Both ERα and ERβ contain activation functions (AFs) that contribute to the ER's transcriptional activity. AF-1 is located in the amino-terminal region within the A/B region and is believed to be constitutively active and ligand-independent in ERα80,81 (Fig. 1). AF-2 is present at the carboxy-terminus, and is believed to be ligand dependent. Mutational analyses demonstrate the importance of this region for ER transcription because AF-2 can interact with a number of transcriptional coactivators in a ligand-dependent manner.82,83 AF-1 and AF-2 can each activate transcription independently but in most cases they synergize with one another as promoters within the specific cell context. The ER activates gene expression through binding to estrogen response elements (EREs) in responsive genes through the action of AF-1 and AF-2.84 ERβ also activates transcription of target genes through EREs.85 Additionally, estrogen can induce an AP-1 site in a reporter construct through ERα, but it is inactive through ERβ. Interestingly, ER antagonists activate ERβ to induce activity through an AP-1 site.86 These two receptors, ERα and ERβ, form functional heterodimers on DNA and stimulate transcription of a target gene. The existence of two rather than one ER portends a more complex mechanism of action than previously thought and remains a primary therapeutic target for new and existing SERMs.
|
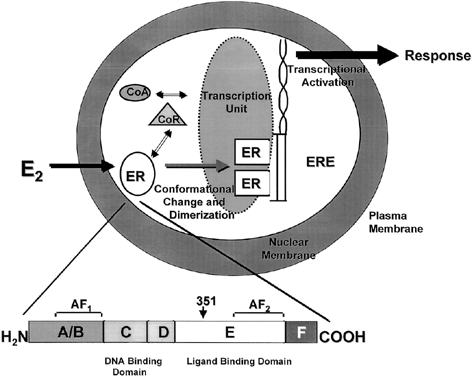
One of the functional areas of the ER, the E region, is the steroid-binding domain that undergoes a conformational change on binding with estrogen, locking the ligand into the hydrophobic pocket of the receptor. This allows the ER to dissociate from heat shock proteins, dimerize, and bind to specific DNA sequences of estrogen responsive genes. Changes in conformation of the ER allow for interaction with coregulatory proteins (coactivators or corepressors) that act as signaling intermediates between the ER and the transcriptional machinery.87 The crystal structure of the ligand binding domain of ERα was determined with estradiol88 and DES.89 A key feature of the agonist-receptor complex is the availability of a portion of the ligand binding site of ERα, helix 12, to lock the steroid in the hydrophobic pocket and allow for recruitment of coactivators to the AF2 site. The repositioning of helix 12 after ligand binding is an important mechanism for full estrogen action at ERα.90,91
The nuclear steroid receptors must associate with other nuclear proteins to form a transcription complex. Several coactivators, such as ERAP16092 and RIP140,93 have been identified based on their ability to interact with the agonist-bound receptor and not to the antagonist-bound receptor at AF-2. However, the SRC-1 protein has been identified and it also interacts with AF-194 and with ERb through phosphorylation of AF-1 by the MAP-kinase signaling cascade.95 It has been demonstrated that SRC-1 and another protein, p300/CBP, contain intrinsic acetyltransferase activity and can interact with other histone acetyl transferases (HATs). Acetylation by SRC-1 of histones bound at specific promoters could be a mechanism by which the AFs of ER and associated coactivators activate transcription of specific genes by enhancing formation of a stable preinitiation complex.96
Antiestrogens are competitive inhibitors of estrogen action; the shape of the ligand is essential to reducing the estrogenic efficacy. The side chain of the antiestrogen prevents helix 12 from closing so that activation cannot occur.88,97,98 It is known that tamoxifen silences AF-2 whereas AF-1 remains constituitively activated.81,99 Other antiestrogens, such as raloxifene, silence both AF-1 and AF-2.100,101 This is a possible explanation for the promiscuous estrogen like actions of tamoxifen compared with raloxifene.