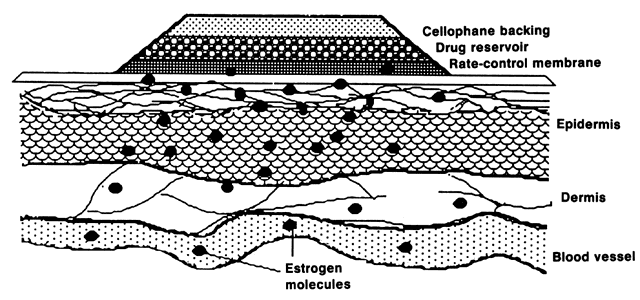
E2 availability to intracellular receptors is modulated by plasma-protein binding; estrone sulfate (E1S) conjugation; formation of metabolites, such as catechol estrogens, which may modulate estrogen-receptor interaction
by acting as antiestrogens in some tissues, such as the endometrium, and
estrogens in others, such as the nervous system; estrogen conjugation; microvascular extraction in target organs; and autoregulation in target cells. The roles of these metabolic regulators are reviewed next. Plasma Protein Binding Steroid hormones cross target tissues and microvascular membranes by passive
diffusion.10 Their rate of transfer is regulated by the availability of nonprotein-bound
steroid, which must compete between estrogen target tissue receptor
proteins and plasma-binding proteins.10,11 E2 interaction with sex hormone binding globulin (SHBG), a high-affinity
plasma glycoprotein, is about 38% of the total E2. Circulating E2 is also bound to albumin for another 60%. Of the total, only 3% is unbound
and fully available.9 E1 is not strongly bound to plasma proteins and has a much higher MCR than
E2. E1S is bound with high affinity to albumin, with 90% of E1S circulating in bound form.12 MCR of El and E2 is greatly influenced by protein binding. Whenever serum-binding capacity
is increased, tissue extraction and MCR are proportionally decreased. Although
SHBG is probably regulated primarily by growth hormone, which
promotes its synthesis in the liver, and somatomedin C, which stimulates
its extravasation and uptake in tissues, sex steroids are believed
to have a modulating influence that is indirect and quantitatively
somewhat less important.13 Accordingly, androgens decrease SHBG production in the liver such that
the binding capacity in men is lower than in women. Estrogens and thyroid
hormone increase SHBG production.13 Binding capacity is further increased in women with hyperthyroidism. in
pregnancy. by exogenous estrogens. and by smoking.13 Thus, in patients with polycystic ovarian disease, SHBG is suppressed
by excessive androgens, resulting in elevated free estrogen as well as
elevated free testosterone levels, all of which act in concert to aggravate
further the underlying pathophysiology of this disorder.14 Excess E2 bioavailability may be important in the pathogenesis of breast carcinoma
because women who develop this disease have higher levels of unbound
E2 and lower levels of SHBG than non-cancer-afflicted controls.15 E1S is also affected by protein binding. Very high binding affinities between
E1S and albumin are responsible for the slow turnover of the E1S pool, low E1S MCR, and low E1S glomerular filtration.8 Estrone Sulfate Conjugation and the E1 S Pool The interconversions between E2, El, and E1S are depicted in Figure 3. Transfer constants are given for each of these reversible interconversions. Although
E2 is the biologically active hormone, E1 is the first step to biologic inactivation of E2. It is an obligatory intermediate leading to the formation of either E1S or to ring A (catechol) and ring D (estriol and epiestriol) estrogen
metabolites (see Fig. 3) through nonreversible transformations.8 Although E1 binds with estrogen receptors, much of its biologic activity may involve
intercellular conversion to E2.16 E1S becomes hormonally active after cleavage of the sulfate radical by the
liver or other peripheral sites and in target tissues, which may have
sulfatase activity that permits autoregulated conversion to El and E2 from E1S. Such autoregulated activity, occurring at the cell level in the endometrium, breasts, and
neoplastic tissues, may vary with the time of the
menstrual cycle.16 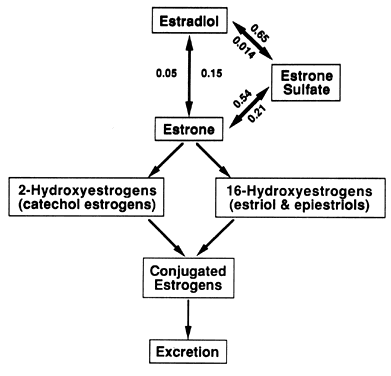 Fig. 3. Estradiol (E2) and estrone (El) and their metabolites. The E2, El, and estrone sulfate (E1 S) equilibrium is described by the individual transfer constants (PBBAB ). The metabolism of E2 passes through El into the E2, E1, E1 S equilibrium or irreversibly into the ring A 2-hydroxyestrogens (catechol
estrogens) or the ring D 16-hydroxyestrogens (estriol and epiestriols) pathways. These
metabolites are conjugated and excreted. Fig. 3. Estradiol (E2) and estrone (El) and their metabolites. The E2, El, and estrone sulfate (E1 S) equilibrium is described by the individual transfer constants (PBBAB ). The metabolism of E2 passes through El into the E2, E1, E1 S equilibrium or irreversibly into the ring A 2-hydroxyestrogens (catechol
estrogens) or the ring D 16-hydroxyestrogens (estriol and epiestriols) pathways. These
metabolites are conjugated and excreted.
|
E1S is quantitatively the single most important plasma estrogen metabolite. Its
relative importance is described by the respective transfer constants (pBBE1-E1S 0.54; pBBE2-E1S 0.65), which means that 54%. of E1 and 65% of E2 entering the blood is converted to E1S.8 Although these transformations are reversible, the transfer constants
describing the opposite reaction (pBBE1S-E1 0.21; pBBE1S-E2 0.014) indicate that a smaller percentage of E1 is formed from E1S than El to E1S, and an almost insignificant amount of E2 is formed from E1S. E1S apparently is not secreted by any endocrine tissue because virtually
all of its blood production can be accounted for by the conversions El-ElS and E2-ElS.8 The E1S pool is conceptualized as a large, slowly metabolized estrogen reservoir
with 90% of its mass albumin bound with consequently low MCR (~150 liters/24 hr) and
low renal clearance.8,12 PBE1S fluctuates with the menstrual cycle much as do its major precursors, El and E2; follicular phase PBE1S is ~95 μg/24 hr, whereas luteal phase PBE1S is ~ 180 μg/24 hr.8 ElS plasma concentrations are higher than any other known estrogen, with
representative follicular phase values of 1000 pg/ml and luteal phase
values of 1800 pg/ml.17,18,19 When plasma ElS concentrations (Fig. 4) are compared with E1 and E2 during the menstrual cycle, ElS concentrations appear to lag slightly behind the large rises and decreases
of E2 by about 24 hours, an observation that probably results from the large
size of the ElS pool, its low MCR, and its slow turnover.19 Even though ElS is not itself biologically active, E1 and E2 formed at the target tissue from E1S may be of major importance. Endometrium, for example, converts ElS to E1 and E2.11 Target tissue metabolic activity of this type cannot be measured by in vivo isotope dilution techniques, and as such the actual contribution of E1S to overall estrogen effect is difficult to assess. Substantial quantities of ElS and estradiol sulfate are found in breast tissues of patients with mammary
carcinoma (Fig. 5).20 ElS is converted at high percentage to E2 in different hormone-dependent mammary cancer cell lines, with very little
or no conversion in hormone-independent mammary cancers.20 Progesterone provokes a decrease in E2 uptake in vitro.20 Accordingly, the control of the sulfatase and 17β-hydroxysteroid
dehydrogenase activity may be important regulators in estrogen-sensitive
neoplastic tissues.20 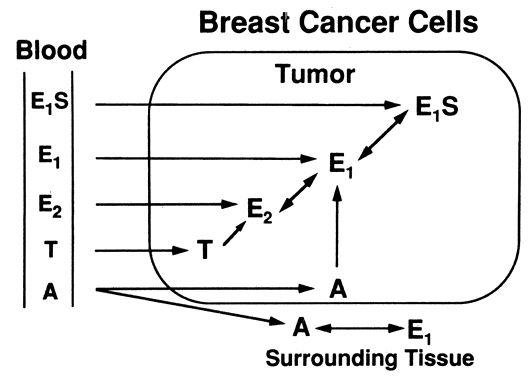 Fig. 5. Sources of intratumoral estrogens in breast carcinoma. A major source is
believed to be interconversion of El and E2 from El S extraction by tumor tissue from the circulation, but other sources are
active as well.(Modified from Lonning PE, Dowsett M, Powles TJ: Postmenopausal estrogen
synthesis and metabolism: Alterations caused by aromatase inhibitors
used for the treatment of breast cancer. J Steroid Biochem 35:355, 1990) Fig. 5. Sources of intratumoral estrogens in breast carcinoma. A major source is
believed to be interconversion of El and E2 from El S extraction by tumor tissue from the circulation, but other sources are
active as well.(Modified from Lonning PE, Dowsett M, Powles TJ: Postmenopausal estrogen
synthesis and metabolism: Alterations caused by aromatase inhibitors
used for the treatment of breast cancer. J Steroid Biochem 35:355, 1990)
|
Estrogen Metabolites: Ring A (Catechol) and Ring D (Estriol and Epiestriol) Estrogens Virtually all estrogen metabolites flow through El (Fig. 6) as the parent precursor. After formation of El, further metabolic transformation takes place through one of two major
pathways: the ring A hydroxylated compounds (catechol estrogens)21 or ring D hydroxylated compounds (estriol and the epiestriols).22 The relationships are shown schematically in Figure 6. Quantitatively, these pathways are of approximately equal significance. They
are mutually exclusive--that is, they do not convert into one
another, and there are relatively few compounds metabolized concurrently
as both ring A and ring D compounds. The hormonal effects of the ring
A compounds are limited primarily to the central nervous system (CNS) and
are antiestrogenic in other estrogen-sensitive tissues.21 The ring D compounds retain their estrogenic attributes but are less active
than E2.23 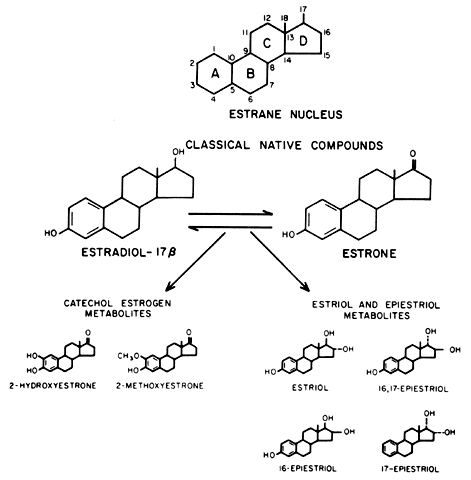 Fig. 6. Molecular structures of estrone (E1) and estradiol (E2) and the major estrogen metabolites. The estrane nucleus is shown for
reference. Fig. 6. Molecular structures of estrone (E1) and estradiol (E2) and the major estrogen metabolites. The estrane nucleus is shown for
reference.
|
CATECHOL ESTROGENS. The parent catechol estrogen compound is 2-hydroxyestrone, and its principal
metabolite is 2-methoxyestrone. The term catechol is derived from the presence of two hydroxyl groups on the aromatic A
ring, a structural feature in common with the catecholamines.21 A group of 4-hydroxyestrogens, also catechol estrogens, has been identified
but is of comparatively minor quantitative importance compared with 2-hydroxyestrones. Although the conversion of El to 2-hydroxyestrone takes place primarily in the liver, it also occurs
in the hypothalamus and other CNS tissues.24,25 The 2-hydroxylation of the parent classic estrogens in the CNS is of major
importance because the derived 2-hydroxy estrogens compete with catecholamines
for the enzyme catechol-O-methyltransferase.24,26 As highly efficient competitive inhibitors of catechol-O-methyltransferase, catechol estrogens interfere with metabolic degradation
of catecholamines at specific CNS foci, resulting in prolonged catecholamine
activity with the consequent release of hypothalamic releasing
and inhibiting factors that are catecholamine modulated.26 An alternative CNS activity for the catechol estrogens is competition
for CNS E2 receptors; in this role, the catechol estrogens act as antiestrogens.21 The catechol estrogens behave as antiestrogens in peripheral estrogen-sensitive
tissues. The 2-hydroxy compounds have virtually no uterotropic
activity yet have 20% to 50% the affinity for uterine estrogen receptors
as does E2 itself.21 Thus, direction of E2 metabolism through the catechol pathway may greatly suppress the peripheral
estrogenic activity of secreted E2.21 Plasma concentrations of 2-hydroxyestrone are slightly lower than E2, with a range of approximately 50 to 95 pg/ml.27 Urinary excretion of 2-hydroxyestrone observed over the menstrual cycle
ranged from approximately 15 μg/24 hr, with highest values observed
at midcycle and during the luteal phase (Fig. 7).28 A shift of estrogen metabolites to the catechol pathway occurs in patients
with hyperthyroidism or anorexia nervosa, female athletes, and in
women who smoke.29,30,31,32,33 A shift to ring D metabolites occurs in hypothyroidism30 and in obesity.31 Increased metabolism through catechol estrogens has been believed to be
important in breast cancer pathogenesis,34 uterotropic functions,35 cigarette smoking,33 and exercise-induced amenorrhea.32 In competitive female athletes, serum levels of catechol estrogens are
increased during strenuous exercise, suggesting that conversion to this
non-uterotropic steroid could be a factor in the amenorrhea of these
women.32 Premenopausal female smokers show significantly increased estrogen 2-hydroxylation
expressed as increased 2-hydroxyestrone urinary excretion.33 This shunting of estrogen metabolites through the catechol pathway may
be an important mechanism for their association with the antiestrogen
effects of cigarette smoking.33 Thus, female smokers have an early natural menopause, a lower rate of
cancer of the endometrium, and an increased risk of osteoporotic fractures.33,36 They have a reduced risk of benign breast disease, endometriosis, and
uterine fibroids.37,38 RING D PATHWAY (ESTRIOL AND THE EPIESTRIOLS). The principal ring D compound, estriol (E3), is derived by 16α-hydroxylation of E1 to form the intermediate, 16α-hydroxyestrone. The epiestriols comprise
various 16- and 17-hydroxylated estrogens arrayed into an assortment
of α and β configurations (see Fig. 6).22 Although E3 is the most abundant urinary estrogen, unconjugated E3 circulates in plasma at very low concentrations (7 to 11 pg/ml) because
much of it is conjugated to glucuronides.39 Although unconjugated E3 is believed to have an estrogen potency comparable to E1, its very low concentration indicates that its hormonal effect is not
substantial. Estrogen Conjugates and Their Excretion Estrogen metabolites formed from either the catechol pathway or ring D
pathway are conjugated in the liver and kidneys with a glucosiduronate
radical at various hydroxyl groups to form highly polar (water-soluble) estrogen
glucuronides, which are excreted into the urine. E1 and E2 can also be conjugated directly and excreted by the same process. Some
estrogen is excreted in feces.40,41 The details of these reactions have been reviewed extensively.40,41 Microvascular Extraction Bioavailability of biologically active estrogen is affected by the distribution
and permeability properties of the microvascular circulation
in estrogen-responsive organs. Extraction efficiency in estrogen-sensitive
tissues may thus be much higher than in non-estrogen-sensitive tissues. Thus, when
human uteri are perfused in vitro with a lactated Ringer's solution containing physiologic concentrations
of E1, E2, and E1S and compared with perfusion with human female serum, endometrial and
myometrial estrogen extraction is much higher from the Ringer's solution
because it is free of binding proteins. Also, endometrial extraction
of unbound E1S and E1 greatly exceeds myometrial extraction.10 Extraction is most efficient in the luteal phase and in specimens with
endometrial hyperplasia. 10,42 This is thought to occur because influxes of steroids are related to the
surface area of the microcirculation, which changes during the menstrual
cycle. Cellular Autoregulation The final common pathway of estrogen hormone action is incorporation of
E2 into the nuclear receptor complex. Target tissues (e.g., myometrium, endometrium, and CNS structures) modulate their response
to circulating estrogen at the cell level by self-regulated reversible
conversion of biologically active estrogens to relatively inactive ones
before receptor interaction. Thus, human endometrial cells convert
E2 to El by progesterone-stimulated 17β-estradiol dehydrogenase, which increases
markedly in activity during the progesterone-dominant luteal phase.43 Conversely, endometrial sulfotransferase augments conversion of unconjugated
E1 and E2 to their respective and inactive 3-sulfates.44 Like 17β-dehydrogenase, sulfotransferase activity increases during
the luteal phase, when it produces increasing E1S, which does not bind to intracellular estrogen receptors. E1S is excreted back into the circulation and is associated with decreased
nuclear estrogen-receptor complex formation during the luteal phase.43 A similar activity occurs in myometrial tissues.44 In breast carcinoma tissue, this interconversion may modulate the estrogen
dependency of this disease.20 Analogous autoregulatory functions involving the catechol estrogens modulate
catecholamine metabolism in the CNS. Although catecholamine formation
is modified by various physiological and pathological conditions, the
regulatory impact of catechol estrogens is not well understood.21 |
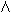 4A] to E1).
4A] to E1).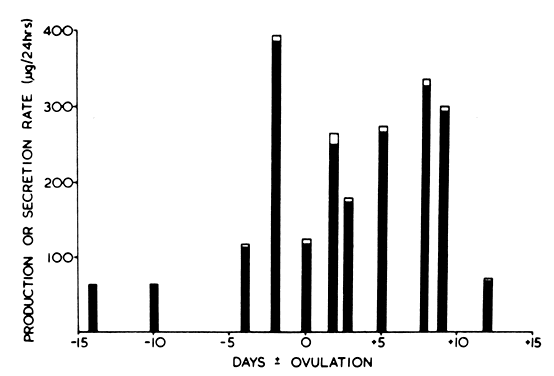
 SovE1;
SovE1;  E1 derived from E2; ·El, derived from other sources.
E1 derived from E2; ·El, derived from other sources.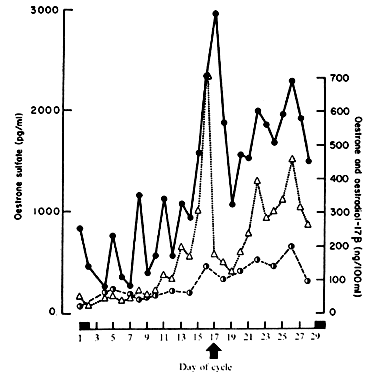
 , the day of maximum urinary excretion of luteinizing hormone;
, the day of maximum urinary excretion of luteinizing hormone;  ---
---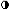 ---
---
 ---
---