Attempts to refine the sensitivity and specificity of chromosomal screening and to reduce the overall costs of the screening programs per se have been the focus of more recent efforts.1,2 The goal is to reduce the need for expensive invasive testing that follows a positive screening and, although not commonly mentioned, to reduce the cost of the care of abnormal newborns who as a result of screening might be detected and the pregnancy terminated at the wishes of the parents.3,4 Work surrounding such screening and reducing the incidence of birth defects falls into essentially four categories:
- The use of preconceptual and early pregnancy folic acid to reduce the incidence
of neural tube defects
- The use of biochemical and ultrasound markers in the second trimester to
increase the detection of Down syndrome
- The expansion of biochemical markers into the first trimester to allow
for screening at earlier gestational ages
- The development of biophysical-ultrasonic characteristics of fetal structure
in the first and second trimesters.
Neural Tube Defects
In the 1970s, Brock and Sutcliffe5 first described the use of AFP in amniotic fluid and later in maternal serum6 for the prenatal detection of neural tube defects. Routine prenatal screening has been accepted in the United Kingdom since the mid-1970s and in the United States since the mid-1980s. Evaluation of the impact of such screening has shown that the birth rate of children with neural tube defects has declined from 1.3 per 1000 births in 1970 to 0.6 per 1000 births in 1989.7 In some sections of the United States, such as the Southeast, which had higher than average rates, the decline has been even more dramatic.
Several changes in the epidemiologic characteristics of neural tube defects have been observed: (1) The proportion of spina bifida cases has increased. (2) The proportion of neural tube defects combined with other unrelated defects (i.e., syndromes) has increased. (3) The incidence in the white population has decreased relative to the incidence in other races. (4) The incidence of isolated neural tube defects in females has decreased. All of the aforementioned findings are consistent with increased use of maternal serum AFP screening, particularly in the white population. A similar study in South Australia by Chan and colleagues8 from 1966 to 1991 found that the overall prevalence of neural tube defects (including prenatally diagnosed cases) had not varied between the two years. There was an 84% reduction in births, however, from 2.29 per 1000 in 1966 to 0.35 per 1000 in 1991. The fall was 96% for anencephaly and 82% for spina bifida. Approximately 85% of open and closed defects were detected before 28 weeks’ gestation by AFP or ultrasound. Likewise, the proportion of terminations in prenatally detected cases has risen steadily, from an average of about 20% in 1980 to greater than 80% in 1991.
It has long been appreciated that there are racial, geographic, and ethnic variations in the incidence of neural tube defects and that patients are at increased risk based on other medical conditions. Diabetics are known to have an increased risk of neural tube defects, as are women taking antiepileptic drugs.9 Conversely, a 1992 study concluded that patients undergoing ovulation induction do not have higher than background rates of neural tube defects.10
An important question has been the association of folic acid, its deficiency, and the incidence of neural tube defects. Experimental, clinical, and embryologic studies have investigated the role of vitamins as a causative factor. Special attention has been focused on the essential B vitamin, folic acid, which serves as a methyl donor involved in nucleic acid synthesis, purine-pyrimidine metabolism, and protein synthesis.11 Because of increased folic acid needs in pregnancy, pregnant women are particularly prone to develop relative deficiencies. Other possible influences include insufficient diet; physiologic hemodilution of pregnancy; increased plasma clearance; and genetic disorders that might affect production, transport, and metabolism.12
Throughout the 1990s, there have been many studies attempting to supplement folic acid in women at high risk for neural tube defects that generally have shown a decrease in the recurrence risk for such women who already have had an affected child. The U.S. Food and Drug Administration mandated, beginning in 1998, folic acid supplementation of breads and grains. Our group has shown a dramatic drop in the proportion of high maternal serum AFP values in the United States after the introduction of routine supplementation of breads and grains.22 Another published study showed a 20% drop in neural tube defects based on certifiable data.23 Several others likewise have confirmed a remarkable decrease in incidence.24
Screening for Chromosome Abnormalities
In 1984, Merkatz and coworkers25 first published the association of low maternal serum AFP with an increased risk of chromosome abnormalities, particularly Down syndrome. In subsequent years, there has been gradual acceptance of the association and an understanding that Down syndrome is not the only aneuploid condition associated with low maternal serum AFP. Trisomy 18 has even lower AFP values.26
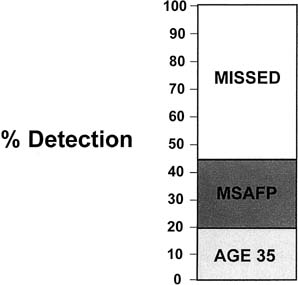
The adoption of wide-scale screening with maternal serum AFP effectively doubled the potential detection of chromosome abnormalities in the population. Even with an aging reproduction population, only 25% to 30% of Down syndrome infants are born to women older than age 35 (Fig. 1). The addition of a well-coordinated maternal serum AFP screening program can detect approximately 30% of the 80% of cases that are born to women younger than age 35. The mechanics of biochemical screening (i.e., with adjustments for gestational age, race, diabetic status, multiple gestation status, and maternal weight and adjustments via a different database or correction factors for maternal race) have been published previously and are not repeated here.27
|
In 1988, Wald and associates28 suggested that a combination of parameters including AFP, β-human chronic gonadotropin (β-HCG), and unconjugated estriol could significantly increase the detection frequency of Down syndrome to approximately 60% of the total (Fig. 2). Multiple studies have corroborated the increased efficacy of multiple marker screening as opposed to AFP alone in detecting chromosome abnormalities, particularly Down syndrome.29,30,31,32,33,34
|

Despite overwhelming data and recommendations of national organizations such as the American College of Obstetrics and Gynecology that multiple marker screening be offered, nearly 20% of patients in the United States who have screening still have AFP alone.35 There is essentially universal agreement that among the three common parameters used, AFP, β-HCG, and unconjugated estriol, that if one could choose only one of the above, that β-HCG is the best. There is a virtual tie for second place in efficacy between AFP and unconjugated estriol. Because AFP already is used in North America and much of Western Europe for the detection of neural tube defects, however, the only real remaining question is whether adding unconjugated estriol as a third parameter is cost beneficial.
The debate over the use of unconjugated estriol (i.e., double screening versus triple screening) has become intense and emotional with staunch proponents on both sides. We believe the studies as a whole suggest there is no real effectiveness of adding the third marker. The literature is divided between several studies that say the third marker helps and others that say it does not. Since 1991, when Crossley and associates first proposed the β-HCG-to-AFP ratio be used as a marker, the question of how the data are interpreted has been added into the overall equation of sensitivity and specificity. Our data suggest that the differences may be due to higher variability among unconjugated estriol assays rather than to the other parameters. This variation may explain in part the incongruity among studies.37 Cuckle38 raised the question of whether screening should be offered to all patients or, for example, only to patients age 27 or older and showed that if screening were offered only to women older than age 27, more than 50% of the population would be excluded, and there would be an approximate 9% lower detection of affected pregnancies. Although not reaching the conclusion that this was cost beneficial, Cuckle38 stated that such rationing of services can be considered when resources are scarce.
Evans and colleagues37 investigated many of the “dogmas” of 3 decades of biochemical screening and found that many of these are no longer valid. We believe the wide variance in results reported from around the world is largely due to subtle and sometimes not so subtle differences in laboratory methods.37 The bitter arguments about double versus triple screening are, in part, answered by the fact that there is a much wider variability in estriol measurements than other parameters. Such variation helps explain why labs have such widely diverging experience with estriol. When the methods are standardized, much of the variability disappears and allows for an “apples versus apples” as opposed to “apples versus oranges” comparison. Similarly, much of the reported variation in the literature likewise disappears with standardization, and the diabetic correction factor becomes unnecessary with proper accounting for the fact that diabetic patients are of higher maternal weight.39,40
Many articles in the past several years have looked at the various constituents in the marker regimen, with the most important being touted being use in the second trimester of free β-HCG as opposed to intact β-HCG. Wald and Hackshaw41 reported in 1993 that the use of free β-HCG compared with total β-HCG would increase the detection frequency by about 4% for a given false-positive rate used in conjunction with maternal age, AFP, and unconjugated estriol. Other studies have suggested that, particularly at earlier gestational ages (e.g., 14, and 15 weeks) free β-HCG may have better sensitivity and specificity than the intact molecule.
Another promising marker has been the search for fetal cells in maternal circulation. Studies throughout the 1990s suggested that isolation and analysis of fetal cells may become practical and useful as a screening test.42,43,44 The current state follows essentially 2 decades of various starts and stops that alternatively have looked promising and frustrating since Hertzenberg and coworkers45 first showed detection and enrichment by fluorescent activated cell sorting. Much of the work of the past 2 decades has focused on ways to improve the efficacy of detection methods primarily centered on the need to increase the enrichment of fetal cells from the maternal blood circulation whose prevalence has been estimated to be approximately 1 in 10 million cells.46,47,48
Three types of fetal cells have been sought extensively—trophoblast, lymphocytes, and nucleated fetal red blood cells. The cell type most likely to be successful is thought to be nucleated red blood cells. Bianchi and colleagues46 were the first to use flow sorting to isolate nucleated fetal erythrocytes using an antibody of the transfer interceptor. More recent studies have focused on two general approaches—fluorescent activated cell sorting and magnetic activated cell sorting.43 Trisomic conceptions subsequently confirmed by invasive testing have been found by both methods.46,47,48 Although the results are encouraging, further work needs to be done before the screening test becomes practical. Analysis of progress through the millennium suggested that the magnetic activated cell sorting approach seemed to have a better sensibility than fluorescent activated cell sorting to isolate fetal cells.43 Another approach of looking at freeDNA (i.e., not cellular) is developing as a promising area of study.49 Although the overall sensitivity of fetal cells was not an improvement over current screens, the specificity of fetal cells might be much better. If so, a two-step approach might emerge in which a higher percentage of patients—perhaps 10% using double or triple testing—would be called positive to raise the sensitivity to around 80%. Then these 10% would undergo fetal cell testing to reduce that risk group to 2% to 3% but not lose sensitivity.
Another area of potential applicability of fetal cells in maternal blood is for the isolation of molecular diagnosis of mendelian disorders. Lo and associates50 were able to determine fetal Rh status in women known to be sensitized and married to heterozygous men. Geifman-Holtzman and colleagues51 determined fetal Rh status using polymerase chain reaction fetal nucleated red blood cells sorted from maternal blood. The next several years ultimately will determine how successful fetal cell sorting is as a screening test. It originally was hoped that it could be a diagnostic test and replace the need for invasive testing; however, as of this writing, fetal karyotypes cannot be obtained from cells that are isolated, and only fluorescence in situ hybridization–related results are possible. Although such is good as a screening test for aneuploidy, our experiences show that approximately one third of abnormal karyotypes seen in prenatal diagnosis programs are not ones that would be detected by the standard probes for chromosomes 13, 18, 21, X, and Y.52 Until and unless complete karyotypes can be obtained, fetal cells will not replace invasive testing but may be an important addition to the armamentarium of screening technologies.
Many studies have suggested dimeric inhibin A as an excellent marker that may raise the sensitivity by 3% to 7% for a given screen-positive rate.53 Some have suggested quadruple screening. There are also paradigms that include different parameters at different times combined. Although preliminary data suggest a high sensitivity with improved specificity, hiding results from patients for up to 1 month is ethically problematic in our opinion. No doubt there multiple approaches to screening will emerge, and there will be no one uniform standard approach.
Another two-step approach has been the so called integrated test.54 This is a combination of first-trimester blood and ultrasound. The results are not communicated to the patient, who then waits for second-trimester blood results before a risk assessment is made. Preliminary data suggest a reduced false-positive rate for comparable sensitivity, but the tradeoff is the need for patients to wait 6 weeks for start to finish of the screening process. For patients who do not particularly care about the results, the delay may be fine, but many patients would find such delay intolerable.
Trisomy 18
Although screening generally has focused on trisomy 21, our data and those of others always have shown a varied pattern of anomalies detected by screening.55 A different pattern of analytic levels has been observed in trisomy 18. The values of AFP, β-HCG, and unconjugated estriol appear to be low.56 These low values suggest a different pathophysiology than for Down syndrome. In Down syndrome, the low AFP and unconjugated estriol and high β-HCG can be explained as reflecting inappropriate immaturity or dysmaturity of the fetus (i.e., all values are consistent with a younger gestational age). In trisomy 18, that explanation does not work. We previously showed that there are different patterns of genomically directed intrauterine growth retardation in different aneuploidies, but how this translates into serum markers is unclear. Nevertheless, some reports have shown that an algorithm can be used to identify most trisomy 18 cases, while adding about 0.75% to the population being offered amniocentesis.