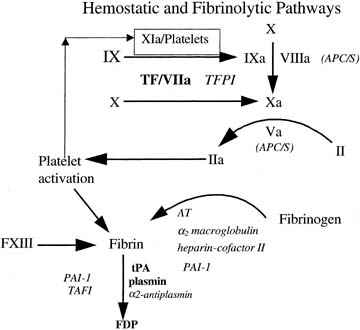
Deep Vein Thrombosis The diagnosis of deep vein thrombosis (DVT) has typically been
made on the clinical grounds of a tender and acutely swollen lower extremity
in the absence of trauma. Homan's sign, which is pain in
the calf when the foot is passively dorsiflexed, may be demonstrated. Unfortunately, many
of these same symptoms are present in women with
a normal pregnancy. More than 50% of patients exhibiting the classic
presentation of calf or thigh tenderness, pain, erythema, and edema
do not have a DVT.65,66 An objective evaluation is required in all clinically suspicious cases. Although venography is considered the definitive test, noninvasive testing
has emerged as the initial step in the diagnostic management of these
patients (Fig. 2). 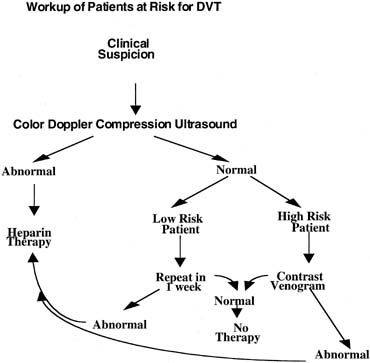 Fig. 2. Workup of patients at risk for DVT. Fig. 2. Workup of patients at risk for DVT.
|
ULTRASOUND Color Doppler may be used to evaluate vessel anatomy, blood flow, augmentation
of blood flow with muscle activity, and vessel compressibility. Ultrasound
is both highly sensitive (92%) and specific (98%) in popliteal and femoral vein thrombosis. Doppler
flow studies are less effective for evaluating calf vein thrombosis
with a sensitivity of only 50%.67,68 If the patient is placed in the left lateral decubitus position, Doppler
flow variation with respiration can assist in diagnosing isolated iliac
vein thrombosis.6 If the sonographic findings are abnormal, venous thrombosis can be diagnosed
and treatment initiated. If the sonographic findings are normal
and the patient has no other risk factors (e.g., no history of VTE, inherited thrombophilia, plasma D-dimers, or
clinical progression), the study can be repeated in 7 days.69 If the sonographic findings are normal but there is a high index of suspicion (positive
personal or family history, D-dimers, and
clinical progression), a contrast venography procedure should be
performed. IMPEDANCE PLETHYSMOGRAPHY Rarely used today, impedance plethysmography (IPG) was the first
noninvasive test for DVT in pregnancy. It measures the electrical
impedance between two electrodes wrapped around the calf. A pneumatic
cuff applied to the upper leg is used to reduce venous outflow. Characteristic
changes in impedance are noted on deflation of the occlusive
cuff if no obstruction to outflow is present.70 IPG is reliable for evaluating for thrombus formation from the iliac vein
to the knee. Compression by the enlarging gravid uterus may produce
false-positives.71,72 Placing the patient in the left lateral decubitus position increases specificity.73 IPG was shown to be inferior to ultrasound in a comparison study of 985 symptomatic
patients.72 MAGNETIC RESONANCE IMAGING Although the safety of magnetic resonance imaging (MRI) in pregnant
women has yet to be proven, no adverse effects have been noted.73 It is useful for detecting thigh and pelvic vein thrombosis.74 As more experience with this diagnostic modality is gained, it may find
increasing use in the obstetric population. VENOGRAPHY Contrast venography remains the gold standard for diagnosing DVT. Contrast
agents are injected into lower extremity veins and the venous system
of the leg and pelvis are evaluated radiographically. Venography with
an abdominal lead shield exposes the fetus to very low levels of radiation (.0005 grays).75 This exposure is below that associated with childhood cancers and teratogenicity.76,77 The incidence of chemical phlebitis is 3%.78 Doppler ultrasound is recommended as the initial test in pregnant women
with suspected DVT. If this noninvasive test is positive for a DVT, then
treatment should be started. With equivocal test results, contrast
venography or MRI (for pelvic thromboses) should be performed (see Fig. 2). Pulmonary Embolism Similar to the diagnosis of DVT, the clinical diagnosis of pulmonary embolism (PE) lacks sensitivity and specificity. It occurs in 1 in 2500 pregnancies
and results in obstruction to pulmonary arterial
blood flow, vasoconstriction of small arterial vessels, and a progressive
loss of surfactant. The triad of dyspnea, pleuritic chest pain, and
hemoptysis occurs in only 25% of patients with documented PE.79 Other clinical manifestations include cyanosis, tachypnea, syncope, diaphoresis, fever, a
pleural friction rub, and a fixed S2. Although arterial
blood gases may show hypoxia, one in six patients with a PE will
have a normal PaO2.80 It also should be remembered that the supine position may lower PaO2 by as much as 15 millimeters of mercury in the third trimester of pregnancy.81 An electrocardiogram may reveal right bundle branch block, right axis
shift, Q wave in leads III and aVF, S wave in leads I and aVL more than 1.5 millimeters, T
wave inversions in leads III, and aVF, or new-onset
atrial fibrillation.82 Echocardiographic findings may include right ventricular dilation and
hypokinesis, tricuspid regurgitation, and pulmonary artery dilation. Ventilation/perfusion (V/Q) scanning remains the initial
approach to the investigation of patients with suspected PE.83 The scans must be interpreted in the context of clinical probability. Only
normal (negative) and low-probability scans in the
setting of a low clinical probability, and high probability scans in
the setting of high clinical probability are considered diagnostic. Nondiagnostic
scans (i.e., intermediate probability) or low-probability scans in high-risk
patients (e.g., thrombophilics, positive D-dimer, ECG, and echocardiogram) should
be followed-up with a color Doppler compression ultrasound
study of the lower extremities. The presence of a DVT in conjunction
with a nondiagnostic lung scan mandates therapy. However, fewer than 30% of
unselected patients with PE have radiographic signs of
DVT at the time of presentation.84,85 Thus, a negative compression ultrasound in a high risk patients warrants
pulmonary angiography69 (Fig. 3). 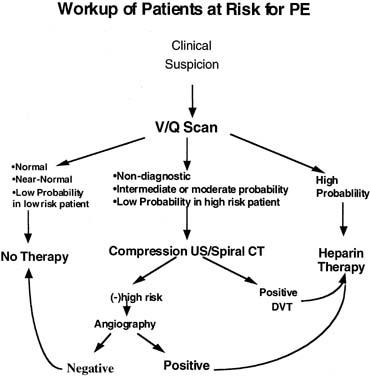 Fig. 3. Workup of patients at risk for PE. Fig. 3. Workup of patients at risk for PE.
|
MRI or spiral volumetric computed tomography pulmonary angiography (SVCT) may
be used as a noninvasive substitute in specialized centers.69,86 However, the reported sensitivities of SVCT compared with the gold standard
pulmonary angiography vary widely (64%–93%). Moreover, while
SVCT may be relatively sensitive and specific
for diagnosing central pulmonary artery thrombi, it is insensitive
for diagnosing subsegmental clots. Therefore, SVCT appears to have a role
as a rule-in test for large central emboli, but cannot exclude smaller peripheral lesions. Thus, like
compression ultrasonography, in the setting of an equivocal
V/Q scan in a high-risk patient, a positive spiral
CT should mandate therapy but a negative spiral CT should prompt pulmonary
angiography. Although the amount of radiation exposure from a V/Q scan is less than .005 Gy
of radiation, in a 1998 survey, only 67% of respondents
reported that they perform V/Q scans in pregnant patients.87 One may further reduce the radiation exposure performing a perfusion scan
first. A normal perfusion study requires no further testing. Spiral
CT scans and pulmonary angiography, with appropriate abdominal shielding
and using minimal fluoroscopy, expose the fetus to less than 50 millirads
for the entire examination. Septic Pelvic Thrombophlebitis An uncommon complication of pelvic infection, septic pelvic thrombophlebitis (SPT) typically occurs in the postpartum; however, it
has been reported to occur after gynecologic procedures. It is more common
after cesarean than vaginal delivery. Thrombus formation in the pelvic
veins is a result of pelvic infection. Multiple infected emboli
may result from thrombolysis. Physical findings are nonspecific. The typical
presentation is a patient who has spiking fevers that persist despite
adequate antibiotic coverage. CT or MRI imaging may aid in the
diagnosis. Although these techniques may be specific, a nonocclusive thrombus
in the iliofemoral veins may not be detected.88 Some authorities89,90 suggest that patients with a presumed diagnosis of SPT should start on
a course of therapeutic anticoagulation. In many cases, the clinical
response is considered therapeutic and diagnostic. Defervescence is expected
in 48 to 72 hours. In the absence of a clinical response, the cause
of the fever should be reassessed for other possible intra-abdominal
complications such as an abscess or perforated viscus. The
duration of treatment has been somewhat arbitrary. Some authors advocate
therapy for 7 to 10 days, while others recommend a full 6 weeks of
anticoagulation.91,92 A recent study has called into question the role of anticoagulation in
SPT.93 In a study of 14 patients with CT-documented SPT, eight received
continued antibiotic therapy alone (ampicillin, gentamicin, clindamycin) and
six received a combination of heparin and antibiotic
therapy. The overall incidence of SPT in this population of 44,922 women
was 1 in 3000 deliveries (1 in 800 for cesarean deliveries, 1 in 9000 for
vaginal deliveries). The duration of fever was not
different between the two groups. Ovarian Vein Thrombosis The incidence of ovarian vein thrombosis is 1 in 4000 deliveries.94 Some question whether ovarian vein thrombosis is a distinct entity from
SPT. Several clinical manifestations distinguish the two processes. Ovarian
vein thrombosis (OVT) typically presents with acute
pain 2 to 3 days postpartum (with or without fever). It occasionally
can be mistaken for appendicitis in a postpartum patient. It
also can occur in the absence of infection and has been described in antepartum
patients.95 Thrombosis can be diagnosed with CT or MRI and is most commonly noted
in the right ovarian vein.91 Treatment for OVT is the same as outlined for SPT. Treatment of VTE in Pregnancy The treatment of VTE in pregnancy mirrors that of the nonpregnant patient
with the exception of avoiding warfarin during the antepartum period. Anticoagulation
with heparin is the therapy of choice for DVT with
or without PE. Supplementary therapy with leg elevation and application
of warm moist heat may decrease edema and provide some symptomatic relief. In
the case of PE, careful attention should be placed on maintaining
the maternal PaO2 more than 70 millimeters of mercury or O2 saturation of more than 95%. Heparin Heparin is a complex glycosaminoglycan that potentiates antithrombin activity, increases
levels of factor Xa inhibitor,96 and inhibits platelet aggregation.97 Heparin does not cross the placenta, appears safe for the fetus, and does
not enter breast milk.98,99 Side effects of heparin therapy include hemorrhage, osteoporosis, and
thrombocytopenia. Hemorrhage is more common with concomitant aspirin use, recent surgery, thrombocytopenia, and liver disease, and is less
common when using low-molecular-weight heparin preparations.69 Heparin-induced thrombocytopenia occurs in 3% of patients
and has two forms: the more common early-onset, transient, heparin-induced
platelet aggregation for which therapy need not be
interrupted and the rare IgG-mediated, heparin-induced
thrombotic thrombocytopenia that occurs within 2 weeks of initiating
therapy and mandates cessation of therapy.69 The occurrence of heparin-associated and induced thrombocytopenia
appears far less common with low-molecular-weight heparin.100 In any case, platelet counts should be followed for the first 3 weeks
of therapy. Heparin-induced osteoporosis is far more common when
doses of more than 15,000 units per day are administered for more than 6 months.101 The goals of initial therapy are to maintain heparin levels of between .2 to .4 U/mL, or
antifactor Xa activity between .6 and 1.0 U/mL
or the aPTT between 1.5- and 2.5-times control. The dose
required to achieve these goals will vary between individuals because
of biological variability in heparin-binding proteins such
as vitronectin, fibronectin, von Willebrand factor, platelet factor 4, and
histidine-rich glycoprotein.69 Heparin should be administered with an intravenous loading dose of 150 U/kg
for PE and 100 U/kg for DVT, followed by an infusion rate
of 20 U/kg per hour. Doses are adjusted until the target aPTT, heparin, or
antifactor Xa level is reached. The aPTT, heparin, or antifactor
Xa level should be evaluated every 4 hours in the early stages of
therapy. Note that the aPTT is not reliable in LAC patients and, therefore, heparin
concentrations should be followed-up. Intravenous
heparin should be administered for at least 5 days or until clinical
improvement is noted.102 Thereafter, heparin may be administered subcutaneously 10,000 to 15,000 units
every 8 to 12 hours, titrating to an aPTT of 1.5- to 2- times
control (or heparin level of at least .2 U/mL) obtained 6 hours
after the morning dose.98,103 Alternatively therapeutic low-molecular-weight heparin may
be used (e.g., dalteparin sodium [Fragmin] 200 antifactor Xa units/kg administered subcutaneously
in divided doses twice per day −5000 μg q.b.i.d. in
a 50-kg woman). Alternatively, enoxaparin (Lovenox) 1 mg/kg
twice per day may be used. In the absence of data that monitoring
therapeutic efficacy is not required during pregnancy, it is our
practice to titrate the dose of low-molecular-weight heparin
to a target therapeutic antifactor Xa levels of .6 to 1.0 U/mL
assessed 4 to 6 hours after administration. Low-molecular-weight
heparin appears safe and efficacious in pregnancy.104 However, given concerns with epidural hematoma formation, epidural and
spinal anesthesia are contraindicated within 18 hours of low-molecular-weight
heparin administration. For this reason, we recommend
stopping therapy at 36 weeks, or earlier if preterm delivery is
anticipated, and using unfractionated heparin until delivery. After 4 months of either therapeutic unfractionated or low-molecular-weight
heparin therapy, the therapy may be switched to a prophylactic
dose of unfractionated heparin (5000 units subcutaneously
every 12 hours adjusted to produce an antifactor Xa level of .1 to .2 U/mL 6 hours
after the injection), or low-molecular-weight
heparin (e.g., dalteparin sodium [Fragmin, Upjohn] 2500 antifactor Xa units subcutaneously twice
a day, or enoxaparin [Lovenox, Aventis] 30 mg subcutaneously
twice daily titrated to maintain antifactor Xa levels of .1 to .2 U/mL 4 hours
after administration). Again, we recommend stopping
therapy at 36 weeks and using unfractionated heparin to permit epidural
and spinal anesthesia for delivery. Management of the delivery period can be difficult in high-risk
patients. Vaginal or cesarean delivery should not be accompanied by significant
risks of treatment-related bleeding if the procedure
occurs more than 4 hours after a prophylactic dose of unfractionated heparin. If
the patient has been using therapeutic doses of unfractionated
heparin an aPTT should be checked preoperatively. Protamine sulfate
may be administered to reverse a markedly prolonged aPTT at the time
of vaginal or cesarean delivery. Similarly, vaginal or cesarean delivery
should not be accompanied by significant risks of treatment related
bleeding if it occurs more than 12 hours after a prophylactic dose of
low-molecular-weight heparin or more than 18 to 24 hours
after a dose of therapeutic low-molecular-weight heparin. Antithrombin
concentrates can be used in antithrombin deficient patients
in the peripartum period. Heparin or low-molecular-weight
heparin should be resumed 4 to 6 hours after a vaginal delivery
and 8 to 12 hours after a cesarean delivery. Prophylactic therapy should be continued during the postpartum period for 6 to 12 weeks
after a DVT and 4 to 6 months after a PE or complex iliofemoral
DVT. Oral anticoagulant therapy can be initiated postpartum
by titrating the warfarin dose to maintain the patient's INR at
approximately 2 to 2.5. Note that warfarin has a more rapid inhibitory
effect on levels protein C than on many of the clotting factors because
of the former's shorter half-life (6 hours vs. 24 to 48 hours). Therefore, heparin should always be maintained during
the initial 4 days of warfarin therapy and until a therapeutic INR
is achieved to avoid warfarin-induced skin necrosis and paradoxical
thromboembolism. Calcium supplementation (1500 mg orally every day) should be
provided all patients treated with heparin or low-molecular-weight
heparin during pregnancy. In patients receiving more than 15,000 units
of unfractionated heparin for more than 6 months, bone densitometry
studies can be performed in the postpartum period. Evidence
of significant osteoporosis should prompt referral to a reproductive or
medical endocrinologist familiar with treating osteoporosis in premenopausal
women. As noted previously, the benefit of anticoagulation in
patients with SPT has been questioned.92 Warfarin Warfarin gains its therapeutic anticoagulant effect form its ability to
inhibit the action of vitamin K, which is a cofactor in the synthesis
of the final molecular forms of factors VII, IX, X, and prothrombin.105 Warfarin, a small molecule loosely bound to albumin, crosses the placenta. A 33% risk
of embryopathy is associated with exposure between 7 and 12 weeks' gestation. Stigmata include nasal hypoplasia, stippled
epiphysis, and central nervous system abnormalities. The latter
include dorsal midline dysplasia characterized by agenesis of the
corpus callosum, Dandy-Walker malformation, midline cerebellar
atrophy, and ventral midline dysplasia characterized by optic atrophy.104 Fetal and placental hemorrhage is also a major complication with warfarin
use during pregnancy and especially with delivery.106 Vitamin K or fresh-frozen plasma can be used to reverse the effects
of warfarin in the rare pregnant patient using this anticoagulant. The
prothrombin time begins to normalize within 6 hours of a 5-mg
dose of vitamin K (orally or subcutaneous). While larger
doses may work more quickly, they will render the patient resistant
to re-anticoagulation with warfarin. One clinical situation that may warrant the use of warfarin in pregnancy
is the rare patient with a mechanical heart valve in pregnancy. Because
of the lack of adequate controlled trials, no consensus exists to
guide the management of these patients. Older studies using heparin with
predominantly older-generation prosthetic heart valves have
shown and increase in thrombogenic complications including fatal maternal
valve thrombosis.107,108,109,110 No large clinical studies exist to guide in the use of low-molecular-weight
heparin in pregnant patients with mechanical heart
valves.108 However, the manufacturer of Lovenox (Aventis) specifically recommends against its use in this setting
based on reports to the FDA of valvular thrombosis in pregnant women
treated with Lovenox. The use of low-dose aspirin in adjunct to warfarin or heparin has been advocated based on a study
of antithrombotic therapy in high-risk patients with mechanical
valves.109 The risk of fatal maternal valve thrombosis may outweigh the risks of
warfarin to the fetus between 12 and 36 weeks' gestation. A full
discussion of these risks should precede conception in this select population. Women
with mechanical heart valves who choose either unfractionated
or low-molecular-weight heparin during the first
trimester should realize that they will be at higher risk for thrombosis. These
women should be maintained on an intravenous dose of unfractionated
heparin sufficient to prolong the aPTT two- to three-times
the control. After the first trimester and until near-term, the
therapy for these women can be switched to warfarin. Because
warfarin does not accumulate in breast milk and does not induce an
anticoagulant effect, it is not contraindicated in breastfeeding mothers. Thrombolytic Therapy and Surgery Thrombolytic therapy with plasminogen activators (tPA, urokinase, and
streptokinase) is relatively contraindicated in pregnancy because
of the theoretical risk of massive abruption. No controlled studies
examining the efficacy and safety of thrombolytic therapy in pregnancy. In
a review of 172 pregnant patients treated with thrombolytic therapy, the
maternal mortality rate was 1.2%, the fetal loss rate
was 6%, and maternal complications from hemorrhage occurred in 8%.111 Currently, massive PE with hemodynamic instability should be the only
indication for thrombolytic therapy in pregnant patients.112 Surgery, including embolectomy, should be reserved for life-threatening
settings. Inferior Vena Cava Filters Although rarely used in pregnant patients, inferior vena cava filters may
be used in pregnant patients with the same indications as the nonpregnant
population. These indications include patients with recurrent PE
despite adequate anticoagulation, cases of pulmonary embolism, or ileofemoral
DVT in a patient with a contraindication to anticoagulation
such as recent surgery, hemorrhagic stroke, or active bleeding, the development
of serious hemorrhagic complications with anticoagulant therapy.113,114,115,116 Retrievable vena cava filters may prove ideal for pregnant patients requiring
this therapy. Currently, they remain investigational.117 Prevention of Venous Thromboembolism Prevention of VTE should be the goal of the obstetrician. It is important
for the clinician to define the true risk of VTE in a particular pregnant
patient. Among pregnant patients who have had a previous VTE during
pregnancy, there is a 10% risk of recurrence.118 However, in general, among patients with a history of VTE before their
current pregnancy, the risk of recurrence will vary with the presence
of a thrombophilia or risk factors at the time of the previous VTE. Brill-Edwards
and colleagues prospectively studied 125 pregnant
women with a previous episode of VTE and withheld heparin until the post-partum
period.119 They observed that three of the 125 women (2.4%) had
a recurrent VTE event (95% confidence interval [CI]: .2%–6.9%). However, there were no recurrences
in the 44 women who had no evidence of thrombophilia and whose previous
VTE was associated with a temporary risk factor (e.g., oral contraception, surgery, and pregnancy). In contrast, among the 51 women
with a thrombophilia or unexplained previous VTE episode, or
both; three (5.9%) had a recurrent VTE (95% CI: 1.2%–16.2%). Other authors have also
observed that thrombophilic patients with a previous VTE or in whom
the VTE was unexplained (i.e., not associated with nonrecurring risk factors) have a substantially
increased risk of recurrent VTE in pregnancy.65,120 We recommend that patients having a previous DVT associated with a nonrecurring
nonpregnant state (e.g., high-estrogen dose oral contraceptives or after orthopedic procedures
or surgery) who are without other risk factors (e.g., identifiable thrombophilia, need for prolonged bedrest, obesity, superficial
thrombophlebitis) do not appear to need prophylactic heparin
therapy during pregnancy but should receive postpartum prophylaxis
because 80% of pregnancy-associated fatal PEs occur in the
postpartum period. In these patients, the individual risks and benefits
must be weighed. Obviously thrombophilic patients or those with previous
unexplained DVT or other major risk factors for VTE should have
both antenatal and postpartum unfractionated or low-molecular-weight
heparin prophylaxis. We would include a VTE occurring in
a previous pregnancy as unexplained risk factor for VTE. Historically, mini-dose heparin has been effective in preventing
DVT in patients at risk. As noted, the standard dose of unfractionated
heparin is 5000 units administered subcutaneously every 12 hours. However, some
authors recommend following-up heparin levels to guide
VTE prophylaxis during pregnancy.121 In a small study by Barbour and associates, heparin doses appropriate
for prophylaxis varied from patient to patient and from one trimester
to the next. They suggested following-up heparin levels in order
to obtain adequate prophylaxis.122 Adequate heparin levels for prophylaxis have been defined as .1 to .2 U/mL. Studies
also exist that document the safety and effectiveness
of low-molecular-weight heparin for thromboprophylaxis. A
dose of 40 mg subcutaneously per day of enoxaparin was adequate to
prevent VTE in a prospective study of 69 pregnancies (61 women).123 Patients with antithrombin deficiency or homozygotes for prothrombin or
FVL mutations should receive therapeutic unfractionated heparin or low-molecular-weight heparin therapy during pregnancy and
full anticoagulation in the puerperium regardless of their history of
previous VTE. Nonpharmacologic therapies aimed at preventing VTE in pregnancy include
left-lateral decubitus positioning during the third trimester, graduated
elastic compression stockings, and pneumatic compression stockings. The
graduated elastic compression stockings have been shown to
increase femoral vein flow velocity in late pregnancy124; however, their role in decreasing VTE in pregnancy has yet to be defined. Pneumatic
compression stockings improve blood flow and decrease stasis
by using sequential cephalad compression. In a study by Nicolaides
and associates, pneumatic compression of the lower extremities increased
blood flow in the femoral vessels by 240%.125 By increasing plasminogen activator, pneumatic compression stockings also
increase fibrinolysis.126 In a meta-analysis of moderate-risk surgery, they were shown
to decrease the incidence of DVT by 60%.127 Because pneumatic compression stockings have no hemorrhagic risk associated
with use and they have been shown to be an effective means of DVT
prophylaxis in gynecologic oncology surgery,128 it stands to reason that they would be an ideal device for prophylaxis
in at-risk pregnant patients at prolonged bed rest or who are
undergoing a cesarean delivery. |