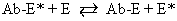The endometrium, or uterine mucosa, responds promptly to changes in the hormonal milieu, notably estrogen and/or progesterone. The hormonal concept of the endometrial cycle dates from the mid 19th century and has been reviewed by Novak and Novak2. Pfleuger, in 1863, first ascribed menstruation to the presence of the ovaries and it was believed to be caused by a reactive pelvic hyperemia secondary to a spinal reflex originating from the ripening follicles in the ovary. The transplant work of Knauer negated the necessity for a neural linkage in this system, which reaffirmed a hormonal role for the ovary in the endometrial cycle. In 1903, Frankel demonstrated the endocrine importance of the corpus luteum, which previously had been thought to be functionless. Hitschman and Adler in 1908 described the histochemical cycle of the endometrium, and it then became possible to integrate the ovarian and endometrial cycles.
In 1917, Frank called attention to the estrus-producing effects of a hormone contained in ovarian follicular fluid. Allen and Doisy in 1923 made more elaborate demonstrations of the müllerian-stimulating effects of this hormone. After Zondek demonstrated large amounts of this substance in the urine of pregnant women, two groups simultaneously in 1929 (Doisy, Veter, and Thayer from the United States and Butenandt from Germany) isolated and characterized the purified urinary extract. This urinary extract was later to be known as estrogen. The term estrogen has now been broadened to include all of the natural and synthetic derivatives of this female sex hormone.
Long before either the follicular or corpus luteal hormones were isolated, it seemed certain that two separate and distinct ovarian hormones must exist, simply on the basis of the histologic changes of the endometrium. Frankel's demonstration of the importance of the corpus luteum in 1903 was followed by the isolation and purification of progesterone by Butenandt, Allen, and others in 1929. Entwined in these discoveries was the identification of the pituitary gonadotropic hormones (follicle-stimulating hormone [FSH] and luteinizing hormone [LH] and their role in the ovarian cycle and the ultimate control of endometrial growth, maturation, and menstruation.
Research in the next 4 decades was involved with further purification and identification of the various natural estrogens and progesterone, their synthesis for experimental and therapeutic use, the development of highly sensitive and specific assays applicable to small samples of biologic fluid, and the development of molecular analogues which not only could be used orally but also possessed varying degrees of differing biologic effects. Simultaneous with these advances in steroid endocrinology were further explorations into the natural history of endometrial adenocarcinoma and hyperplasia. Endometrial hyperplasia, which can arise after prolonged estrogen stimulation, was found to be associated with and perhaps a precursor of endometrial cancer.
Effects of Estrogen
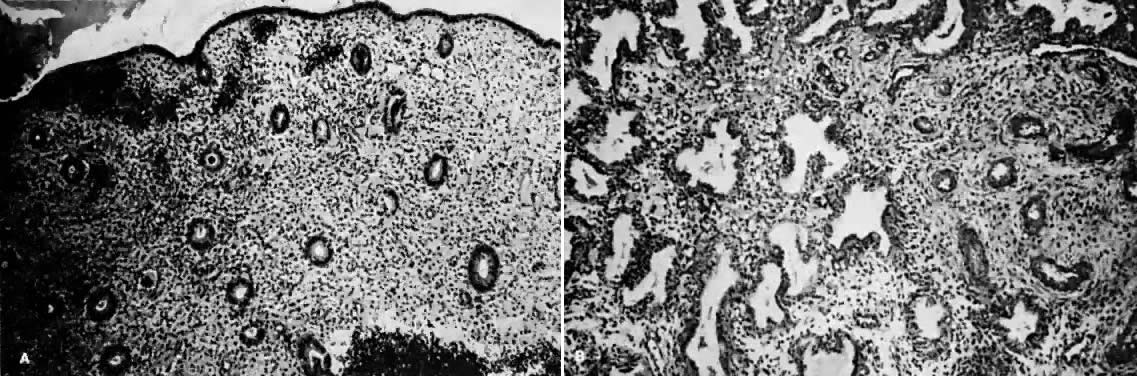
Estrogens are growth hormones for tissues derived from the müllerian ducts, the endometrium being one of the most sensitive of these tissues. During the follicular phase of the endometrial cycle (proliferative) the endometrium regenerates from perivascular “collars” about residual vessels remaining after menstruation. This epithelium gradually thickens, becomes taller and more dense, and is characterized in this phase by increased mitotic activity in glands and stroma. This estrogenic stimulation results in straight tubular glands and a compact stroma when follicular phase tissue samples are microscopically examined (Fig. 1A).
|
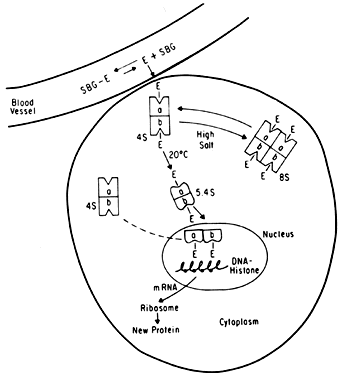
It is now known that estrogens exert their end-organ effect by activating a complex intracellular mechanism. Tissues which respond to estrogen possess intracytoplasmic proteins (receptors) that preferentially bind specific steroids (Fig. 2). For instance, a cell from the uterus will possess 5,000 to 15,000 estrogen receptors whereas a cell from the spleen will have none. These receptors recognize estrogens by their three dimensional and chemical characteristics and bind it with high affinity (KD =10-10), specificity, and saturability. The estrogen molecules present in the circulation are relatively loosely bound to intravascular carrier proteins (sex-steroid-binding globulin [SBG]) (KD = 10-8) or to albumin. In excess of 95% of the estrogen in the circulation is found in the bound form. The estrogen readily diffuses across the cell membrane in its active free form due to a concentration and a binding gradient. The estrogen molecule is relatively small (molecular weight is 300) and lipophilic and probably passes through the cell membrane by simple diffusion. Once in the cell, the estrogen is promptly bound to the intracellular (intracytoplasmic) receptor protein, which then undergoes a series of complex spatial changes prior to intranuclear transport. This nuclear transport occurs within 30 to 45 minutes after the target tissue is exposed to estrogen. The following system of nuclear interactions between receptor and DNA is a model that has been proposed by McCarty3. The activated receptor-estrogen complex then nonspecifically binds to the DNA and protein of dispersed chromosomes (euchromatin) and stimulates acetylation of the histone protein. This acetylation of the histones in nucleosomes causes the nucleosome to “open up” and expose specific DNA segments for transcription. The “estrogen message” is transcribed into new messenger RNA which then migrates back into the cytoplasm and activates various cellular processes including new protein synthesis. The now “freed” receptor protein is probably recycled back into the cytoplasm for further use3,4. The estrogen receptor recognizes a molecule as being “estrogen” if its size, three-dimensional configuration, and charge are similar to the parent molecule. Therefore, the nonsteroidal synthetic estrogens may not resemble the “prototype” estrogen (estradiol-17β) on paper diagrams but are very similar in shape and other properties as seen by the cellular receptor. Estrogen receptors are perhaps the determinants of potency for estrogenic substances. The estrogen receptors preferentially bind estradiol over estriol (2x) and estrone (3x)5. This receptor also discriminates among the estrogens by binding estradiol within the cellular nucleus longer than the weaker estrogens estriol and estrone4,6. Therefore, estradiol is the most potent of the natural estrogens probably because of the greater affinity and duration of its receptor-binding compared with the other available estrogens. Receptors for estrogen and other steroid hormones can now be accurately quantified and studied. Estrogen in physiologic concentrations stimulates the synthesis of estrogen receptors and of progesterone and testosterone receptors7. Progesterone and testosterone, however, inhibit estrogen receptors. Progesterone inhibits its own receptor population in the secretory phase of human endometrium8. Thus, it is apparent that for estrogen to bind and influence a tissue, the specific estrogen receptors must be present. The potency of a particular estrogen in a tissue roughly parallels and is probably dependent on the quantity of the estrogen receptor in the cells of that tissue. Studies with estrogen receptors in breast cancers are being successfully utilized to predict the responsiveness of these tumors to hormonal manipulation9,10.
|
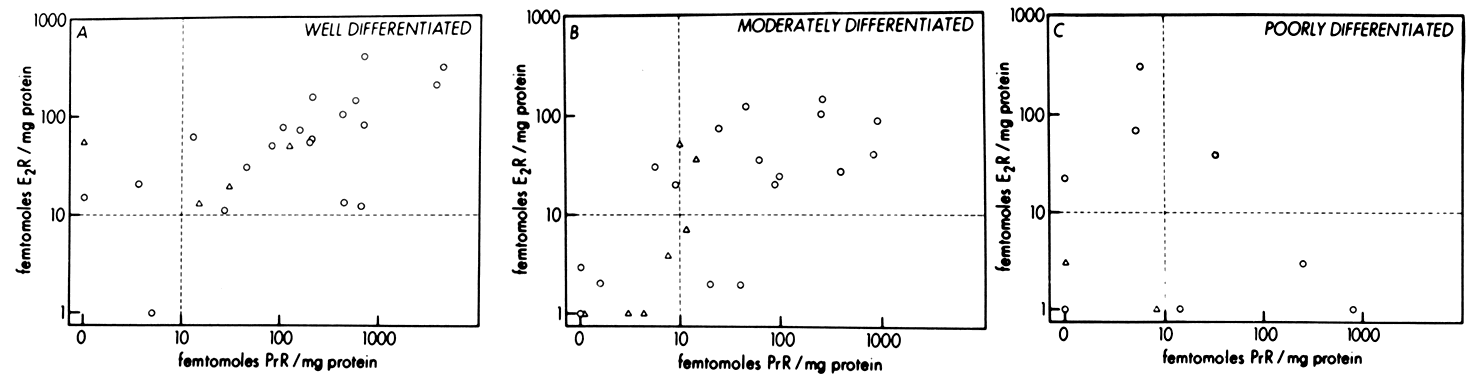
Endometrial carcinoma is another malignancy that can be hormonally dependent and manipulated by endocrine therapy. Both normal endometrium and endometrial carcinoma possess active estrogen receptors. There have been few reports showing qualitative or quantitative changes in estrogen receptors in hyperplastic endometrium or endometrial cancer, but the potential exists and detailed investigation is beginning.
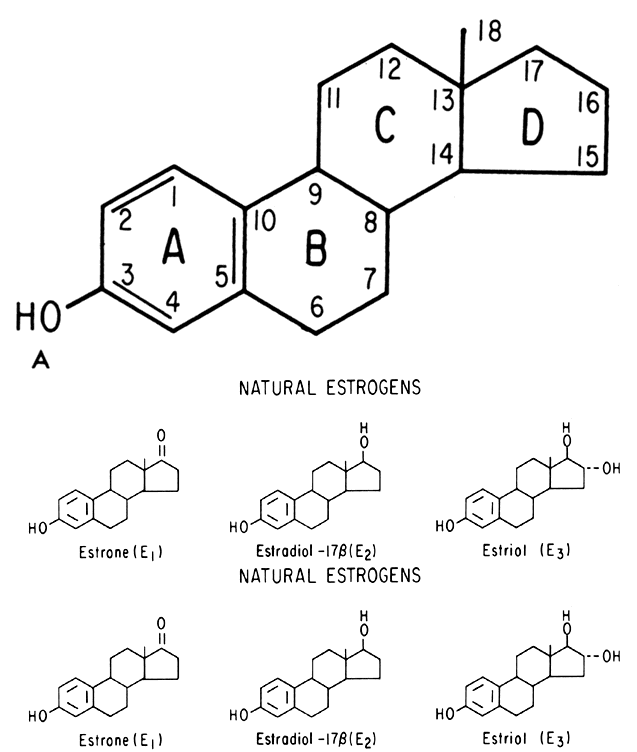
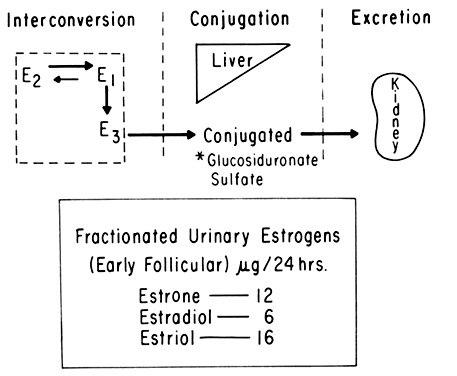
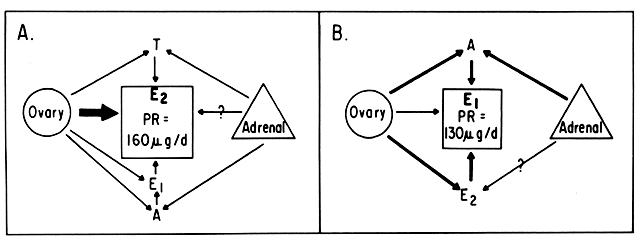
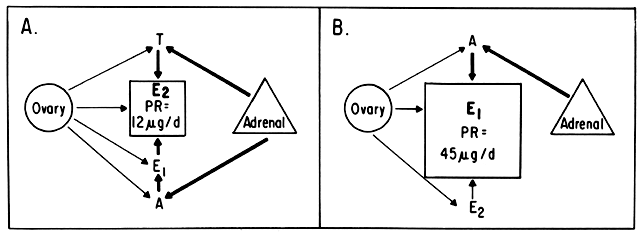
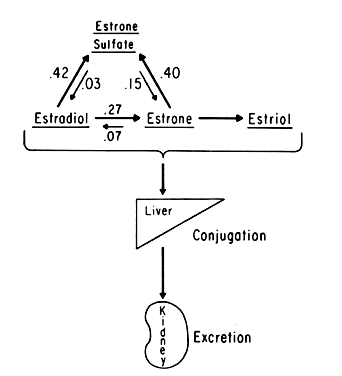
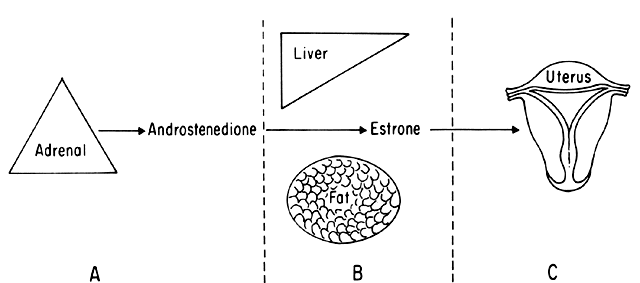
An intriguing theory has been proposed linking endometrial carcinoma to the relative levels of estrone and estriol in the postmenopausal female11. This theory ascribes a protective effect to estriol over estrone. Clearly, estrone is the dominant estrogen in postmenopausal women and also in younger women with polycystic ovarian disease; therefore, it has been implicated in the etiology of endometrial carcinoma. A protective effect from cancer for estriol was proposed when epidemiologic studies demonstrated higher estriol-estrone ratios in control groups as compared with groups with endometrial or breast cancer12,13, but other studies have failed to demonstrate these findings14,15. As noted, the receptor data show estrone and estriol competing for binding on about an equimolar basis, with similar decreases in duration of nuclear binding as compared with estradiol. There is no evidence that estriol has a different or less carcinogenic induced cellular response than estrone. Therefore, in postmenopausal patients, the unopposed levels of estrone and estriol are probably inducing the same cellular response. The concept that estriol binds to the receptor and produces a “benign” product or that it occupies the receptor for a long period of time and blocks estrone binding is not substantiated by current data. Therefore, the theory of a protective effect of estriol must rest on epidemiologic data which is conflicting and weak.
Effects of Progesterone
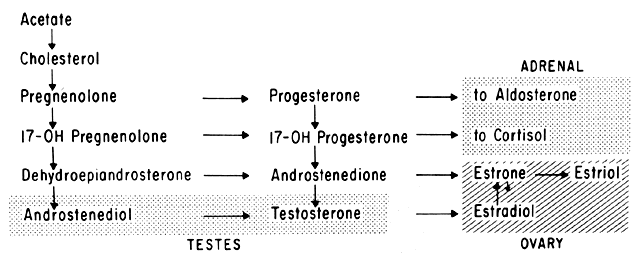
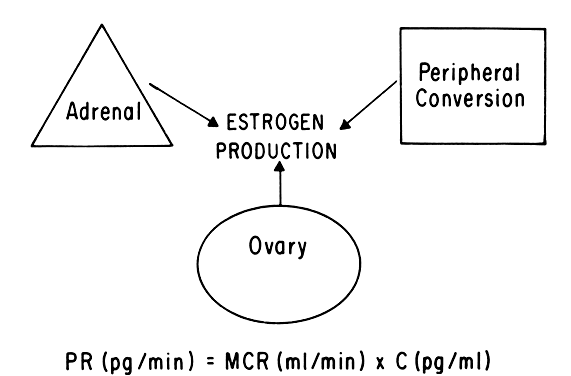
Progesterone is produced and secreted by the adrenal cortex, ovary, and placenta. From the ovary, the progesterone secretory rate is negligible during the follicular phase and increases dramatically after ovulation, yielding 30 mg per day between the 18th and 24th day of the cycle. Greater than 90% of the progesterone is removed by the liver within 25 minutes16. Progesterone acting alone has little effect on unstimulated endometrium. However, if the endometrium has been “primed” by estrogen (proliferated), then progesterone causes a secretory change in the glands, stromal edema, regression and stromal pseudodecidual formation (Fig. 1B). Under the influence of progesterone, the total thickness of the endometrium decreases due to fluid loss and three layers become defined: the basalis next to myometrium, the compact layer immediately beneath the endometrial surface, and the spongy layer between the other two layers. The basal layer undergoes little if any histologic alteration during the menstrual cycle, but mitoses are found in the glands of this layer. The spongy layer comprises a lacy labyrinth with little stroma between the glands, which are tortuous and serrated. Little change occurs in the compact layer except that gland lumens are filled with secretion. By late in the secretory phase, the endometrium has become extremely vascular and succulent, ripe for implantation. If no implantation occurs, then 2 to 3 days before menstruation the reticular framework of the endometrium begins to disintegrate and vascular compression of the coiled spiral arteries occurs with vasoconstriction and decreased blood flow. Subsequently, vascular relaxation occurs at which time bleeding and endometrial sloughing begin. Thus, after endometrial proliferation induced by estrogen, progesterone ultimately causes secretion, regression, and sloughing. Progesterone acts through specific intracellular receptors, as described for estrogen.
Abnormal Endometrial Changes
The histopathologic picture of endometrial hyperplasia differs from that of normal proliferative endometrium as a result of sustained and unopposed estrogen stimulation. It is characterized by an increased endometrial thickness, on occasion threefold or greater, and both qualitative and quantitative changes in glandular patterns which exceed similar changes in the stroma. A number of types of endometrial hyperplasia have been reported and include cystic and adenomatous hyperplasia. A further subdivision of adenomatous hyperplasia is the atypical variety. It is thought that endometrial hyperplasia represents uninterrupted growth of a tissue which normally should undergo cyclic menstrual degeneration and sloughing1.
Endometrial hyperplasia is usually found in women who experience 3 to 6 months or more of uninterrupted (by progesterone) estrogen stimulation. Continuous estrogen stimulation most often occurs in anovulatory women who maintain significant estrogen production. Women with chronic anovulation include the adolescent (in whom Frasier17 has reported that up to 50% of early cycles are anovulatory), the perimenopausal female, patients with the polycystic ovary syndrome, obese women, and a large group of females who idiopathically fail to ovulate. Another group of patients exposed to unopposed estrogen is women receiving estrogen replacement therapy without added progesterone. The clinical impact of endometrial hyperplasia falls into two basic areas: dysfunctional uterine bleeding and, perhaps, the subsequent development of adenocarcinoma of the endometrium. It has been estimated that 1% to 2% of patients with cystic endometrial hyperplasia will ultimately develop endometrial cancer1, while 5% to 15% of patients with adenomatous hyperplasia18 and 20% to 25% of women with atypical adenomatous endometrial hyperplasia19 will also ultimately develop this malignancy. While the precise incidence and mechanism of progression of carcinoma of the endometrium following in situ carcinoma is unknown, it is certainly significant.
Thus, it appears that tonic,. prolonged, and/or higher concentrations of unopposed estrogen can at least initiate hyperplasia of the endometrium. Whether the next step to endometrial malignancy can be linked to estrogen is unclear. Certain animal data support the contention that estrogen can be carcinogenic in terms of the endometrium, but the human data are less certain. Perhaps the best representation of these changes was based on data by Sommers20 and presented by Kistner21 (Fig. 3), which illustrates endometrial polyps progressing to cystic hyperplasia and then to adenomatous hyperplasia under the influence of exogenous estrogen. All three of these steps are clearly capable of being stimulated by estrogen. The linkage of estrogen to the remaining step from hyperplasia to neoplasia is less certain, but it appears to be related in some manner.
|