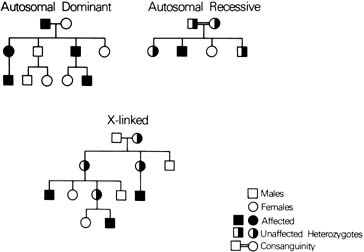
Autosomal Dominant Conditions SKELETAL DYSPLASIA Until recently, the diagnosis of skeletal dysplasias in children was dependent
on radiographic and physical features. Because of the often-subtle
variations between categories and the limitations of ultrasonography, prenatal
detection was limited to only the most severe types. With
the discovery of the molecular basis for many of these conditions, a
precise prenatal diagnosis can now be determined as early as the
first trimester via chorionic villus sampling (CVS). ACHONDROPLASIA Achondroplasia is the single most common skeletal dysplasia in humans. Inherited
in an autosomal-dominant fashion, a person with achondroplasia
has a 50% chance of having a child with this disorder. Because
the gene is 100% penetrant, a person will have the disease
if the gene is inherited. Parents of normal height who have a child
with achondroplasia would normally be expected to have a nearly 0% chance
of having a second child similarly affected; however, the
risk is actually 5% to 6% because one parent (usually
the father) has the mutation in more than one germ cell, known
as germ cell mosaicism (Fig. 8). Achondroplasia results from a mutation in a gene known as fibroblast
growth factor receptor 3 (FGFR-3). Identification
of a mutation in this gene enables a precise diagnosis in subsequent
pregnancies, either by amniocentesis or by CVS.4,5 Moreover, the evaluation is simplified by the fact that all mutations
to date have been point mutations, and almost all of the mutations have
involved the same amino acid substitution. 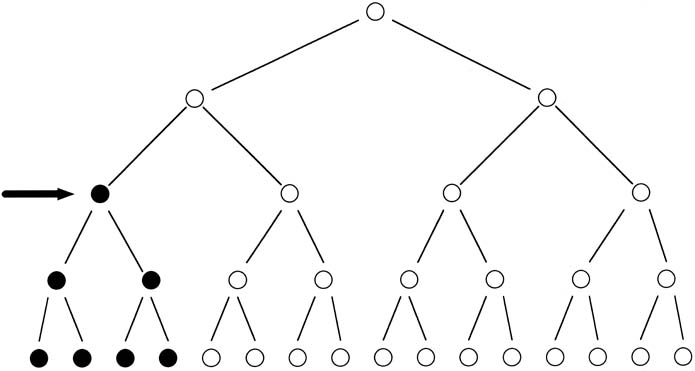 Fig. 8. Schematic representation of a mutation in the formation of sperm (arrow), resulting
in germline mosaicism for a new mutation. Fig. 8. Schematic representation of a mutation in the formation of sperm (arrow), resulting
in germline mosaicism for a new mutation.
|
FGFR-3 mutations also have been found to cause lethal dwarfism known
as thanatophoric dysplasia.6 Like achondroplasia, it represents a new dominant mutation with an empiric
risk for recurrence of approximately 5%. As important, however, is
the ability to use molecular testing to confirm the diagnosis
in cases in which severe short-limb dwarfism is diagnosed on the
basis of ultrasound examination. Using amniotic fluid cells, one can
make a precise diagnosis and therefore provide the family with more
exact prognostic information. OSTEOGENESIS IMPERFECTA Osteogenesis imperfecta (OI) describes a constellation of disorders
characterized by brittle bones and defects in type I collagen. The
form of OI of significance for the obstetrician–gynecologist
is the perinatal lethal form, OI type II. Also a new dominant mutation, it
can most easily be diagnosed by assays of collagen structure.7 If these assays are performed on fibroblasts from the affected child, prenatal
diagnosis is straightforward in any subsequent pregnancy. There
is, however, one major caveat: because this analysis is of protein
structure, not DNA sequence, future pregnancies must be evaluated by CVS, not
amniocentesis. Collagen defects can be studied in chorionic villi, but
not in aminocytes. NEUROLOGIC DISEASES The onset of many of the autosomal-dominant neurologic diseases
occurs in adulthood, and these diseases are generally more familiar to
the neurologist than to the obstetrician–gynecologist. However, two
of these disorders, myotonic dystrophy and Huntington disease, are
discussed to illustrate the necessity of having a basic knowledge of
the mechanisms of inheritance and the clinical implications of these
disorders. Myotonic dystrophy (MD) is a condition characterized by myotonia, cataracts, and
other variable features, such as male-pattern
baldness. The onset of symptoms usually occurs in the third or fourth
decade of life. In addition to the adult form, there is a neonatal
form of the disorder known as congenital myotonic dystrophy. This disorder
is characterized by severe hypotonia, respiratory compromise, and, often, death
in the newborn period. Those infants who do survive commonly
have severe developmental delay. The MD gene is characterized by a repeated sequence of cytosine-thymidine-guanine (CTG), which in normal persons is repeated
between 5 and 30 times. This "trinucleotide repeat" is expanded
to greater than 100 repeats in persons affected with adult-onset
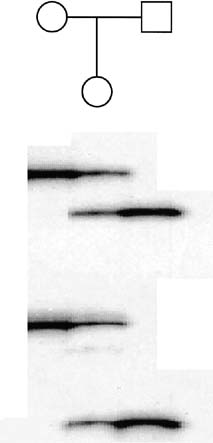
MD.8 Neonates with congenital MD may have more than 2000 copies of the repeat. Figure 9 presents a pedigree of a family that would typically present to the obstetrician–gynecologist. The patient (proband) presents
at 10 weeks' gestation with a history that her sister has adult-onset
MD. Neither of her parents, both in their late 1950s, have
any symptoms to suggest that either is affected with MD. Based on the
absence of any symptoms in the patient or her parents, we might assume
that the sister's disease represents a new dominant mutation. Molecular
testing of the family, however, indicates a much different
scenario. The sister has 200 repeats, consistent with her clinical symptoms. Her
mother has 50 repeats, which is in the range of what is called
a premutation (35–100 repeats). The proband also has 200 repeats and
can be expected to experience MD symptoms in the future. Molecular evaluation
reveals that the fetus has 2000 repeats, consistent with the
diagnosis of congenital MD. 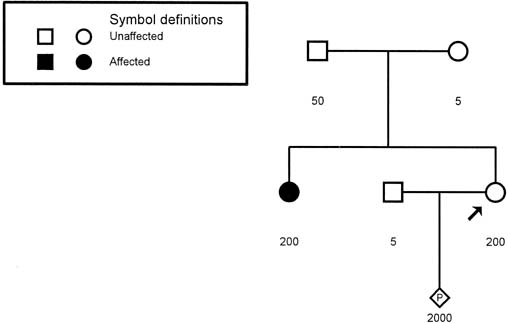 Fig. 9. Pedigree of a family with an apparently isolated case of myotonic dystrophy (shaded
circle). Molecular studies for trinucleotide repeats
are depicted under each symbol. The number of repeats in the proband (arrow) is similar to that of her affected sister, and the proband can be
expected to experience symptoms in the future. Fig. 9. Pedigree of a family with an apparently isolated case of myotonic dystrophy (shaded
circle). Molecular studies for trinucleotide repeats
are depicted under each symbol. The number of repeats in the proband (arrow) is similar to that of her affected sister, and the proband can be
expected to experience symptoms in the future.
|
Trinucleotide repeat disorders are characterized by the presence of unstable
premutations that may remain unchanged or may amplify during spermatogenesis
or oogenesis. This process of expansion of the trinucleotide
repeat is known as anticipation. In the case of MD, marked amplification can occur when the gene is passed
from the mother, but not from the father. Therefore, the congenital
form of MD is seen only when the gene is passed from the mother to
the fetus. Determination of the status of a person with a family history
of MD (i.e., normal, premutation, full mutation) is quite
precise with the use of modern molecular techniques. Huntington disease (HD) is a late-onset, progressive, fatal
disease inherited in a classic autosomal-dominant fashion. The
early symptoms of the disease are subtle loss of muscle coordination, forgetfulness, and
personality changes. The disease progresses in
stages, from choreiform movements (hence the older name of Huntington's
chorea) to hypokinesis and then to rigidity. Ultimately, the
patient is bedridden with dysphagia, dysarthria, and impairment
of gait and coordination. Onset of the disease occurs most commonly
between the ages of 30 and 50 years.9 HD is also caused by a mutation involving a trinucleotide repeat sequence. Normal
persons have between 11 and 31 copies of a CAG repeat. The
full mutation range is between 38 and 100 copies. There is an intermediate
range of 32 to 38 repeats, and there are examples of both affected
and unaffected persons with this number of repeats.10 Therefore, there does not appear to be a true premutation in HD. Unlike
MD, inheritance of HD from the father is associated with expansion of
the repeats and an earlier age of onset. In approximately one third
of cases in which the father passes on the gene to his offspring, there
is an expansion resulting in juvenile-onset HD.11 The ability to diagnose HD precisely by molecular techniques offers the
possibility of presymptomatic (predictive) testing and prenatal
detection of an affected fetus. Although predictive testing can offer
freedom from the psychological burdens associated with being at risk
if the person does not carry the mutation, the impact on persons found
to have the gene can be devastating. Those found to carry the gene
face the inevitability of a disease for which there is currently no
treatment. More commonly the obstetrician--gynecologist will be presented
with the request for prenatal testing by an at-risk patient. It
must be emphasized to the couple that testing will determine precisely
whether the fetus has the HD gene. Thus, a positive result gives
an at-risk patient a presymptomatic diagnosis of HD. Therefore
direct prenatal testing should not be performed unless an at-risk
patient has already undergone predictive testing. An alternative to prenatal testing is available for at-risk couples
who do not wish to undergo predictive testing. This method is known
as exclusion testing.12 The HD gene is located on chromosome 4, and DNA markers (restriction
fragment length polymorphism [RFLP]) are available
that allow chromosome 4 to be traced through generations. By testing the
grandparents of the fetus, it can be determined whether the fetus received
a chromosome 4 from the affected grandparent or the unaffected
grandparent. Because the chromosome 4 received from the affected grandparent
could carry either the HD gene or the normal gene, the fetus has
the same risk as the parent for having HD (i.e., 50%). If
the fetus receives a chromosome 4 from the unaffected grandparent, the
risk for having HD is 0%, and the patients can be assured
that the gene will not be passed on to the next generation. Because of the significant ethical, psychological, and medical issues associated
with predictive testing, such testing should take place only
at institutions where protocols are in place for pretesting and posttesting
counseling and support. Autosomal Recessive Disorders CYSTIC FIBROSIS This autosomal recessive condition is the most common recessive condition
affecting whites, with a carrier frequency of one in 29. Cystic fibrosis
is caused by a mutation in the CFTR gene, a chloride ion channel
in epithelial cells. Mutations in this gene cause severe lung disease
and pancreatic insufficiency in the classic form but may result in milder
phenotypes, including only congenital bilateral absence of the vas
deferens in some affected males. The most common mutation is delta-F508, the
deletion of a phenylalanine residue at position 508, which
accounts for 70% of the mutations in the white population. To
date, however, there are more than 1000 known mutations throughout
the gene that can cause disease. Because of the large number of known
mutations in the gene, and because the phenotype is variable, prenatal
testing may be problematic as complete gene sequencing is expensive, cumbersome, and
impractical. Screening for the most common mutations
in a population is more practical. Several laboratories now offer panels
testing for the most common mutations in the white population. In 1997 the
National Institutes of Health (NIH) Consensus Development
Conference on Genetic Testing for Cystic Fibrosis recommended that
genetic screening for cystic fibrosis be offered to individuals with
a family history of cystic fibrosis, to partners of individuals with
cystic fibrosis, to couples planning a pregnancy, and to couples seeking
prenatal care.13 In 2001 the American College of Obstetricians and Gynecologists (ACOG) adopted
these recommendations and set forth the following testing
guidelines: in addition to couples who have a family history of
cystic fibrosis or when one partner is affected by cystic fibrosis, all
couples in which one or both of the partners are white (including
those of Ashkenazi Jewish descent) and are planning a pregnancy
or are currently pregnant be offered carrier screening for cystic fibrosis.14 ACOG recommended that these couples be offered a prenatal screen that
includes the 25 most common mutations and any mutation with a population
incidence of more than1%. Although prenatal identification of a fetus affected with cystic fibrosis
gives couples the option of pregnancy termination, cystic fibrosis
is not a condition that is currently treatable in utero, nor does prenatal detection of the disease appear to improve outcomes
for affected individuals. Rarely, an affected pregnancy may present with
ultrasonographic abnormalities such as echogenic bowel (caused
by meconium ileus). HEMOGLOBINOPATHIES Rapid progress has been made in the area of antenatal detection of the
hemoglobinopathies, and they serve as the prototype for the available
diagnostic methodologies. From initially requiring a fetal blood sample
obtained by the relatively high-risk procedures of fetoscopy
or placental aspiration, we have moved to methods that require only uncultured
amniotic fluid cells, or chorionic villi. Hemoglobin is a tetrameric protein consisting of four globin chains. In
humans, there are six structurally different types of globin chains: alpha (α), beta (β), gamma (γ), delta (δ), epsilon (ε), and zeta (ζ).15 The synthesis of various chains is switched on and off during the process
of differentiation and may represent an adaptive process for the developing
fetus. The embryonic chains (ε and ζ) are
rapidly replaced as the fetus develops. Each of the different hemoglobins
found in humans is formed by the combination of two α-chains
and two non-α–chains (γ, β, δ). The messenger RNA (mRNA) for each globin gene is transcribed
from a varying number of genes, which may differ among populations. In
general, there are four genes controlling α-chain synthesis
and two controlling β-chain synthesis. Genes for the α-chains
are located on chromosome 16, and those for β, γ, and δ are
located on chromosome 11. In the hemoglobinopathies, the
specific disorder may be considered recessive, because heterozygous
persons have half the genes (either one or two) functioning
to make the normal protein and half functioning to make no protein
or an abnormal one. Therefore, either the homozygous state or a double
heterozygous state (i.e., two different deleterious mutations) can
result in a hemoglobinopathy. Sickle cell disease is caused by a single nucleotide change (AT) in
codon six of the b gene. A similar point mutation (GA) in
the same codon results in sickle C disease. As discussed, sickle
cell disease can result from the homozygous state (SS or CC) or
from the double heterozygous state (SC). Sickle S disease
is unique in that the mutation modifies the recognition site for two
restriction endonucleases, Mst II and DdeI. As outlined in Figure 2, this change results in a change in length of the DNA fragment with the
sickle gene. In Figure 10, a Southern blot technique is shown, indicating that it is possible to
distinguish sickle trait (both βS and βA), sickle disease (only βS), and normal hemoglobin (only βA) by
the size of the fragments produced. However, the sickle C mutation
does not alter this specific cut site; therefore, it could not
be detected by this specific methodology. 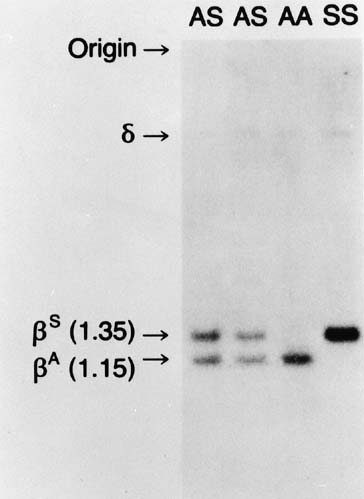 Fig. 10. Autoradiograms of DNA fragments after Mst II digestion and hybridization
with radioactive β-globin complementary DNA probes. The βA-specific
fragment is 1,150 bases long and the βS specific
fragment 1,350 bases long. A faint δ-band is seen because
of cross-hybridization with the δ-globin gene sequence. Fig. 10. Autoradiograms of DNA fragments after Mst II digestion and hybridization
with radioactive β-globin complementary DNA probes. The βA-specific
fragment is 1,150 bases long and the βS specific
fragment 1,350 bases long. A faint δ-band is seen because
of cross-hybridization with the δ-globin gene sequence.
|
Because the gene sequence for both the S and the C mutation is known, it
is possible to develop specific oligonucleotide probes that recognize
both the normal and the mutant DNA sequence.16 As depicted in Figure 5, the technique of ASOH can provide a precise diagnosis. DNA is extracted
from blood, amniotic fluid cells, or chorionic villi; amplified by
PCR; and then dotted onto the filter paper. Oligonucleotides for both
the normal gene and the mutant gene are then used in the hybridization. Analysis
of the pattern of dots allows precise identification of normal
persons, heterozygous carriers, and affected persons. α-Thalassemia is characterized by a deficiency in α-globin
chain synthesis. Normal α-chain production is the
product of four functioning a genes (two on each chromosome 16). Therefore, either
one or both genes can be deleted on a chromosome 16, resulting
in four clinical states according to the number of functional α-globin genes (3, 2, 1, or 0). Of clinical
significance is the circumstance in which there is one functional
gene, known as hemoglobin H disease, or 0 functional genes, known as hemoglobin
Barts disease, which results in fetal hydrops. Although a number of mutations have been described in the α-gene, α-thalassemia results from deletion of the α-gene
cluster on chromosome 16. At least 14 different deletions have
been characterized.17 Because the mutations represent deletions, standard restriction enzyme
analysis is the preferred method for both carrier detection and prenatal
diagnosis. Of importance to the obstetrician–gynecologist is the clinical presentation
of the homozygous form of the disorder. The mother may present
with severe preeclampsia in the early third trimester, and ultrasound
evaluation reveals hydrops fetalis (mirror syndrome). Because
of the lethal nature of this condition and the potential maternal
morbidity, all patients of Asian ancestry should be screened for the
carrier state of α-thalassemia. A simple screening tool is
a complete blood count with red cell indices. Carriers of the α-thalassemia
mutation have a mild anemia and a mean corpuscular volume
of less than 80 femtoliters (fL). Currently, the simplest
method for the diagnosis of α-thalassemia carriers is to
exclude iron deficiency anemia by appropriate studies and to exclude
the carrier state for β-thalassemia by hemoglobin electrophoresis. Because
both parents must be carriers to have an affected fetus, the
next step at this point should be to test the woman's partner. If
he also appears to be a carrier, appropriate molecular studies
can be performed to confirm the diagnosis in both parents. When both
parents are confirmed to be carriers, prenatal diagnosis is possible
either by CVS or by amniocentesis. β-Thalassemia is defined as either reduced (β+) or
absent (βO) β-chain synthesis. β-Thalassemia
is prevalent in areas of the world where malaria
is endemic (Mediterranean area, Africa, Middle East, India, Southeast
Asia, and Southern China). Although more than 170 mutations
have been characterized,18 only 25 different mutations account for the majority of mutant alleles
in at-risk populations.19 Based on knowledge of the mutations that are specific to a geographic
region or ethnic group, one can use a PCR-based screening protocol
to detect approximately 80% of the common mutations and another 15% of
rare mutations.20 As with α-thalassemia, the simplest initial approach to screening
for β-thalassemia is to obtain a complete blood count
with red cell indices. When the mean corpuscular volume is less than 80 fL, hemoglobin
electrophoresis is indicated. Carriers of the β-thalassemia
gene will have an elevated level of hemoglobin A2 (>3.5% of
total hemoglobin will be A2). If the partner
is also found to be a carrier, appropriate molecular studies must
then be performed to determine the exact mutations before prenatal diagnosis
can be offered. An algorithm for the screening for hemoglobinopathies
is depicted in Figure 11. 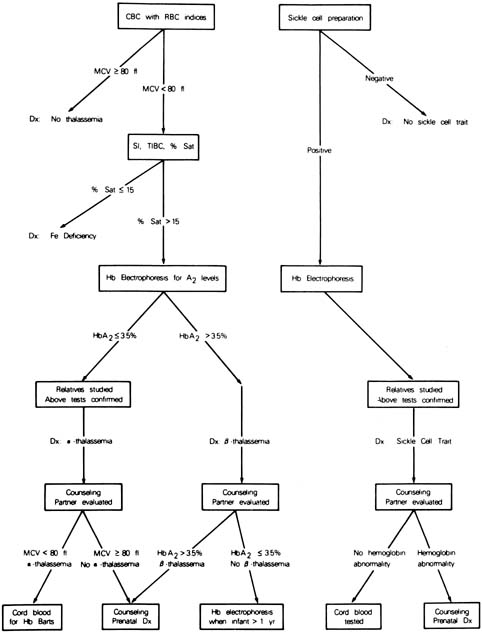 Fig. 11. Scheme for screening obstetric patients for hemoglobinopathies. Fig. 11. Scheme for screening obstetric patients for hemoglobinopathies.
|
X-Linked Disorders DUCHENNE'S MUSCULAR DYSTROPHY Duchenne's muscular dystrophy (DMD) is an X-linked
recessive disorder affecting one in 3,500 males. The disease is caused
by a mutation in the dystrophin gene, the largest (2.5 megabases) gene
known. Deletions within the gene account for 60% to 65% of
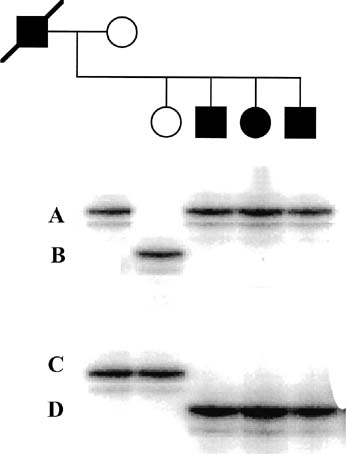
the dystrophin mutations.21 An additional 6% to 10% of mutations are duplications.22 Therefore, using the techniques of multiplex PCR (Fig. 12), one can detect approximately 70% of all mutations. In Figure 12, a multiplex STR polymorphism analysis for the four repeat elements found
within the dystrophin gene is shown for a DMD family. In this example, females
have two alleles at each locus (X-linked), each
allele differing by a few base pairs. This type of analysis is
beneficial in determining the carrier status of II-1 and II-3. Affected
son II-2 shows a deletion by the absence of a
peak at STR-45. From this information, the mother's haplotype
can be accurately determined, thus identifying the disease haplotype (a-aa). Inspection of the analysis of II-1 and
II-3 shows that they are both DMD carriers. Therefore, during
a subsequent pregnancy, DNA derived from either chorionic villi or
amniocytes would show whether a son was affected (a-aa) or
unaffected (acab). Because the affected males may no longer
be alive, determining carrier status may be complicated, and in
some cases impossible, in families with a single affected person. An algorithm
for evaluation of a potential carrier is outlined in Figure 13. Because of the significant amount of work required before prenatal testing
can be offered, it is essential that patients be evaluated whenever
possible before conception, if all options for prenatal testing are
to be available to them. 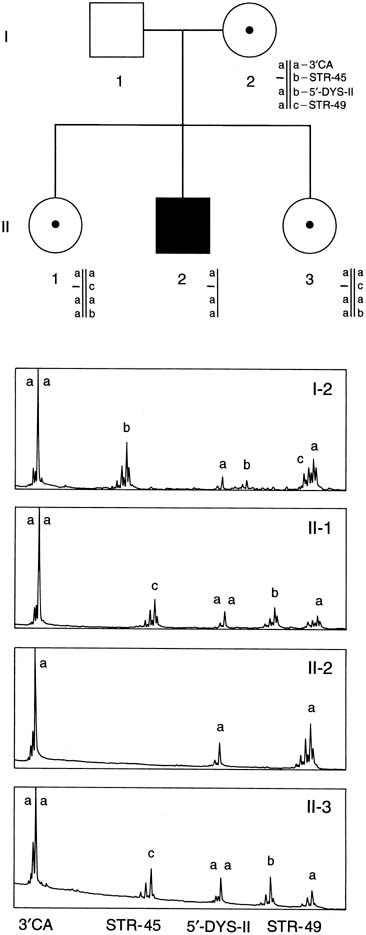 Fig. 12. Multiplex PCR-based deletion mapping of Duchenne's muscular
dystrophy. In this pedigree, there is a deletion encompassing the STR-45 CA
repeat. This can be seen in the affected son by the absence
of the second peak in his trace. Fig. 12. Multiplex PCR-based deletion mapping of Duchenne's muscular
dystrophy. In this pedigree, there is a deletion encompassing the STR-45 CA
repeat. This can be seen in the affected son by the absence
of the second peak in his trace.
|
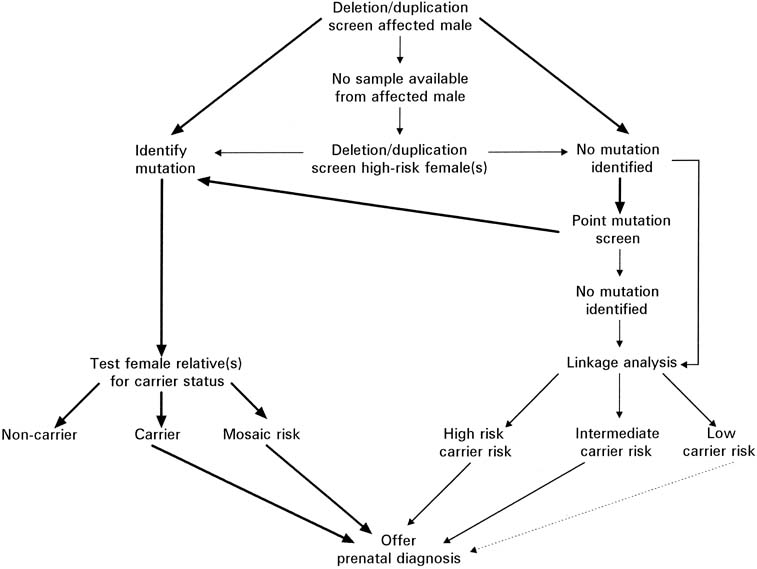 Fig. 13. Flow chart depicting the evaluation necessary for possible prenatal testing
for Duchenne's and Becker's muscular dystrophy. (Modified from
Abbs S: Prenatal diagnosis of Duchenne and Becker muscular dystrophy. Prenat
Diagn
16:1187, 1996) Fig. 13. Flow chart depicting the evaluation necessary for possible prenatal testing
for Duchenne's and Becker's muscular dystrophy. (Modified from
Abbs S: Prenatal diagnosis of Duchenne and Becker muscular dystrophy. Prenat
Diagn
16:1187, 1996)
|
Despite the availability of these molecular techniques, there remain a
number of families in which the fetal risk for DMD remains unresolved. However, in
those cases in which the potential for DMD transmission is
significant, the alternative is to assay directly for dystrophin, the
protein product of the gene, via an immunofluorescent technique; this
assay requires a sample of muscle obtained by fetal muscle biopsy.23 Although associated with a higher risk for fetal loss, this approach does
offer the possibility of accurate prenatal diagnosis when molecular
studies are uninformative. FRAGILE X SYNDROME Fragile X syndrome is the most common inherited form of mental retardation, affecting
approximately one in 4,000 men and one in 8,000 women.24 Therefore as many as one in 259 women in the general population may be
carriers of the gene.25 Fragile X syndrome has a wide range of clinical presentations, including
moderate disabilities in females, autism, and other psychiatric disorders. Unlike classic X-linked disorders (e.g., DMD, hemophilia), fragile
X syndrome affects both males and females; it is important
to note that there are males who carry the gene but have normal intelligence
and no physical stigmata of the disorder. This variable clinical
phenotype reflects the novel mutation in the gene known as fragile
X mental retardation 1 (FMR1). The FMR1 gene is characterized
by a repeated sequence of the trinucleotide cytosine-guanine-guanine (CGG).26 In unaffected persons, between six and 50 repeats of this sequence are
found. Intellectually normal carriers of the mutation have between 50 and 200 CGG
repeats, called a premutation. In carrier females, premutations
are unstable and may undergo further expansion during oogenesis. If
the CGG sequence expands beyond 200 repeats, it is considered a full
mutation, and all males with this number of repeats will show the
clinical features of fragile X syndrome, and approximately one-half
of females likewise will have mental retardation.27Males with the premutation do not have expansion during spermatogenesis
but will pass the premutation to all of their daughters, each of whom
will be at risk for having an affected child. Table 1 outlines clinical situations that should prompt screening for fragile
X syndrome, either in a pregnant patient or in the patient presenting
for preconception counseling. Two of these recommendations are straightforward, but
the latter two require explanation. We traditionally have
assumed that X-linked disorders in the patient's father's
family (paternal side) would not place the pregnancy
at risk. In the case of fragile X syndrome, however, the gene is nonpenetrant
in 20% of males with the fragile X gene (i.e., they
do not show clinical symptoms). Therefore, a patient's father
who has a family history of mental retardation may carry the fragile-X
premutation, which he will pass to all of his daughters. In
the circumstance in which the patient's paternal uncle has unexplained
mental retardation, her risk of being a carrier of the fragile
X gene is approximately one in 100. Therefore any history of unexplained
mental retardation should prompt testing for fragile X carrier status. Table 1. Patients for Whom Fragile X Screening Is Indicated
| Women with a known family history of fragile X syndrome |
| Women who have learning disabilities or mental retardation |
| Women with a family history of unexplained mental retardation |
| Women with a family history of premature ovarian failure | Of particular interest to the obstetrician–gynecologist is the association
of fragile X premutation and premature menopause.28,29 Preliminary studies have suggested that fragile X premutation carriers
are three-times more likely than other women to experience premature
menopause. Based on this information and the ease of carrier screening, testing
would seem appropriate in circumstances in which there
is a family history of premature menopause. Prenatal testing for fragile-X syndrome by either amniocentesis
or CVS is quite precise because of the consistent mutation present in
all families. There are, however, two caveats. The finding of a full mutation
in a male fetus is indicative of an affected child. However, the
presence of a full mutation in a female fetus gives an empiric risk
of approximately 50% that the child will be affected with the
clinical features of fragile X syndrome. Although prenatal testing is
possible using either CVS or amniocentesis, the difference in methylation
patterns of the gene found in early gestation requires that a laboratory
providing prenatal testing have experience with the nuances of
testing that are associated with CVS.30 |