Metabolic Clearance Rate The rate of metabolism of a steroid by the whole body can be expressed
as the volume of blood completely cleared of that steroid per unit time, usually
a 24-hour day. This rate is referred to as the metabolic clearance
rate (MCR) for that steroid and represents the summation of the
clearance rates of the individual organs (e.g., liver, kidney, lung).33,34 The blood production rate of a steroid can be calculated from these data
according to the formula:
PB = MCR × PC
where PB is the amount of steroid entering the circulation from all sources and
PC is the plasma concentration. It must be emphasized that this method gives
only an approximation of the daily production rate, since glandular
secretion and MCR can both vary independently during the day.33
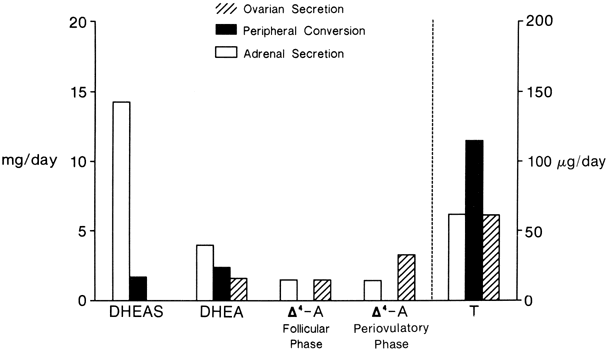
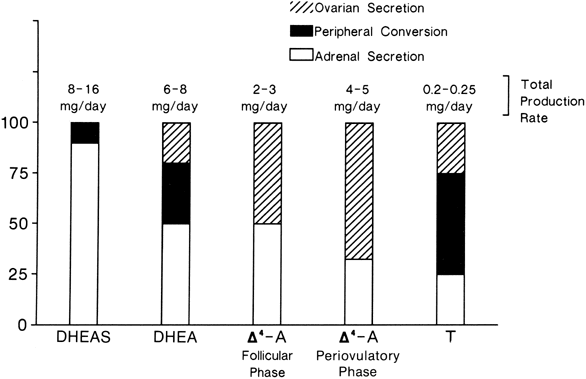
The MCRs of the major androgens in healthy women and men are listed in
Table 4.11,30,31,32,33,34
The MCR of a steroid is directly correlated with the free fraction of
that steroid and, for steroids with a strong affinity to SHBG, inversely
correlated with SHBG levels.31,35
Thus, in situations in which SHBG is decreased, as in hyperandrogenism
or obesity, the MCRs of T and DHT are increased. In situations in which
SHBG is increased, as in hyperthyroidism or estrogen administration, the
MCRs of T and DHT are decreased.36,37
The MCRs of Δ4-A and E are also decreased in hypothyroidism,
but this is believed because of a decrease in blood flow to splanchnic
tissue.38 The MCRs of DHEAS, DHEA, and Δ4-A
remain relatively constant despite changes in SHBG.39
However, it should be noted that irrespective of SHBG levels, obesity
per se is associated with an increase in MCR for all steroids.40
The MCR of T can also be increased by the induction of metabolizing enzymes
in the liver, as seen with medroxyprogesterone administration.41
Table 4. Metabolic Clearance Rates in Liters per Day of the Androgens in
Normal Women and Men
| DHEA | DHEAS | DHT | T | Δ4-A |
Women | 1820* | 13.8 | 315 | 485 | 1840 |
Men | 2080 | 15.2 | 650 | 780 | 2390 |
DHEA, dehydroepiandrosterone; DHEAS, dehydroepiandrosterone sulfate; DHT, dihydrotestosterone; T, testosterone; Δ4-A, androstenedione.
*Data from multiple sources.
Androgen Metabolism in the Liver The splanchnic tissues are the major sites of androgen metabolism, and 65%to 90% of
the total metabolism occurs in these tissues, of which the
liver carries out the major portion. The major sites involved in the
metabolism of androgens are the C-3, C-5, and C-17 (Figs. 5 and 6). In the liver, the 5α:5β ratio of reduced androgens can be
altered in a number of situations. In hyperthyroidism, the ratio is increased, and
in hypothyroidism, it is decreased.42 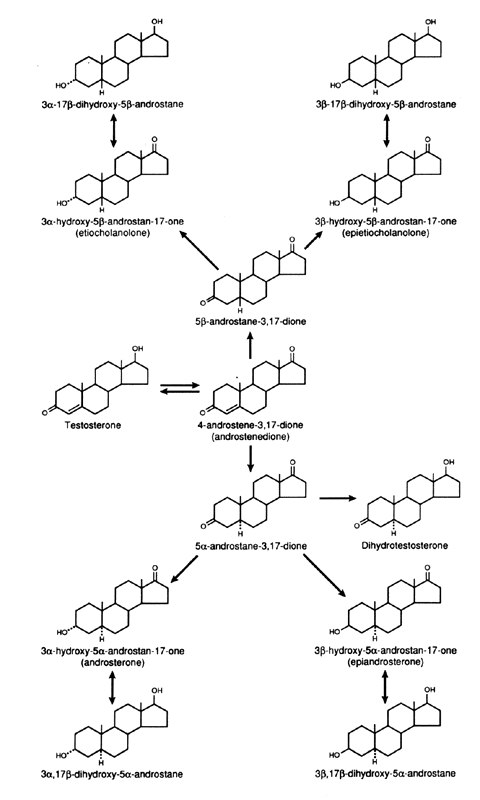 Fig. 5. Metabolism of Δ4 -androstenedione in liver and peripheral tissues. Fig. 5. Metabolism of Δ4 -androstenedione in liver and peripheral tissues.
|
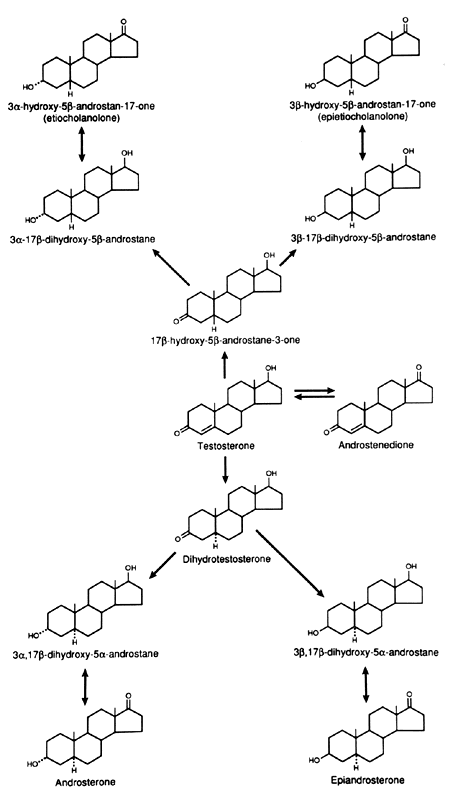 Fig. 6. Metabolism of testosterone in liver and peripheral tissues. Fig. 6. Metabolism of testosterone in liver and peripheral tissues.
|
The conjugation of androgens to form sulfates or glucuronides occurs primarily
at the C-17 and C-3 sites. Because conjugation is rapid, androgens
leaving the liver in hepatic vein blood are conjugated primarily
as glucuronides, but sulfates are also present. Because there is little
cleavage of the glucuronide in tissues, the androgens formed in the
liver do not contribute to the blood or tissue pool of unconjugated androgens.15 Androgen glucuronides do not bind to the androgen receptor and are, therefore, biologically
inactive. There is some degree of sulfurylation
of androgens that occurs in the liver, and the androgen sulfates, especially
DHEAS, can be hydrolyzed in other tissues and contribute to the
pool of active androgens. The extraction, or uptake, of androgens by the liver is high and close
to 100% for those androgens not bound to SHBG. The binding to SHBG decreases
the extraction, and only 40% to 60% of T and 30% to 40% of DHT
are extracted by the liver.43,44 However, because of extensive metabolism in the intestinal wall and the
liver, the oral administration of T or DHT is an inefficient way to
increase overall androgenicity. Skin and Genital Structures As noted previously, the free hormone, which is inversely related to the
SHBG concentration, is available for entering the cells of the target
tissues.23 Recently, T was considered to be the male hormone. However, with advancement
of the knowledge of cellular biology, particularly the intracellular
events associated with biologic responses to androgen, it was evident
that in many androgen-responsive tissues, DHT rather than T mediated
the intracellular molecular events associated with responses to androgens.8 As noted previously, because of different binding affinities, DHT is a
stronger androgen than is T.5 In tissues with relatively high levels of 5α-reductase, such as
the hair follicle and sebaceous gland, the amount of DHT formed from precursors
is relatively large and androgenic events are mediated by DHT
rather than T. Thus, DHT is responsible for the male differentiation
of the prostate and external genitalia, but virilization of the wolffian
ducts is due to T.5,45 The levels of 5α-reductase are low in muscle;therefore, muscle development
is under T control.8 The concept that steroids bind to and result in the nuclear translocation
of a cytoplasmic receptor has altered in the past few years.46-48 It has been shown that estrogen receptors exist almost entirely in the
nucleus, irrespective of steroid binding, and that estradiol diffuses
through the cell membranes and cytoplasm to bind to the receptor in the
nucleus. It would appear that the same concept applies to the androgens.48 The receptor is present in the nucleus but not bound to DNA. When androgen
binds to its receptor, the latter can then bind to DNA and initiate
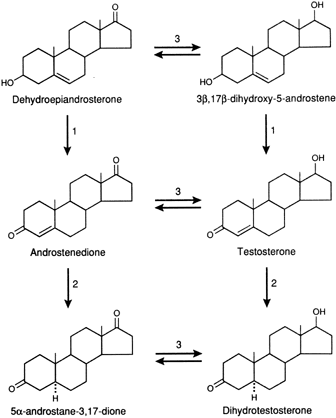
transcription.46,48 In tissues with high levels of 5α-reductase, when a precursor, DHEAS, DHEA, Δ4-A, or T enters the cell, conversion to DHT occurs through the pathways
shown in Figure 1. DHT is the androgen that is primarily responsible for stimulating the
androgenic activity. In tissues that have minimal 5α-reductase
activity, the same precursors are converted only to T, and it is that
steroid that stimulates the androgenic activity.49,50 In either case, the degree of androgenic expression is dependent on the
amount of active steroid or precursor steroid in the circulation, the
blood flow to the tissue, the extraction of steroid by the tissue, and
the enzyme activities of the tissue. In vitro studies have documented that human skin has the capacity to metabolize
androgens and is an androgen-responsive tissue.8 The skin from the genital area of the fetus at 12 to 22 weeks of age has
a greater capacity than nongenital skin to convert T to DHT, and the
external genital skin of male and female fetuses can transform T to
DHT. Thus, 5α-reductase activity in the urogenital tubercle, fold, and
swelling is acquired very early in embryogenesis in both male and
female fetuses and is not, itself, androgen dependent.45 The production of T by the testes of the male fetus and the in situ conversion to DHT stimulate these structures to form the external male
genitalia. In the absence of T production or conversion to DHT, female
external genitalia develop. The 5α-reductase activity persists
after birth in those structures derived from the urogenital tubercle, fold, and
swelling in both sexes. The clitoris is, therefore, androgen
responsive. Valuable knowledge concerning the role of androgen metabolism in sexual
differentiation of the internal and external genitalia has been acquired
from studies of the skin and genital structures in male pseudohermaphroditism.51,52 In testicular feminization (i.e., complete male pseudohermaphroditism), the
intracellular androgen receptor is absent but Müllerian inhibiting
factor is fully active. Consequently, both male internal and external
genitalia fail to develop, male secondary sex characteristics
are not acquired, and the phenotype is female. In the type 2 incomplete
form of male pseudohermaphroditism, an autosomal-recessive disorder, there
is deficiency of 5α-reductase activity, resulting in deficiency
of DHT formation, despite normal T and Δ4-A production. As a consequence of the inadequate DHT formation, there
is incomplete differentiation of the male external genitalia, although
the internal male genitalia develop normally.53 These findings indicate that 5α-reductase activity and DHT are required
for differentiation of the male external genitalia but not the
male internal sexual structures. The increased secretion of T at puberty
stimulates enlargement of the phallus and induces the male secondary
sex characteristics, including an increase in muscle mass, but beard
growth and acne are minimal, indicating that these events are DHT dependent. One of the most interesting aspects of androgen metabolism concerns the
growth of body hair.54 Not all hair growth is androgen dependent or androgen responsive. Androgen-dependent
hair, or sexual hair, is located on the face, chest, and
abdomen (center of the body). Ambisexual hair is stimulated by low levels
of androgens and includes axillary hair and the hair on the lower
pubic triangle. Nonsexual hair (e.g., eyebrows) is independent of androgen
effects. The control of 5α-reductase activity is different in the external
genitalia and skin containing sexual hair.54 The ability of the skin containing sexual hair to form DHT from T increases
during puberty as T production increases. Thus, 5α-reductase
activity in these areas is induced by T.54 Male secondary sex characteristics (loss of hair, increased sebaceous
gland activity) are induced by T during puberty and are dependent on acquisition
of 5α-reductase activity necessary for the conversion
of T to DHT . There is a direct relationship between the extent of hair
growth in the skin of sexual areas and the amount of 5α-reductase
activity.54 Many hirsute women have increased production of androgens (DHEA, A, T, or
all three), and these are transformed to DHT, especially in hair follicles
and sebaceous glands.54 The amount of DHT formed will be increased primarily because of increased
precursor production. There is, however, an increase in 5α-reductase
activity in most hirsute women.54 In certain instances of hirsutism and male pattern baldness, there is
increased formation of DHT from precursors in the hair roots,51,52,54 even though the circulating precursors DHEAS, DHEA, A, and T production
are normal in these women. It is believed that an increased 5α-reductase
activity is the cause of the hirsutism since the increased 5α-reductase
activity would result in increased DHT production from
normal amounts of precursors within the hair follicle. Then, after
stimulating hair growth, the DHT is metabolized to androstanediol that
is conjugated before leaving the hair follicle as 5α-androstanediol-3α, 17β-diol glucuronide(3α-Adiol gluc).54 The latter steroid is believed to be a marker for the increased 5α-reductase
activity in these women.51,54,55 The sebaceous glands are also androgen-responsive organs and have the capability
of converting precursors to DHT. Sebaceous gland secretory activity
is increased in hyperandrogenic women, and seborrhea usually accompanies
hirsutism. There is hypertrophy of the sebaceous glands in
the skin with acne and an increased 5α-reductase activity.56 There is poor correlation between the plasma level of T, or other androgens, and
the severity of hirsutism or acne. However, plasma-free T correlates
better than total T with the degree of androgenization,57 and it now appears that many hirsute women have increased androgen production.58 In such women, there is good correlation between the production rate of
T and the degree of hirsutism or virilism. As noted, many women with
hirsutism but normal androgen production rates have increased 5α-reductase
activity and increased circulating levels of 5α-Adiol
gluc.54 It has also been suggested that other conjugated androgens, specifically
androsterone sulfate and androsterone glucuronide and 3α-diol
sulfate, may reflect peripheral androgen activity.59 One of the major causes of hyperandrogenism in women is polycystic ovarian
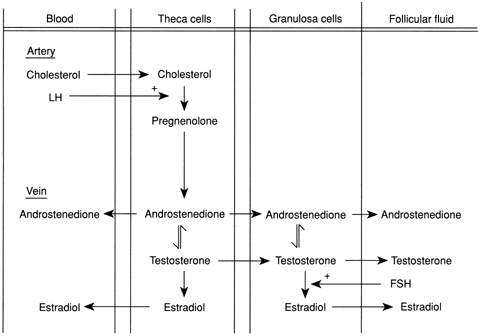
syndrome (PCOS).60,61 There may be overproduction of DHEA, A, T, or all three in this disorder, and
the cellular origin of the excess androgens includes the stromal
cells, those cells lining atretic follicles, and luteinized stromal
cells. Most women with PCOS have elevated LH levels,62 although this may not be obvious in a single blood sample. A major component
of PCOS is insulin resistance,54,63,64 which leads to glucose intolerance and diabetes mellitus.65 Androgen-secreting tumors of the ovary include thecomas, luteomas, hilar
cell tumors, and arrhenoblastomas.66 Androgen-secreting ovarian tumors may be gonadotropin dependent and responsive
and occur with increased frequency in the LH-stimulated polycystic
ovary. T levels are usually markedly elevated (greater than 2 ng/mL; greater
than 7 nmol/L) in association with ovarian tumors but only
occasionally reach such levels in PCOS.67 Adrenal disorders causing excessive androgen production include the various
forms of congenital adrenal hyperplasia (CAH), the most common of
which is that associated with 21-hydroxylase deficiency.68 This can present in a variable manner from abnormal genitalia and adrenal
insufficiency at or soon after birth to postpubertal hirsutism.68 The less-common forms of CAH are those associated with deficient 11β-hydroxylase69 and 3β-hydroxy-Δ5— steroid dehydrogenase deficiency.70 In the former, the deficiency of 11β-hydroxylase activity can cause
hypertension and signs of androgen excess and is associated with an
elevation of 11-deoxycortisol.69 In subjects with a deficiency of the 3β-hydroxy-Δ5— steroid dehydrogenase enzyme system that converts 3β-hydroxy-Δ5— steroids to 3-keto-Δ4-steroids, there is an increase in the secretion of 17α-hydroxy pregnenolone
and DHEA and a relative decrease in the secretion of progesterone
and Δ4-A.70 The increased DHEA results in signs of hyperandrogenism, presumably because
the enzymes in the peripheral tissues are not defective to the same
degree as in the adrenals and ovaries. The signs of hyperandrogenism
depend on the degree of enzyme deficiency. The classic type presents
at or near birth, usually with abnormal external genitalia and severe
symptoms of adrenal insufficiency. The nonclassic type presents at late
childhood or adolescence with varying signs and symptoms of hyperandrogenism. The
diagnosis depends on finding an increase in the levels
of 17α-hydroxypregnenolone or DHEA.71 The frequency with which this syndrome is seen is variable depending on
the clinic population. The nonclassic 21-hydroxylase deficiency is transmitted as an autosomal-recessive
trait and becomes apparent in the affected person in later
childhood or adolescence.68 The 21-hydroxylase deficiency gene is located on chromosome 6, and there
appears to be an increased frequency of the HLAB14,DR1 haplotype. In
Ashkenazi Jews, the frequency of the disorder has been reported to be
as high as one in 30, but in mixed non-Jewish whites, the frequency
is much less(1:1000).68 The incidence in clinical series is, thus, dependent on the population
base of that clinic. Affected persons have an increased level of 17α-OH
progesterone, but this is often demonstrable only after ACTH
stimulation.72 Interestingly, there is a cryptic form of the disorder in which the person
has the genetic and biochemical defect but is asymptomatic. Benign or malignant tumors of the adrenals may also be a source of excess
androgens. The functioning adrenal tumors usually have relative deficiency
of 3β-hydroxy-Δ5— steroid dehydrogenase activity; and, consequently, most produce primarily Δ5— steroids, particularly DHEA and DHEAS. However, a few T-secreting adrenal
tumors have been reported. As noted earlier, the population base of clinics is variable so that reports
on the most frequent cause of hyperandrogenism (adrenal, ovarian, or
idiopathic) also are variable. However, PCOS is believed by many
to be the most common cause. This is confounded because androgen excess
itself can result in PCOS, and thus a combined adrenal ovarian defect
can be demonstrable.73 Adipose Tissue The capability of adipose tissue to metabolize androgens is well recognized. Foremost
among the androgen metabolic activities of adipose tissue
is its ability to aromatize androgens to estrogens,74,75 and obese men and postmenopausal women can have elevated levels of estrogens. Although
adipose tissue does have the 17β-hydroxysteroid
dehydrogenase enzyme system, the interconversion of Δ4-A and T is not increased in obesity as is the aromatization.76 In obese persons, the location of the adiposity appears to play a role
in their androgen metabolism.77 In women with most of the adipose tissue in the abdominal area, there
is a tendency for more signs of hyperandrogenism than in women with the
obesity in the hip and buttocks area.78 The amount of aromatization also differs depending on the waist-hip ratio. Those
women with high ratios have lower aromatization.78 As noted previously, the production rates of androgens are usually increased
in obese women, but this is primarily because of an increase in
the adrenal secretion of precursors. The conversion rate of A to E1 increases with age and is the major source of estrogen in postmenopausal
women.75,79,80 The excessive production of E1 in the fat tissue of postmenopausal obese women can result in abnormal
uterine bleeding. This pathologic state may also result from excessive
A production with a normal A-to-E1 conversion rate. The physiologic significance of other known aspects of
androgen metabolism in fat tissue remains largely speculative. Brain Masculinization of the brain in males is induced by androgens early in
fetal life.8,49 Female animals exhibit male psychic and sexual activity after an injection
of T during a critical neonatal period. Data indicate that this effect
is motivated by way of the estrogen produced from the peripheral
conversion of T. Finally, 5α-reductase activity is present in the
hypothalamus and the anterior pituitary gland. Although in men androgens
play a role in the control of gonadotropin secretion, in women the
estrogens are the steroids that are most important. Aromatization occurs
in the hypothalamus, but in healthy women, most of the estrogen
controlling gonadotropin release comes from the circulation. |