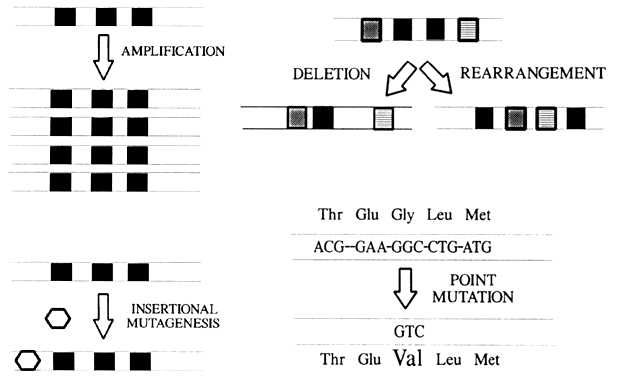
Proto-oncogenes are deoxyribonucleic acid (DNA) sequences found in the genomes of normal and neoplastic tissues that are quite similar to the transforming genes of ribonucleic acid (RNA) tumor viruses. Proto-oncogenes exhibit striking evolutionary conservation, being found in a large number of phylogenetically diverse organisms ranging from Drosophila flies to humans. More than 40 proto-oncogenes have been described, and this list will undoubtedly continue to expand. Protooncogenes encode growth factors, growth factor receptors, and proteins that regulate cell growth and differentiation. Proto-oncogenes are usually grouped according to the primary function or sequence homology of their protein products (Table 1).
TABLE 1. Proto-Oncogene Classification
| Gene Product or Function | Proto-Oncogene |
Growth factors | FGF growth factor | fgf-5, hst, int-2 |
| PDGF B-chain | sis |
Transmembrane receptors | EGF receptor | erbB |
| EGF-related-receptor | HER-2/neu |
| CSF receptor | fms |
| Stem cell receptor | kit |
| Nerve growth factor receptor | trk |
Inner membrane receptors | Plasma membrane signal transducer | bcl-2 |
| GTP-binding/GTPase activity | Ha-ras, Ki-ras, N-ras |
| Other | fgr, lck, src, yes |
Cytoplasmic messengers | Tyrosine/kinase activity | crk |
| Serine/threonine kinase activity | cot, pim-1, mos, raf/mil |
Nuclear transcription | DNA-binding proteins | erbB1, ets-1, ets-2, fos, gil-1, jun, rel, ski, vav, lyl-1, |
factors |
| maf, myb, myc, L-myc, N-myc, spi-1, evi-1 |
CSF = colony-stimulating factor; EGF = epidermal growth factor; FGF = fibroblast growth factor;
GTP = guanosine triphosphate; GTPase = guanosine triphosphatase;
PDGF = platelet-derived growth factor
Tumor suppressor genes are a second major group of genes that regulate cell growth. These genes, which include the p53 gene and the retinoblastoma gene, encode DNA-binding proteins that normally act to inhibit cellular proliferation.
Proto-oncogenes and tumor suppressor genes encode proteins that play important roles in every step of the intracellular signal transduction system and permit the cell to respond to external stimuli in a specific, coordinated, and controlled fashion (Fig. 1). The net result of the expression of proto-oncogenes and tumor suppressor genes is the regulation of cell growth and differentiation.
|
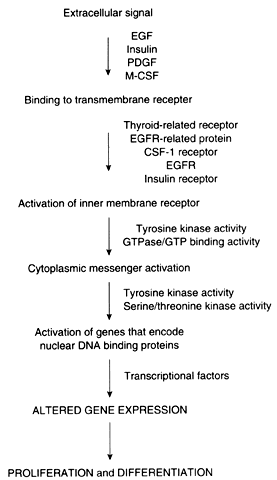
Membrane receptors bind extracellular molecules, such as growth factors and steroid hormones, initiating a cascade of events that culminates in altered gene expression. These receptors are often linked to inner membrane receptors, perhaps the most notable of which are encoded by the ras gene family. The ras genes encode proteins that exhibit guanosine triphosphate (GTP) binding or guanosine triphosphatase (GTPase) activity, or both. These proteins, collectively designated p21, are attached to the inner surface of the cell membrane. These proteins couple membrane receptors to a number of signaling systems, including cyclic adenosine monophosphate and the inositol phospholipid pathway.
Cytoplasmic messenger systems transmit external signals from the cell membrane to the nucleus. This part of the regulatory network involves protein phosphorylation activity and is the target of substances such as platelet-derived growth factor, fibroblast growth factor, epidermal growth factor, colony-stimulating factor, and insulin-like growth factor (see below).
Numerous proto-oncogenes have been identified that encode nuclear DNA-binding proteins that regulate transcription of other genes. Typically, more than one gene acts in concert with several other genes to trigger a change in proliferation. As an example, increased expression of c-jun and c-fos, followed by an increase in c-myc, is required for the transition from the G0 to the G1 phase of the cell cycle. In contradistinction, the p53 tumor suppressor gene encodes a protein that normally acts to inhibit cellular proliferation. Expression of the p53 gene also increases in cells that have sustained DNA damage so that the damage is not passed to daughter cells before repair.
Data derived from in vitro and in vivo models clearly demonstrate that proto-oncogenes, acting as a subset of a larger group of genes, play crucial roles in the regulation of cellular proliferation and differentiation. Studies of the placenta and endometrium are of particular interest to the gynecologist. In the placenta, the trophoblast cell population changes from one dominated by the “pseudo-malignant” cytotrophoblast to one composed predominantly of the highly differentiated syncytiotrophoblast. This transition is accompanied by discrete changes in the expression of several proto-oncogenes. The expression of c-fms, confined to the placenta and extraembryonic membranes, exhibits a progressive increase in expression during prenatal development in the mouse.1 In contradistinction, the expression of c-fos is approximately 15-fold greater in cytotrophoblasts than in syncytiotrophoblasts.2 The transcription of c-myc appears to be directly associated with proliferative activity of the trophoblast.3, 4, 5 Peak c-myc messenger RNA expression occurs at 4 to 6 weeks' estimated gestational age, followed by a rapid decline to barely detectable levels at term. The expression of the Ha-ras proto-oncogene demonstrates little variation with gestational age in placental tissue.
Normal endometrium exhibits cyclic proliferation, differentiation, and exfoliation in response to estrogen and progesterone. These events, which collectively characterize the menstrual cycle, are accompanied by discrete changes in the expression of myc, ras, and fos. Interestingly, the expression of these genes varies not only with the phase of the menstrual cycle, but between the glandular and stromal cell compartments as well.6 Cell- and tissue-specific changes in proto-oncogene expression also have been documented in the developing embryo.7, 8, 9