The sinoatrial (SA) node has characteristics that allow depolarization to occur faster than it does in other myocardial cells, initiating the contraction sequence. When the SA node depolarizes, the atrial myocardium contracts in a synchronous fashion with the lateral walls moving toward the interatrial septum, forcing blood into the ventricular chambers. The electrical impulse generated at the SA node is conducted through the atrial myocardium to the atrioventricular (AV) node. From the AV node, the signal is propagated to the right and left ventricular chambers through the Purkinje fibers and causes ventricular systole.
The fetal heart functions in many ways similar to the adult heart. It is important to understand some of the distinguishing features, however. The fetal heart rate is significantly higher (120 to 160 beats/min) than the adult heart rate (50 to 70 beats/min). The fetal myocardium is less compliant than the infant or adult heart. In addition, sometimes there are aberrant pathways connecting the atrial and ventricular chambers, providing an accessory route for AV depolarization.
Methods of Assessment
In the newborn or adult patient, a 12-lead electrocardiogram (ECG) is used to establish the type of arrhythmia. The prenatal diagnosis cannot rely on an ECG through the maternal abdominal wall because the fetal P waves are rarely seen, and the fetal QRS complex is dwarfed by the signals from the maternal abdominal ECG. Investigators rely primarily on ultrasound.
Fetal studies usually begin with an examination of fetal and cardiac anatomy by the use of two-dimensional ultrasound. The fetus also is examined for collections of fluid in the pericardial sac, the pleural spaces, the abdomen, and the skin. The amount of amniotic fluid is recorded. Cardiac size and wall thickness are evaluated.
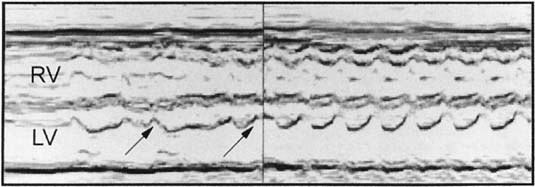
M-mode ultrasound traditionally has been used to establish the type of arrhythmia. The M-mode cursor is positioned so that it crosses one structure that moves with atrial contraction (atrium, AV valves) and one structure that moves with ventricular contraction (ventricle, semilunar valves). In this manner, the relative timing of events can be established.
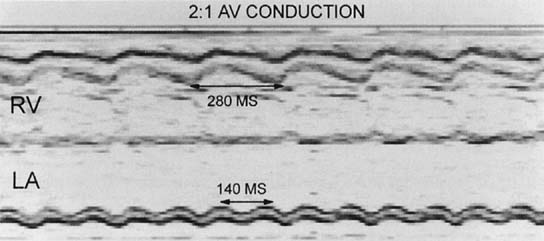
We rely primarily on pulsed Doppler ultrasound for rhythm assessment and detection of flow disturbances (regurgitation, stenosis). The rhythm assessment begins with measurement of the heart rate and its variability. The atrial rate and ventricular rate should be assessed independently because they may not have a one-to-one association.
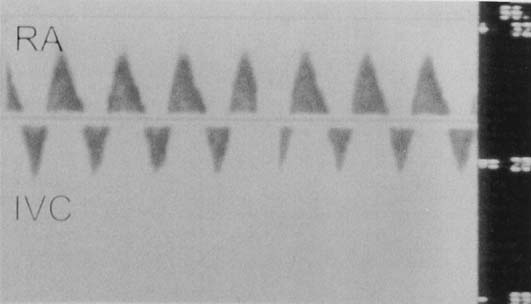
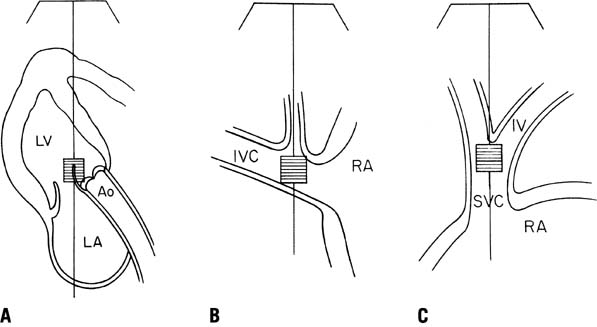
The next phase of the fetal rhythm assessment is determination of atrial and ventricular activation. Our laboratory has used many fetal Doppler interrogation sites for rhythm interpretation (Fig. 1). The venous sampling sites (superior and inferior vena cavae) are useful for determination of atrial rates and rhythm, and the inflow-outflow region of either ventricle is useful for determining the association between ventricular diastolic events and ventricular systolic flow events.1 Normal sinus rhythm is determined on the basis of a normal rate, a stable sequence of atrial to ventricular contractions, and accelerations in heart rate with fetal activity or uterine contractions. Tricuspid or mitral inflow Doppler shows a stable A wave caused by atrial contraction. There should be little (<20%) or no reflux of flow into the systemic veins.2
|
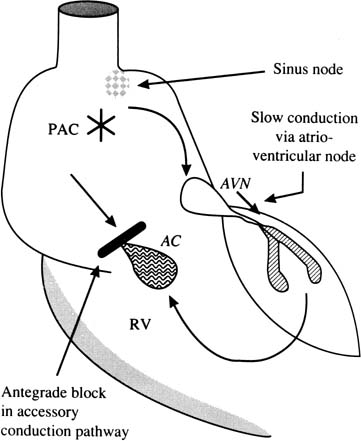
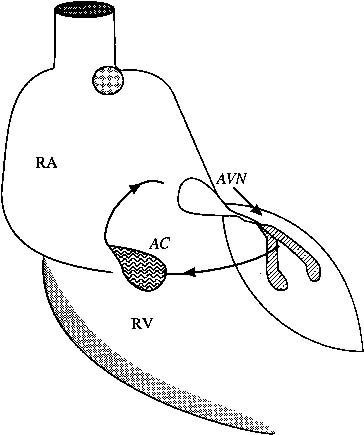
Abnormal atrial activation associated with atrial premature contractions, supraventricular tachycardia (SVT), junctional rhythm with retrograde conduction, or AV dissociation is identified by intermittent or continuous systemic venous reflux noted by Doppler. The so-called cannon A wave is caused by atrial contractions against a closed AV valve. This reversal of flow also is noted when ventricular compliance is abnormal, which can be seen in fetal acidosis and intrauterine growth restriction.3,4,5
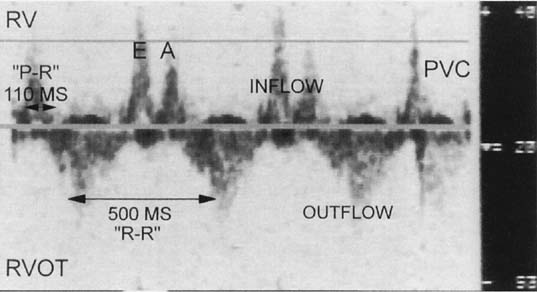
If the sample volume size is increased and the transducer is positioned in the inflow-outflow region of the ventricle, the noninvasive P-R interval can be determined during sinus rhythm (Fig. 2). The inflow-outflow view may not be as predictive of the P-R interval during tachycardia, however, because of changes in diastolic timing.
|
The third aspect of fetal rhythm assessment is determination of the mechanism of arrhythmia.6,7,8 Important issues include the following:
- Are atrial and ventricular contraction rates normal or abnormal?
- If the contraction rates are abnormal, is there a regular or stable pattern
of contraction in the atrium and ventricle?
- If the pattern of contraction is irregular, is the irregularity repetitious
or irregularly irregular?
- What is the association between the atrial and ventricular contractions?
Answering these questions defines the rhythm, and electrophysiologic texts can be used to specify further the diagnosis or differential diagnosis (Table 1).9,10 A full description of the arrhythmia cannot occur, however, until the postnatal period. At our institution, a transesophageal electrophysiologic procedure is performed shortly after delivery in infants with sustained tachyarrhythmias. For bradyarrhythmias and fetal ectopy, an ECG and 24-hour ambulatory recording are usually sufficient to define the rhythm.
TABLE 1. Differential Diagnosis of Fetal Arrhythmias
Atrial Rate | Atrial Rhythm | Ventricular Rate | Ventricular Rhythm | AV or VA Association | Differential Diagnosis |
Normal | Regular | >Atrial rate 180–200/min | Regular | Dissociated or intermittent 1:1 | Accelerated ventricular or junctional rhythm |
Normal | Regular | >>Atrial rate 210–400/min | Regular or minimally irregular | Dissociated or 1:1 | Ventricular tachycardia, junctional tachycardia |
Normal | Regular | <<Atrial rate 40–110/min | Regular | Dissociated | 3rd-degree AV block |
Normal | Regular | < Atrial rate 70–110/min | Irregular | Intermittent dissociated | 2nd-degree AV block |
High 200–300/min | Regular | Same as atrial rate | Regular | 1:1 | AV re-entrant SVT, AV node re-entrant SVT |
High 180–300/min | Regular but varying | Same as atrial rate | Same as atrial rhythm | 1:1 | Permanent junctional reciprocating tachycardia |
High 300–450/min | Regular | < Atrial rate 150–225/min | Regular or irregular | 1:1, 2:1, 3:1, or variable | Atrial flutter |
High 180–220/min | Regular but varying | Same as atrial rate | Same as atrial rhythm | 1:1 | Sinus tachycardia |
High | Irregular “chaotic” | Same as atrial rate | Irregular | Variable | Chaotic atrial tachycardia, atrial flutter/fibrillation |
Low 80–110/min | Regular | Same as atrial rate | Regular | 1:1 | Sinus bradycardia, junctional rhythm |
AV, atrioventricular; VA, ventriculoatrial; SVT, supraventricular tachycardia.
Fetal Magnetocardiography
The fetal magnetocardiogram (MCG) is a more recent addition to the noninvasive tools used to diagnose fetal arrhythmias. The fetal MCG is an alternative signal source for fetal heart rate monitoring that seems to offer significant advantages over other methods. The fetal MCG is the magnetic analog of the fetal ECG but exhibits superior signal quality because of the favorable transmission properties of magnetic signals. Fetal MCG typically has a much greater signal-to-noise ratio and is free of maternal interference compared with fetal ECG monitoring. Because of the expense and sophistication of the instrumentation, only a few groups in the world are working in this area11,12; however, impressive progress has been made. A database of normative fetal conduction findings in several hundred fetuses has been validated against ECG. The heart rate and the waveform obtained by the fetal MCG are particularly useful for the diagnosis of specific types of fetal arrhythmias, such as Wolff-Parkinson-White syndrome13 and long QT syndrome.14