Fig. 2). Although the nucleus remains large and immature, the cytoplasmic mass
decreases normally as it does during maturation. Therefore, the large
macrocytes found in the peripheral blood of those with megaloblastic
anemia represent a delay in nuclear maturation in the erythrocytic series
and are diagnostic by their abnormal nuclear cytoplasmic appearance. Likewise, in
the leukocytic series, the abnormal granulocyte is referred
to as a giant metamyelocyte because of the condensation failure
in the large nucleus. Due to granulocyte marrow turnover, leukopenia
develops and the cells found in the peripheral blood are hypersegmented, with
the nucleus having six or more lobes rather than the normal three
to five. Finally, inadequate thrombopoiesis occurs, which results
in an increase in bone marrow megakaryocyte mass with fewer platelets
in the peripheral blood. The platelets that are present may function poorly, and
a bleeding diathesis may be present if the vitamin B12 or folate deficiency is severe. ETIOLOGY. FAD appears to be the most common cause of megaloblastic anemia. However, vitamin
B12 appears to catalyze the conversion of 5-methyltetrahydrofolate to its
active form, as well as to affect the storage and transport of folic acid
within the body. Therefore, a deficiency in vitamin B12 may lead to megaloblastic anemia singularly or by its effect on folate
metabolism. Causes of folate or vitamin B12 deficiency include decreased intake and conditions in which requirements
are increased (e.g., pregnancy, hyperthyroidism, hyper parathyroidism, and malignancies). Causes
that are not related to vitamin B12 or folate therapy include the purine and pyrimidine synthesis inhibitors (cancer
chemotherapy), pyridoxine responsive megaloblastic anemia, and
Di Guglielmo's syndrome (erythremic myelosis), in which platelet
production is snore severely affected than erythrocyte or leukocyte
maturation. Finally, in various inborn errors of metabolism, such as
Lesch-Nyhan syndrome and hereditary orotic acid deficiency, either a
lack of enzymatic catalases or metabolic blockage to folate incorporation
can cause megaloblastic anemia. All the infrequent causes are difficult
to diagnose, and megaloblastic anemia due to these causes is unresponsive
to folate therapy. Fortunately, vitamin B12 deficiency and FAD are the most common etiologies of megaloblastic anemia, accounting
for 98% of reported cases. FAD is a much more common cause
during pregnancy (99%), and it causes 92% of all cases in any age
group.17 Vitamin B12 deficiency is extremely uncommon during pregnancy (1:8000 pregnancies); the
lack of intrinsic factor almost always occurs in persons beyond
the childbearing age unless significant gastric surgery has been performed. DIAGNOSIS. The major clinical signs and symptoms of megaloblastic anemia are variable
and are usually not detectable until the anemia is severe. As in
other anemia processes, the symptoms increase in severity with the progression
of anemia, from pallor, through weakness, malaise, dizziness, and
shortness of breath, and finally to congestive heart failure. Patients
with megaloblastic anemias may appear jaundiced more frequently
than those with IDA because of the rapid cell turnover and their increased
propensity for bleeding diathesis. If anemia is present, the appearance
of the peripheral smear is usually diagnostic, demonstrating several
hypersegmented granulocytes as well as RBC inclusions (i.e., stippling, Howell-Jolly bodies, Cabot's rings, and nonhemoglobin
iron). The most sensitive indicator of early folate deficiency appears
to be the hypersegmentation of neutrophilic nuclear material (average
low value >3.27 or more than 4% with more than five lobes in 100 consecutive
polymorphonuclear neutrophils). Serum folate values in pregnant
women are usually lower than in the nonpregnant patient, and these
decrease progressively toward term. In the absence of IDA, however, a
low fasting serum folate (<3 ng/ml by radioimmunoassay) is virtually
diagnostic of folate deficiency. In addition, a low erythrocyte folate
activity (<20 ng/ml) is probably the best biochemical index of
FAD. Although the anemia is macrocytic, it is usually normochromic with
normal MCH and MCHC. The MCV, however, is strikingly elevated in contrast
to IDA and may exceed 150 cu μ (see Table 1). The high MCV is helpful because it is in contrast to that found during
pregnancy, IDA, and other microcytic processes. In general, the more
severe the anemia, the more bizarre the erythrocytic changes, with nucleated
RBCs appearing in the peripheral blood smear when the PCV is
less than 20%. Unfortunately, if folate is deficient, there is usually
a concomitant IDA, which may confuse the diagnosis.18 However, the RC is lower in a megaloblastic anemia compared with the level
in IDA. In contrast to findings in IDA, most megaloblastic anemias demonstrate
an elevated serum iron saturation and reduced UIBC. These ferrokinetic
changes are usually combined with elevated plasma bilirubin and uric
acid levels, which demonstrates that the ineffective erythropoiesis is
associated with intramedullary hemolysis, a classic finding in megaloblastic
anemia. This hypothesis is supported by increasing levels of lactic
dehydrogenase (LDH1 and LDH2), serum muramidase, and malic dehydrogenase as the severity of megaloblastic
anemia progresses. Similarly, RBC survival time is only 50 to 60 days
versus the normal 120, denoting a concomitant extramedullary hemolysis. In
contrast, the liver is not involved; serum glutamic oxaloacetic
transaminase (SGOT) and alkaline phosphatase are usually normal. Folic Acid Deficiency. Folic acid (pteroylmonoglutamic acid), like the essential amino acids, cannot
be synthesized by humans and thus must be provided in the diet. Deficiency
of this substance continues to be an infrequent cause of
severe anemia, but more commonly represents a simple deficiency.17 This variable presentation probably reflects the ubiquitous presence of
folic acid in meats and fruits, as well as in dried or fresh vegetables. Although
not a common cause of frank anemia during pregnancy, folate
deficiency has increased in incidence from 1:250 pregnancies in 1960 to 1:50 in 1975. This
increase probably reflects better diagnostic
methods and an increased index of suspicion, since no greater percentage
of women are clinically anemic. However, it demonstrates very graphically
that many pregnant subjects are potential candidates for folate
deficiency. Folic Acid Metabolism. Pteroylmonoglutamic acid, or folic acid, is the parent compound for a
large family of chemicals collectively known as folates. According to
nomenclature developed in 1965, the molecule contains a pteridine derivative, a p-aminobenzoic acid (PABA) residue, and l-glutamic acid (Fig. 8). To be useful in the synthesis of purines and pyrimidines, folic acid
must be converted to 7,8-dihydrofolic acid (FH2) and finally to the active form, tetrahydrofolic acid (FH4). The metabolically important members of the folate family are FH4 derivatives and differ by the addition of one carbon unit to the terminal
nitrogen of the PABA unit. Three oxidation levels of this carbon are
possible: formyl, hydroxymethyl, and formimino (see Fig. 8). The formation of formyl FH4 is catalyzed by the enzyme dihydroglycolate reductase. It is this portion
of the metabolic pathway that the folate analogs (chemotherapeutic
agents) inhibit. Formyl FH4 derivatives are important in DNA synthesis and the production of amino
acids and the enzymatic catalyst citrovorum factor. Without the formyl
FH4 derivatives, thymidylate cannot proceed in DNA synthesis, which ultimately
causes the hematologic manifestations noted in megaloblastic anemia. Interference
with the hydroxymethyl series, in contrast, impairs purine
synthesis, although no clinical manifestations have been related
to this disorder. Finally, inhibition of formimino group synthesis disturbs
histidine catabolism, and while this has no adverse effects, it
does provide the basis for tests useful in the diagnosis of FAD; formiminoglutamic
acid is excreted in increased amounts, particularly when
oral histidine is administered to folate deficient persons. 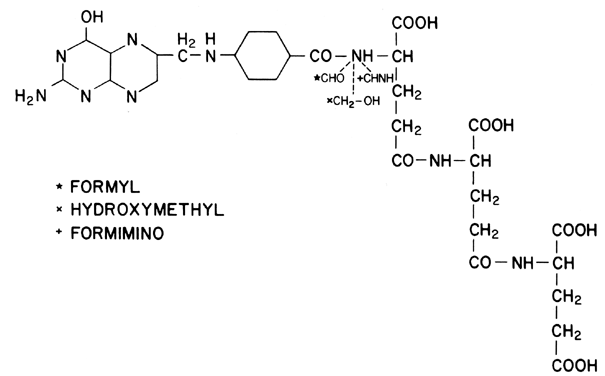 Fig. 8. Structure of folic acid, including its three essential derivatives. Fig. 8. Structure of folic acid, including its three essential derivatives.
|
Folic Acid Requirements. The minimum daily requirement of folic acid or its equivalent is 50 μg/day
in the normal adult or 100 μg/day in those with additional
requirements (e.g., pregnancy or adolescence). Although the average diet contains many times
this amount, most of it is unavailable. In addition, the body is thought
to contain only approximately 5 mg of folate compounds total, so
the reserves are much smaller than those of vitamin B12 or iron. For these reasons, the official recommended daily allowance of
food folate for the adults is 0.4 mg/day; during pregnancy, 0.5 to 1 mg/day
is considered adequate. The richest vegetable sources of folate are asparagus, broccoli, spinach, lettuce, and
lima beans, each of which contains more than 1 mg of folate
per 100 g dried weight. The best fruit sources are lemons, bananas, and
melons. The folate in most vegetables and fruits is conjugated, however, and
must be broken down in its absorptive pathway through the
gastrointestinal tract. Although high concentrations are found in the
liver, kidney, and brain of carnivorous animals, these are poor sources
of usable folate. Several problems may prevent attainment of a positive folate balance. They
include (1) failure to consume adequate amounts of food high in folate, (2) failure
to prepare foods properly (most vegetables and meat
are overcooked, resulting in folate destruction), and (3) increase in
requirements (e.g., in adolescence, successive pregnancies, and multiple gestation). Women
do not appear to have an increased need or an increase in daily loss
compared with men. During pregnancy, however, approximately 100 μg
folate is needed daily to supply the fetus and maintain maternal stores (twice
normal nonpregnant requirements).17 The higher frequency of FAD during pregnancy and the puerperium is attributable
to several factors. Most important is the elevated demand of
this substance by the placenta, the fetus, and the expansion of the maternal
RBC mass. In addition, compromised socioeconomic conditions, which
are usually related to inadequate nutrition, may also be major factors. Finally, complications specific to pregnancy, such as unusual fad
diets, prior gastrointestinal tract surgery or malabsorption states, prolonged
hyperemesis, or the slowed gastrointestinal absorption of
folate (thought to be due to increasing levels of estrogen and progesterone), all
may give rise to an increased incidence of FAD. Chronic infection
of any type, as well as other causes of anemia, the presence of
multiple fetuses, or closely spaced pregnancies, as well as lactation, may
also substantially increase maternal requirements for folic acid. Also, many
drugs, such as ethanol and certain antibiotics (nitrofurantoin, trimethoprim), as
well as the anticonvulsant diphenylhydantoin, may
impair absorption or utilization of folate. Although it is an infrequent
cause of anemia, folate deficiency should be detected and treated, since
megaloblastic anemia will eventually result. Folic Acid Absorption. Although the mechanism of folate absorption in the intestine is unclear, the
proximal jejunum seems to be the principal site of uptake. When
radioactive labeled folic acid is administered in the unconjugated form, only
the monoglutamate form (FH1) appears in the plasma; thus, folic acid or conjugated derivatives in
food products must be broken down before being absorbed. The intestine
contains several pancreatic conjugates to reduce the FH4 to FH1, the site of degradation being within the lumen of the intestine or near
the brush border of the intestinal cell. After entry into the serum
intact, folates, as well as their cleavage products, are excreted by
the liver into the bile. Although some of this folate may be reabsorbed, the
loss of folate by bile secretion may accelerate depletion in patients
with mild absorptive problems. As shown in Fig. 9, once FH1 enters the mucosal cells, it can be absorbed against a concentration gradient, although
a passive transport may also occur. The amount of FH1 absorbed in the mucosal cells depends on the need, which is reflected
by the content of FH4 within these cells. If the level is high, indicating adequate folate stores, little
of the hydrolyzed FH1 will be transported, whereas under conditions of decreased FH4 availability, the FH1 is quickly absorbed into the mucosal cells. Up to 90% of the FH1, even in deficiency states, is reduced to FH4 and methylated in the mucosal cells before it is released into the circulation. Approximately 60% is cleared in one circulation, and 90% to 95% is
cleared within 3 minutes. The rapidity of clearance suggests that
folate is bound quickly by a serum protein that is increased in FAD. The
elevation in this binding protein also occurs in patients with uremia
and in women who are pregnant or are taking oral contraceptives. It
appears that this moiety consists of two proteins, with molecular
weights of 200,000 and 50,000, respectively. The larger fraction is present
in human milk and leukocyte membrane. It appears to have antibacterial
properties similar to those of the β-lactoglobulin. The smaller
portion, however, behaves as a β-globulin similar to transferrin
and participates in the binding and release phenomena. After release
to a metabolically active cell, the folate is held in storage or
used for biosynthesis. 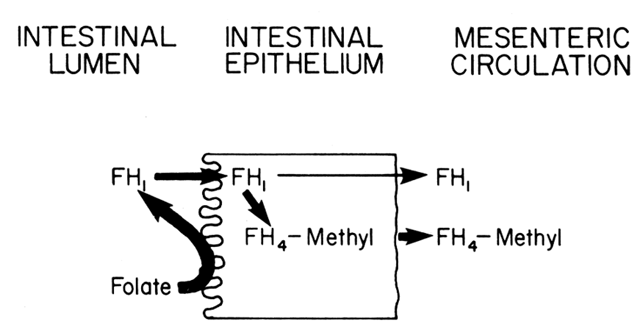 Fig. 9. Absorption of folic acid from the intestinal lumen through the mucosal
epithelium and into the mesenteric circulation. Fig. 9. Absorption of folic acid from the intestinal lumen through the mucosal
epithelium and into the mesenteric circulation.
|
Clinical Features. Clinical syndromes of FAD include all of the nonspecific features described
earlier in the section, Anemia Detection. The most notable features
include roughness of the skin and glossitis, which are seen with both
vitamin B12 and folate deficiency (Plummer-Vinson syndrome). It is important to be
certain that no neurologic deficiency is present, since the neurologic
abnormalities of vitamin B12 deficiency are not remedied by folate supplements. Specific laboratory
diagnosis of folate deficiency includes the use of peripheral blood smear, PCV, hemoglobin
values, and RBC indices. Serum folate assay, a microbiologic
procedure employing the organism Lactobacillus casei, is a reliable method for diagnosis of FAD. Although serum folate is slightly
lower in pregnant subjects (see Table 1) compared with normal subjects, it does not approximate folate levels
found in megaloblastic anemia patients, which are commonly lower than 3 ng/ml. However, since
the serum folate level falls many weeks before
anemia appears, a level lower than 5 ng/ml signifies a deficiency of
folate that leads to anemia if not treated.18 Although white blood cell and RBC folate assays can be performed and may
give a better assessment of tissue folate, the assays are difficult
to perform, hard to standardize, and not widely used. Measurement of
formiminoglutamic acid excretion in response to histidine loads is less
specific than serum folate measurement, but it gives a better measure
of folic coenzyme levels than does the microbiologic assay. Unfortunately, it
is difficult to quantitate because histidine absorption and
excretion are altered during pregnancy. Finally, the response to therapy
is useful clinically. As shown in Table 6 and Table 7, changes in the above parameters occur quite rapidly if anemia is due
to folate deficiency. Since the deficiency of vitamin B12 is rare in pregnancy, folate therapy in the absence of lowered vitamin
B12 levels appears to be justified in most cases. TABLE 6. Changes in Laboratory Values Related to Duration Folate Deficiency
Laboratory Assessment | Duration (wk) |
Low serum folate | 3 |
Abnormal neutrophil | 7 |
Increased formiminoglutamic acid | 14 |
Low folate (RBC) | 16 |
Macrocytosis | 18 |
Anemia | 19 | TABLE 7. Response to Folate Therapy in Folic Acid Deficiency
Response | Response Time |
Reticulocytosis, thrombocytosis | 48 h |
LDH, LDH2 decrease | 96 h |
PCV and hemoglobin increase | 1 wk |
Neutrophil normal | 2 wk |
Normal folate | 6 wk | Differential Diagnosis. Conditions that may result in malabsorptive syndrome include subtotal
gastrectomy, nontropical sprue (gluten-induced celiac disease), tropical
sprue, lymphomatous infiltration of the small intestine, Whipple's
disease, scleroderma, amyloidosis, and diabetes mellitus. Various
drugs, such as oral contraceptives and anticonvulsant medications (hydantoin
derivatives), may also interfere with folate absorption. Conditions
that increase folate requirements and thus can result in folate
deficiency include pregnancy, hyperactive hematopoiesis (hemoglobinopathies
and hemolytic anemias), malignancies, and various skin diseases
such as psoriasis and chronic exfoliative dermatitis. Finally, the activation
of FH4 may be blocked in patients treated with folic acid antagonists and in
patients with scurvy. Therapy. Folic acid is usually administered orally on a prophylactic or therapeutic
basis as a 1-mg tablet, although this dose is greater than the requirement. Moreover, oral
folate is given once a day, and since folate
absorption, even in deficiency states, is only 10 μg to 20 μg every 6 to 8 hours, the
amount of folate absorbed may be minimal. More
recently, the 1-mg tablet has been given as one half tablet (0.5 mg) two
to four times a day for prophylaxis or treatment of a true folate deficiency. Should
neurologic signs be present, serum B12 levels should be measured prior to folic acid therapy, because folate
treatment corrects the anemia but not the neurologic defect. The principal
indication for parenteral folic acid occurs when specific folic acid
antagonists must be used. Toxicity from folic acid has not been observed, even
in doses hundreds of times the usual therapeutic level, and
only rarely have sensitivity reactions been recorded. Indications for
folic acid therapy are (1) actual or anticipated deficiency (e.g., in multiple gestations and adolescent pregnancy), (2) prophylaxis for
women with poor nutrition and other high risk factors, and (3) women taking
certain drugs (e.g., trimethoprim, diphenylhydantoin) on a long-term basis. In the patient with actual megaloblastic anemia, one can expect an elevation
in the RC approximately 48 to 72 hours following therapy (see Table 7). This appears to be the earliest sign of remission and is usually dramatic. The
thrombocytopenia that may be present rapidly clears within
the first few days. The specific lactic dehydrogenase (LDH) isoenzymes
also decrease early in the course of therapy. A slight increase in PCV
and hemoglobin level may be evident by the end of the first week, and
the neutrophils gain their normal appearance after approximately 2 weeks. Serum
iron levels may have been falsely elevated in folate deficiency
because iron stores were not efficiently used for erythropoiesis; these
usually fall to subnormal levels, indicating a coexisting IDA
during the process of folate replacement. Folate Deficiency During Pregnancy. Maternal folate deficiency is common in late pregnancy. In England and
Australia, 95% and 60%, respectively, of women at term have been reported
to have low serum folate levels.19 Sixty percent of women in the United States also demonstrated low serum
folate levels. Although frank megaloblastic anemia of pregnancy is less
frequent, studies in Canada have shown approximately 25% of pregnant
women have folate deficiencies sufficient to produce some megaloblastic
changes.17 Anemia cannot be completely overlooked, since one study reported that
almost 500 women in 3200 obstetric patients had a hemoglobin level below 9.5 g/dL
and 90 of these had frank megaloblastic anemia.17 Embryopathies, especially neural tube defects, have been associated with
folate deficiency and supplementation appears to decrease the percentage
of such complications.20 FAD has been associated with low-birth-weight infants, smaller maternal
blood volume, abruptio placentae, prematurity, and other maternal-fetal
disease processes. However, other studies, particularly those with
specific assay techniques for folate deficiency, have not shown a causal
relationship.19 Indeed, this association tends to occur in women who are, in general, nutritionally
deprived. Since the effects of deprivation are unclear, most
physicians favor supplementation for patients at high risk for folate
deficiency. Vitamin B12 Deficiency. Vitamin B12 is a unique compound, since it is synthesized only by certain microorgansims. High
concentrations of microorganisms producing vitamin B12 are found in liver, seafood, meat, eggs, and milk. Vitamin B12 is enzymatically important as a cofactor in the formation of succinyl-CoA
and in the folate pathway for the production of active FH4. It is in these two enzymatic pathways that vitamin B12 deficiency is clinically apparent. The general clinical features of vitamin
B12 anemia are the same as for any other anemic process, except for the peripheral
neuropathy characteristic of this disorder. Thus, the physical
and laboratory examinations are more informative than the history. Absorption. Intrinsic factor is necessary to facilitate absorption of vitamin B12 in the ileum. Intrinsic factor has been shown to be an alkaline stable
glycoprotein secreted by the parietal cells from the fundic mucosa of
the stomach. Secretion of intrinsic factor parallels that of hydrochloric
acid and therefore is concomitantly reduced in achlorhydric patients. Vitamin
B12 derivatives are bound to intrinsic factor in a complex that attaches to
specific mucosal receptors on the microvilli of the ileum. Other secretions
such as pancreatic enzymes may promote vitamin B12 absorption by protecting the receptor sites and providing a suitable environment
for intrinsic factor—vitamin B12 complex. The complex partially enters the cell by pinocytosis and dissociates
near the membrane, leaving the vitamin B12 in the cytoplasm. Uptake from the mucosal cell into the serum is slow
and requires active transport. Diagnosis. Various laboratory tests, such as measurement of the vitamin B12 level, can be performed to distinguish this deficiency from megaloblastic
anemias due to other causes. It should be remembered that the serum
folate level may be artificially lowered in vitamin B12 deficiency even though folate stores are adequate; thus, serum folate
may be decreased while RBC folate is normal. Laboratory methods for measurement
of vitamin B12 use fecal and urine samples (e.g., the Schilling test), which involves giving a small dose of radioactive
cobalt; thus, it is not used during pregnancy. The measurement of serum
vitamin B12 may be performed by radioactive assay, and although maternal serum levels
fall progressively during gestation to intermediate levels (80 to 120 pg/ml), the
finding of levels below 50 pg/ml is very suggestive of
pernicious anemia. Therapy. Appropriate therapy includes six weekly injections of 1000 μg hydroxycobalamin. Further
diagnostic tests with augmented histamine or the
Schilling test are usually performed during the postpartum period. In
addition, standard prophylactic iron and folic acid supplementation should
be implemented. It is particularly necessary to treat postpartum
women who are breast feeding. Indeed, the first signs of severe vitamin
deficiency may occur in these women. Effects on Pregnancy. Pernicious anemia usually does not complicate pregnancy. The incidence
of vitamin B12 deficiency during pregnancy is 1:6000 to 1:8000. It is usually associated
with a strict vegetarian or other fad diet, incipient pernicious anemia
due to lack of intrinsic factor, or a chronic gastrointestinal disorder
such as tropical sprue. Women who have pernicious anemia are usually
infertile and are usually not of reproductive age. In healthy women, vitamin
B12 deficiency takes 2 to 3 years to develop, even with a severely restricted
diet. Since a pregnancy only lasts 9 months, with a peak requirement
for vitamin B12 of less than 4 months, it is difficult to develop pernicious anemia during
a pregnancy. During pregnancy there is a steady fall in the serum
vitamin B12 level due to the rapid expansion of PV and the increased utilization of
vitamin B12 by the fetus during the latter half of pregnancy, accounting for the reported 15% incidence
of low serum levels of vitamin B12 during pregnancy. Under other circumstances, these women might be considered
to have vitamin B12 deficiency. Nevertheless, if the diagnosis is suspected but cannot be
confirmed, vitamin B12 therapy should still be administered. If the serum level does not return
to normal within 6 weeks after therapy, it is advisable to perform
other investigations, particularly if other adverse symptoms are present. Bone Marrow Failure The failure of bone marrow to produce sufficient erythrocytes is a rare
occurrence in obstetric patients. Nevertheless, the resulting pancytopenia, which
affects all cell lines, is devastating. A selective aplastic
erythropoietic cell line is called pure red cell aplasia. In children, the
inherited type of this disorder is called Diamond-Blackfan syndrome; a
pregnant woman, who had Diamond-Blackfan syndrome as a child, transmits
the trait to 25% of her offspring, who are born with minor
congenital malformations. Acquired red cell dysplasia is associated with
thymic tumors in 30% to 50% of cases and occasionally occurs during
pregnancy. This disorder usually is thought to have an immunologic basis
and presents as a normochromic, normocytic anemia with absolute reticulocytopenia
as well as a normal leukocyte and platelet count. Bone
marrow in this examination is normal except for erythroid hypoplasia. Treatment
is usually by transfusion, although removal of an enlarged
thymus is sometimes necessary. Although there have been few reported
cases during pregnancy, the outcome is usually good. Other disorders associated with bone marrow failure usually involve the
replacement of bone marrow with either fatty, degenerative changes (aplastic
anemia) or granulomatous/tumor infiltration (myelophthisic anemia).21 The development of aplastic anemia when there is fatty, hypocellular bone
marrow is usually caused by exposure to drugs, chemicals, radiation, or
several various diseases. Those agents known to be incriminated
include acetylsalicyclic acid, chloramphenicol, gold salts, phenylbutazone, and
tolbutamide. Both hepatitis virus A and B as well as total body
irradiation have also been incriminated. The diagnosis is made by
the appearance of pancytopenia on the peripheral blood count; normochromic, normocytic
anemia; and reticulocytopenia. Diagnosis is confirmed
by hypocellular or aplastic marrow appearance and fatty replacement of
the bone marrow. The appearance in the blood cell smear on peripheral
smear of nucleated red cells is more compatible with dysfunction rather
than hypoplasia. A total granulocyte count of less than 200/ mm3 correlates closely with poor prognosis, since these patients are very
susceptible to infection. In general, the basic goal of treatment is removal
of the causative agent, if present, and supportive therapy to obtain
remission if no cause can be found. Transfusion with packed red
cells, platelet buttons, and granulocytes to maintain these elements at
near-normal levels is recommended during the gestation. A variable effect
has been noted with steroid administration; therefore, a therapeutic
trial of steroids for 2 to 4 weeks is usually advised. In the nonpregnant
state, stimulation with certain androgens such as oxymetholone (3 to 5 mg/kg/day) has
been helpful. Bone marrow transplantation may
be a therapeutic option in severe cases of this disease, but it depends
on human leukocyte antigen (HLA) compatibility of sibling donors. Aggressive
treatment with antibiotics and early identification of infection
are extremely important, owing to the propensity of these patients
to become infected. For that reason, antibiotic prophylaxis for delivery
or operative procedures is recommended. Exposure to infected persons
or a reduction in an environmental exposure to infections is recommended. In
the hospital, reverse isolation can be used but is generally
recommended only when the absolute granulocyte count is below 200 mm3. In general, the outcome of pregnancy is good, particularly if the aplastic
anemia has been diagnosed before pregnancy and is in a stable state. If
the hemoglobin count is above 8 g/dL, the outlook is good. In addition, if
the granulocyte count is above 600/mm3, the likelihood of infection is reduced. The major causes for maternal
death remain hemorrhage and infection in 90% of patients. In general, the
survival rate of the neonate is about 75%; prematurity is the major
form of morbidity. Antiplatelet antibodies can be transported across
the placenta, and in a large series, six infants were reported to be
born with pancytopenia and a platelet antibody. For this reason, fetal
platelet counts obtained by scalp sampling early in labor may be helpful. Bone marrow infiltration by tumor or granulomatous cells (myelophthisic
anemia) results in severe normochromic, normocytic anemia. The peripheral
smear reveals a large number of fragmented cells with basophilic
stippling reminiscent of a hemolytic process, although the RC is reduced. The
white count, on the other hand, is elevated, and usually a left
shift is noted in the differential. The combination of immature myeloid
cells and normoblasts in the peripheral smear is a hallmark of this
disease. Pregnancy among those with this disorder is rare, and reported
cases have included metastatic disease from ovarian as well as bowel
and hepatic cancer. The fetal and maternal outcome, of course, is dependent
on the primary disease process. Unknown Causes The anemias associated with inflammation are usually chronic and are not
common, since these are unlikely in the reproductive age group. Most
of the disorders in this group occur from chronic suppurative infections
or neoplasia. Patients with these disorders have adequate stores of
iron (present as hemosiderin or ferritin), but the release mechanism
from transferrin to the red cells and the iron pickup from the storage
depot are abnormal. Characteristically, the anemia is mild: a hemoglobin
of 8 to 11 g/dL and a PCV of 25% to 33% are the rule.22 In about 75% of cases, the anemia is normochromic, normocytic, but hypochromia
and microcytosis can be manifest if the iron release is severely
disturbed. As a point of differentiation, the UIBC is increased with
a decrease in transferrin saturation with inflammatory processes. The
opposite findings are seen in those patients affected with neoplasia. Serum
iron levels are reduced in both of these entities. The RC is
usually below 1%, while the sedimentation rate is elevated, frequently
above that seen in pregnant patients. Diagnosis of the specific disorder
is usually based on careful history and physical examination; a high
index of suspicion is needed. Many of these patients, if pregnant, present
to the perinatologists with iron-unresponsive anemia and may have
subtle symptomatology such as a low grade fever or other signs out
of proportion to the mild anemic process. Obviously, the diagnosis is
assured by examination of the storage iron compartment, which will reveal
sufficient quantities of iron; bone marrow assessment may also be
useful in these cases. Therapy should be directed toward the specific
disease process. |
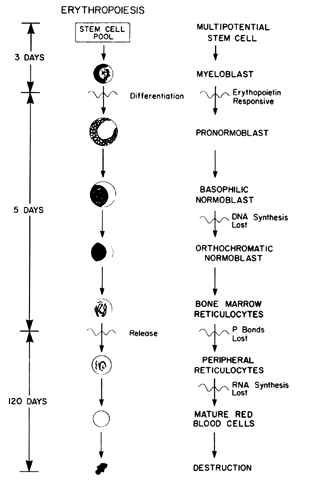
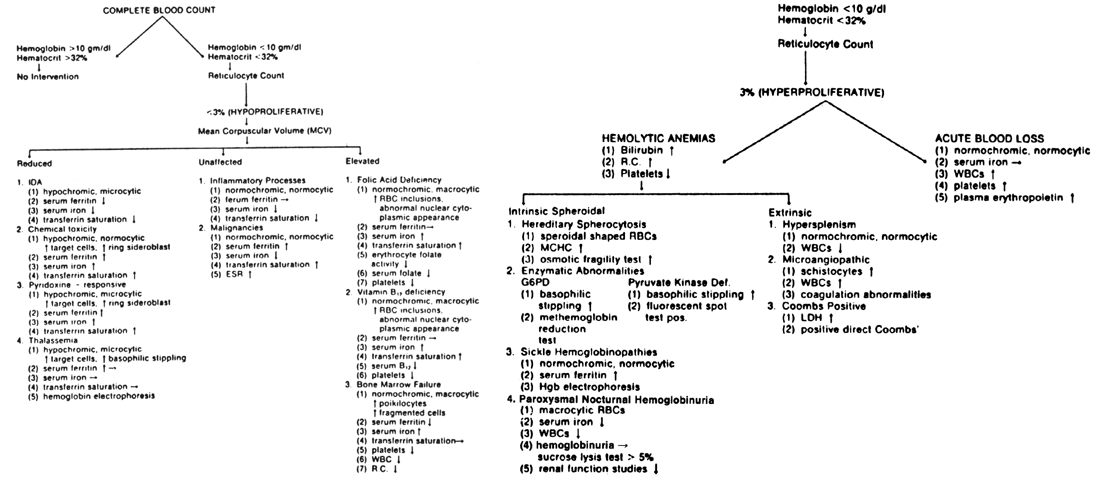
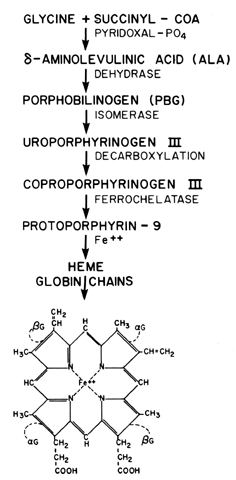
 val).
val).