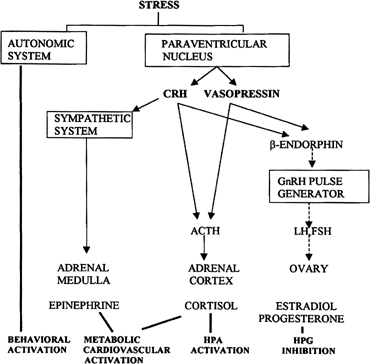
Follicular Phase CYCLIC RECRUITMENT OF A FOLLICLE COHORT. Although mechanisms of initial follicular growth in the ovary are difficult
to investigate because of its protracted process, there is good evidence
that dormant primordial follicles are recruited continuously into
growing that transforms them into primary, secondary, and early antral
follicles, at which stage most become atretic. This process is initiated
already before birth and continues until menopause and seems to
proceed under the control of still unknown local ovarian factors with
gonadotropins perhaps playing a minor modulatory role.37 After puberty and in each menstrual cycle, one cohort of antral follicles
is rescued from going into the atretic process by being recruited
for further growth. This “cyclic” antral follicle recruitment
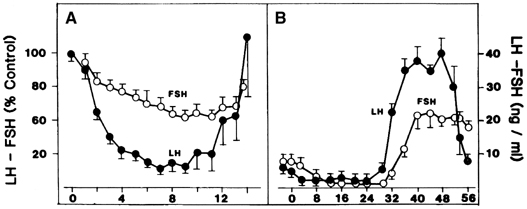
requires an appropriate signal from the pituitary gland ( Fig. 8).38 This signal is FSH and is represented by the small but selective increase
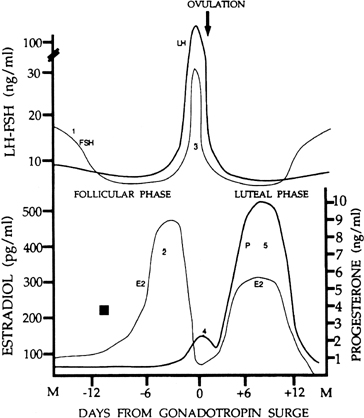
in FSH starting at the end of the preceding luteal phase (see Fig. 2).39 The size of the cohort is dictated by the amount of FSH present at recruitment: A
greater sized cohort can be recruited by increasing FSH amounts, as
shown in gonadotropin-stimulated cycles, in which hormones usually
are administered at supraphysiologic levels. FSH stimulates mitosis
and activates aromatase production in granulosa cells, allowing for
growth and increased local estradiol production. Data also show that
FSH is the major stimulus for inhibin B secretion, which increases at
that time.40 Experimental treatment with a GnRH antagonist at the end of the luteal
phase, which abolishes the early follicular phase rise in FSH, also prevents
the associated increase in inhibin B, whereas the same treatment
followed by exogenous FSH restores the secretion of inhibin B.33 The measurement of circulating inhibin B levels in the early follicular
phase or in the early days of a stimulated cycle provides an early indicator
of the number of recruited follicles and of their activity.41 Studies in premenopausal women suggest that a decrease in inhibin B is
the earliest marker of the decline in follicle number across reproductive
aging.36 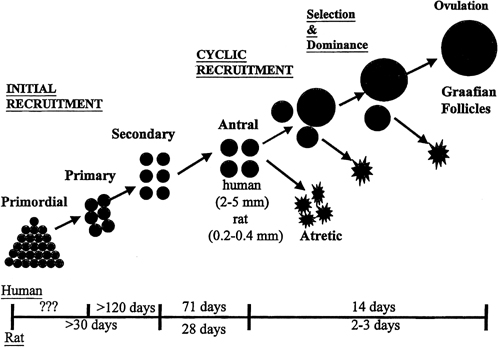 Fig. 8. Follicle recruitment and selection in human and rat ovaries. At the start
of each menstrual cycle, an increase in follicle-stimulating hormone (FSH) allows
a cohort of antral follicles (2 to 5 mm in diameter) to
be recruited (cyclic recruitment) and escape apoptotic demise. From this
cohort, a leading (dominant) follicle emerges (selection), resulting
in the demise of the other follicles in the cohort. This dominant follicle
undergoes final growth and maturation and ovulation.(McGee EA, Hsueh AJ: Initial and cyclic recruitment of ovarian follicles. Endocr
Rev 21:200, 2000; used by permission of The Endocrine Society.) Fig. 8. Follicle recruitment and selection in human and rat ovaries. At the start
of each menstrual cycle, an increase in follicle-stimulating hormone (FSH) allows
a cohort of antral follicles (2 to 5 mm in diameter) to
be recruited (cyclic recruitment) and escape apoptotic demise. From this
cohort, a leading (dominant) follicle emerges (selection), resulting
in the demise of the other follicles in the cohort. This dominant follicle
undergoes final growth and maturation and ovulation.(McGee EA, Hsueh AJ: Initial and cyclic recruitment of ovarian follicles. Endocr
Rev 21:200, 2000; used by permission of The Endocrine Society.)
|
SELECTION OF A DOMINANT FOLLICLE. Although several follicles are recruited in the early follicular phase
as part of the cohort, in mono-ovular species usually only one continues
to grow to become a mature preovulatory follicle. This is referred
to as the selection process, which occurs early in the midfollicular phase.42 Experimental cauterization of the largest follicle at that time in the
monkey results in a delay in the midcycle gonadotropin surge, a reflection
of the absence of a surrogate follicle able to take the place of
the destroyed selected follicle and of the need to start anew and recruit
another cohort of follicles.42
The precise mechanism by which one follicle of the cohort is selected
in primates remains to be elucidated. The selected dominant follicle is
a distinguishable structure, however, in terms of its cellular development
and especially of its vascularization: It possesses a denser microvascular
network than that of lesser developed follicles.43
Experimental evidence has shown an active angiogenic process in this follicle
destined for ovulation and has suggested an active role for regulators
of angiogenesis in cyclic folliculogenesis. Vascular endothelial growth
factor (VEGF), an important angiogenic factor, has been detected in the
developing follicle, with the most intense signal in the mature follicle.44,45
VEGF activity is well correlated with proliferating activity markers in
vascular endothelial cells and is a sign of active vasculogenesis.46
Experimental data in nonhuman primates showed that VEGF receptor inactivation
already in the early follicular phase results in a rapid decrease in inhibin
B secretion, suggesting an arrest in the development of the cohort of
recruited antral follicles and in a delay in the rise in estradiol ( (Fig.
9).47 These data directly show that
normal follicular development cannot occur in the absence of adequate
VEGF stimulation to ensure proper local angiogenesis. Data in the literature
also suggest a role for the gonadotropins in VEGF production by the follicle.
Studies with monkey granulosa cells show that not only large amounts of
gonadotropins representative of the midcycle ovulatory surge, but also
smaller amounts more typical of tonic secretion enhance local VEGF production.48
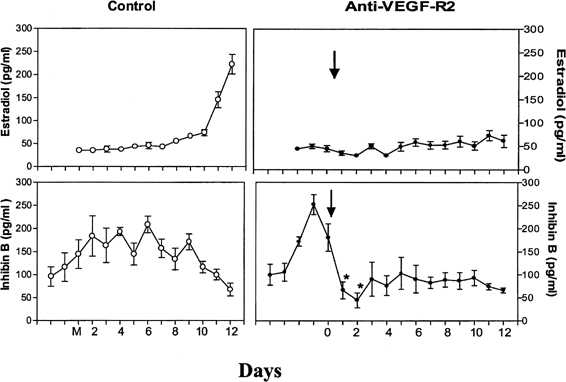 Fig. 9. The role of angiogenesis in follicular development during the menstrual
cycle. Illustrated are estradiol (upper level) and inhibin B (lower level) concentrations during the follicular phase in control monkeys (left panels) and in monkeys treated with antibodies to vascular endothelial growth
factor receptor (anti-VEGF-R2) (right panels). Note the rapid inhibition of inhibin B and the absence of a rise in estradiol
after antibody administration, indicating an essential role of
VEGF and angiogenesis in follicular growth and maturation.Zimmerman RC, Xiao E, Bohlen P, Ferin M: Administration of anti-vascular
endothelial growth factor receptor 2 antibody in the early follicular
phase delays follicular selection and development in the rhesus monkey. Endocrinology 143:2496, 2002; used by permission of The Endocrine
Society.) Fig. 9. The role of angiogenesis in follicular development during the menstrual
cycle. Illustrated are estradiol (upper level) and inhibin B (lower level) concentrations during the follicular phase in control monkeys (left panels) and in monkeys treated with antibodies to vascular endothelial growth
factor receptor (anti-VEGF-R2) (right panels). Note the rapid inhibition of inhibin B and the absence of a rise in estradiol
after antibody administration, indicating an essential role of
VEGF and angiogenesis in follicular growth and maturation.Zimmerman RC, Xiao E, Bohlen P, Ferin M: Administration of anti-vascular
endothelial growth factor receptor 2 antibody in the early follicular
phase delays follicular selection and development in the rhesus monkey. Endocrinology 143:2496, 2002; used by permission of The Endocrine
Society.)
|
GROWTH OF THE DOMINANT FOLLICLE.
The morphologic hallmark of the selected follicle is the acquisition
of LH receptors in response to FSH action49
and the differentiation of an endocrinologically active theca layer capable
of synthesizing androgens in response to LH stimulation. These androgens
are aromatized to estradiol in the granulosa cell layer, which contains
aromatase but which itself does not possess the full complement of steroid
biosynthetic enzymes required for the synthesis of androgens.50
Serum estradiol levels begin to rise as a result of the emergence of the
dominant follicle. The rising estradiol, through the negative feedback
loop, suppresses FSH levels (see Fig.
2) to concentrations that are too low to sustain maturation of the
other follicles in the cohort with the consequence that these undergo
final atresia.39 This process can be prevented
experimentally by neutralizing the rising estradiol levels or by providing
a moderate and continued elevation of FSH during the midfollicular phase:
In these instances, the physiologic process of single follicle dominance
is interfered with, and ongoing growth of multiple follicles is fostered.51,52
By the midfollicular phase, there is an incremental increase in LH receptors, in
aromatizable androgens, and in estrogens, which induce FSH receptors
in the granulosa cells of the dominant follicle in a self-propagating
mechanism. The developing dominant follicle generates its own
estradiol microenvironment, which promotes further granulosa cell growth
directly through its mitogenic activity or indirectly through the
stimulation of local growth factors, for example, insulin growth factor.53 FSH itself also seems to act as a survival factor by stimulating cellular
proliferation and by inhibiting apoptosis, through still unknown genes.54
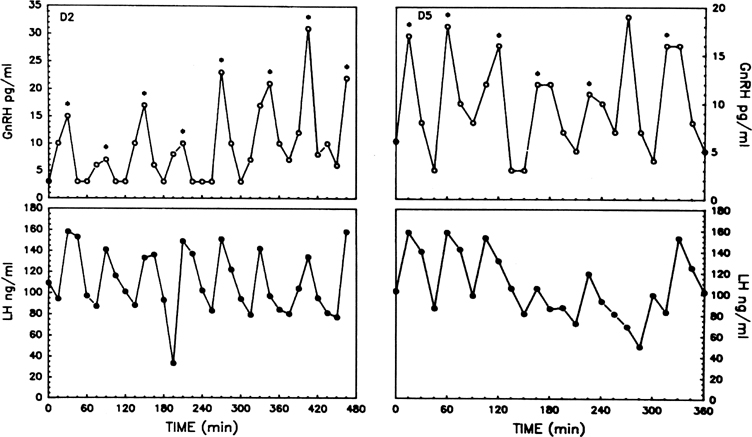
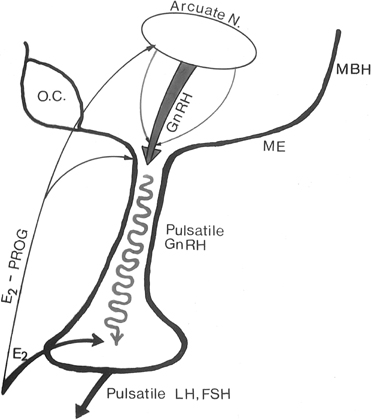
The pattern of pulsatile GnRH-driven LH secretion changes dynamically
across the human menstrual cycle.55 During
the follicular phase, a pulse frequency of about 1 pulse/90 minutes is
about the norm. Adherence to a specific regimen of pulse amplitude and
frequency is crucial for a normal follicular phase, menstrual cyclicity,
and reproductive function, and derangements of episodic LH secretion are
associated with reduced rates of ovulation in humans and nonhuman primates.56,57
LATE FOLLICULAR PHASE. In the late follicular phase, the diameter of the selected follicle increases
exponentially to a final size of 15 to 20 mm, and as a result secretion
of estradiol increases exponentially. Vascular development in
the dominant follicle plays a crucial role in this process because short-term
inhibition of angiogenesis after anti-VEGF antibody administration
during the later growth phase of the dominant follicle interferes
with its normal development, interrupts the characteristic rise in estradiol
secretion, and results in a significant lengthening of the follicular
phase.58 Under this condition, access to peripheral factors needed to support follicle
growth, such as the gonadotropins, is most probably limited. The increased estrogen milieu in the late follicular phase modifies the
genital tract.59 The glandular endometrium proliferates, characteristics of cervical mucus
change (increased secretion, decreased viscosity, and increase in
pH), and cornification of the vaginal epithelium occurs. As the dominant follicle approaches maturity, estradiol secretion reaches
its peak. This peak acts as the crucial ovarian signal that triggers
the ovulatory gonadotropin surge. Experimental neutralization of this
estradiol signal results in a suppression of the gonadotropin surge.60 In humans, an LH surge can be induced experimentally during the early
follicular phase after the administration of estradiol in amounts mimicking
those seen in the late follicular phase (see Fig. 5B).61 The process by which estrogen transients stimulate the release of LH and
FSH is referred to as the positive estradiol feedback loop and represents the crucial process that synchronizes follicle maturity
and ovulation.
Whether gonadotropins are released after an estradiol challenge depends
on the strength and duration of the estrogen signal. Minimal requirements
are an increase in circulating estradiol concentrations to a threshold
level close to that seen spontaneously in the late follicular phase and
for a period of at least 34 hours. Subthreshold (even maintained for a
prolonged period) or short-lived (even larger than threshold) rises in
estradiol levels do not elicit an LH surge experimentally.62,63
As shown in nonhuman primates and in sheep, the preovulatory gonadotropin
surge is preceded by a dramatic and sustained rise in GnRH (Fig.
10).64,65,66 Increased
GnRH secretion drives the preovulatory LH surge in a dose-dependent fashion.66
These data suggest a substantial central action of estradiol on the hypothalamus,
possibly on different neuronal cell populations than those modulating
the estradiol negative feedback loop.68
Experimental evidence indicates that the preovulatory LH surge depends
on GnRH stimulation throughout its entire course, but that the GnRH surge
persists many hours beyond the termination of the LH surge (Fig.
11).65,69 The LH
surge terminates even though there seems to be no change in the biologic
activity of GnRH.70 The functional significance
of this excess of GnRH to midcycle events remains to be determined. Suggesting
a similar role of GnRH in the induction of the preovulatory gonadotropin
surge in humans is the observation that administration of a GnRH antagonist
Nal-Glu acutely inhibits the LH surge and ovulation.71
Because LH responses to identical GnRH stimuli increase during the follicular
phase in parallel with increasing estradiol concentrations, estradiol
also may act to augment the gonadotrope’s responsiveness to GnRH.72
It is logical to postulate that during the normal menstrual cycle, central
and pituitary sites respond to the estradiol positive feedback loop signal
to ensure a timely gonadotropin surge.
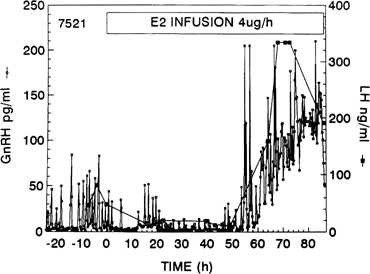 Fig. 10. Gonadotropin-releasing hormone (GnRH) surge in response to the activation
of the positive estradiol feedback loop. GnRH was measured at 15-minute
intervals in cerebrospinal fluid of a monkey given an estradiol infusion. Note
the initial decrease in GnRH pulse amplitude (negative feedback
loop).(Xia L, Van Vugt D, Alston EJ, et al: A surge of gonadotropin-releasing
hormone accompanies the estradiol-induced gonadotropin surge in the rhesus
monkey. Endocrinology 131:2812, 1992; used by permission of The
Endocrine Society.) Fig. 10. Gonadotropin-releasing hormone (GnRH) surge in response to the activation
of the positive estradiol feedback loop. GnRH was measured at 15-minute
intervals in cerebrospinal fluid of a monkey given an estradiol infusion. Note
the initial decrease in GnRH pulse amplitude (negative feedback
loop).(Xia L, Van Vugt D, Alston EJ, et al: A surge of gonadotropin-releasing
hormone accompanies the estradiol-induced gonadotropin surge in the rhesus
monkey. Endocrinology 131:2812, 1992; used by permission of The
Endocrine Society.)
|
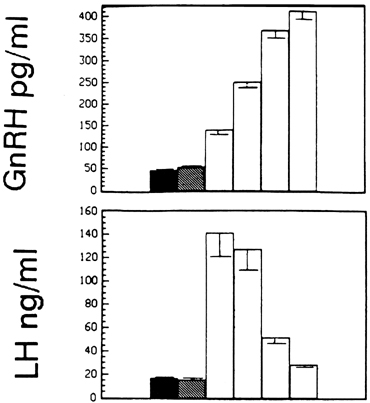 Fig. 11. The estradiol-induced gonadotropin-releasing hormone (GnRH) surge (upper panel) outlasts the luteinizing hormone (LH) surge (lower panel) by several hours. The first bars indicate baseline concentrations, and
the second bars show concentrations during the period of negative feedback
after estradiol administration. Subsequent bars illustrate changes
in concentrations in 12-hour periods after initiation of the surge.(Xia L, Van Vugt D, Alston EJ, et al: A surge of gonadotropin-releasing
hormone accompanies the estradiol-induced gonadotropin surge in the rhesus
monkey. Endocrinology 131:2812, 1992; used by permission of The
Endocrine Society.) Fig. 11. The estradiol-induced gonadotropin-releasing hormone (GnRH) surge (upper panel) outlasts the luteinizing hormone (LH) surge (lower panel) by several hours. The first bars indicate baseline concentrations, and
the second bars show concentrations during the period of negative feedback
after estradiol administration. Subsequent bars illustrate changes
in concentrations in 12-hour periods after initiation of the surge.(Xia L, Van Vugt D, Alston EJ, et al: A surge of gonadotropin-releasing
hormone accompanies the estradiol-induced gonadotropin surge in the rhesus
monkey. Endocrinology 131:2812, 1992; used by permission of The
Endocrine Society.)
|
Although progesterone secretion is minimal during the follicular phase,
there is a small but significant rise in this hormone at the time of initiation
of the gonadotropin surge (see later). It is believed that this increase
in progesterone is required for the full expression of the gonadotropin
surge because administration of a progesterone antagonist at midcycle
in humans results in a delay or in the abolition of the gonadotropin surge.73,74
In small amounts, progesterone facilitates LH release.
OVULATORY PERIOD. The estradiol-induced gonadotropin surge initiates a chain of events that
includes a series of finely orchestrated biochemical events, many of
which are still poorly understood, and that culminates in follicular
rupture and ovulation. Hormonally, drastic changes in ovarian steroid
profiles occur after alterations of enzymatic activity. The large increase
in LH inhibits androgen production, and as a result estradiol concentrations
decrease drastically from the preovulatory peak. Granulosa
cells become “luteinized,” and consequently a small preovulatory
rise in progesterone occurs within 1 hour of the LH surge.
Mechanically, ovulation consists of a rapid follicular enlargement with
subsequent protrusion of the follicle from the ovarian surface. About
24 to 36 hours after the initiation of the gonadotropin surge or 18 hours
after the gonadotropin peak, follicle rupture results in the expulsion
of an oocyte-cumulus complex. Rupture does not seem to be caused by an
increase in intrafollicular pressure; rather the LH surge induces an increase
in follicular volume, which is related to an increase in follicular blood
flow, a decrease in vascular permeability, and subsequent changes in the
properties of the follicular wall.75 These
changes in vascular function most probably reflect the large local concentrations
of angiogenic factors in the mature follicle and a functional VEGF system.76
The LH surge stimulates in the preovulatory follicles a cascade of proteolytic
enzymes, including plasminogen activator, plasmin, and matrix metalloproteinases,
which bring about the degradation of the perifollicular matrix.77,78
Pharmacologic blockage of any of these enzymes results in a reduction
in the ovulation rate. The periovulatory follicle produces prostaglandins
in response to the LH surge, and these seem to be crucial to follicle
rupture also, although their direct role in primates remains to be shown.79
A paracrine role of progesterone in the mediation of LH effects on follicular
rupture also has been suggested in lower species.78
Luteal Phase Shortly after the ovulatory gonadotropin surge, luteal differentiation
is activated. The granulosa cells of the dominant follicle fold and are
transformed into luteal cells. The basal lamina, which separates the
granulosa and theca layers, is disrupted, and capillaries from the theca
interna invade the granulosa layer (which until now had been avascular) to
form an extensive capillary network.80 After ovulation, a new ovarian structure emerges, the corpus luteum. New
key steroidogenic enzymes are activated so that the hallmark of the
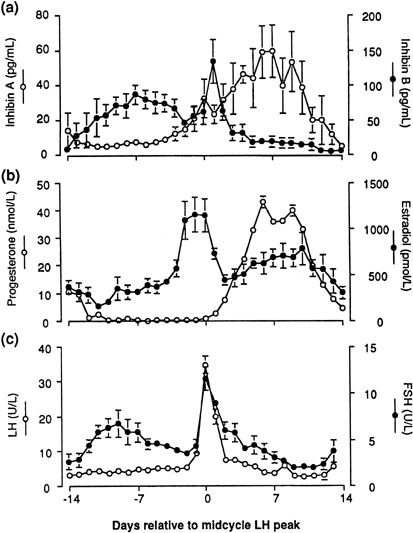
human corpus luteum is the secretion of progesterone and estradiol (see Fig. 2). The corpus luteum also secretes significant amounts of inhibin A (see Fig. 8).81
It is well documented that corpus luteum function depends primarily on
pituitary LH secretion throughout the luteal phase.82,83,84
Studies in hypophysectomized women in whom ovulation was induced by LH
treatment indicated that continuous administration of small amounts of
LH is essential to maintain the viability of the corpus luteum.28
GnRH antagonist treatment disrupts luteal cell morphology and suppresses
plasma progesterone.85 These experimental
results may reflect effects of gonadotropin stimulation on angiogenesis
in the corpus luteum. The mammalian corpus luteum is an exceptionally
dynamic organ in which growth and development occur rapidly. As for cyclic
folliculogenesis, vascular growth plays a central role in this process.86
Angiogenic factors, such as VEGF, also are present in high quantity in
the forming and developing corpus luteum. Experimental treatments in monkeys
that interfere with normal VEGF activity in the early and in the mid luteal
phase suppress the intense luteal endothelial proliferation and vascular
development. As a result, luteal function is compromised, as indicated
by a marked fall in plasma progesterone levels.87,88
These data indicate that gonadotropin and local VEGF support are as crucial
to corpus luteum function as they are to follicular function.
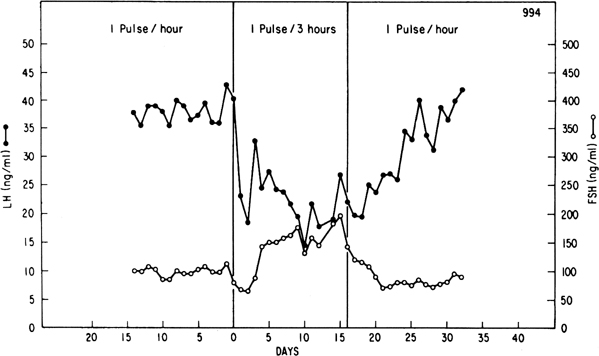
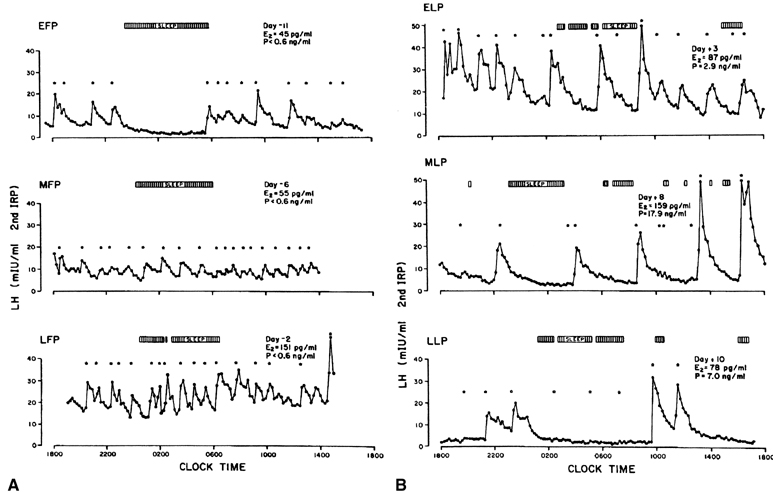
Progesterone dominance in the luteal phase results in a significant decrease
in GnRH/LH pulse frequency throughout this stage of the cycle (see Fig.
7).9,89 There is good
experimental evidence to indicate that this effect of progesterone is
mediated by central opioid peptides.90 Administration
during the luteal phase of competitive antagonists to endogenous opiates,
such as naloxone, is particularly effective in increasing LH pulse frequency
(Fig. 12B).91,92,93
A relevant endogenous opiate known to inhibit pulsatile LH secretion is
β-endorphin.94 Its neuronal cell
bodies are preferentially concentrated in the arcuate nucleus, an area
known to be involved in the control of gonadotropin secretion. β-Endorphin
release from the hypothalamus reflects the ovarian endocrine milieu: In
the absence of significant concentrations of ovarian steroids, such as
after ovariectomy or at menstruation, β-endorphin release is lowest,
whereas it is highest in the presence of estradiol and progesterone, such
as during the luteal phase (Fig.
12A).95,96 The naloxone
test is an indirect indicator of central opiate activity.
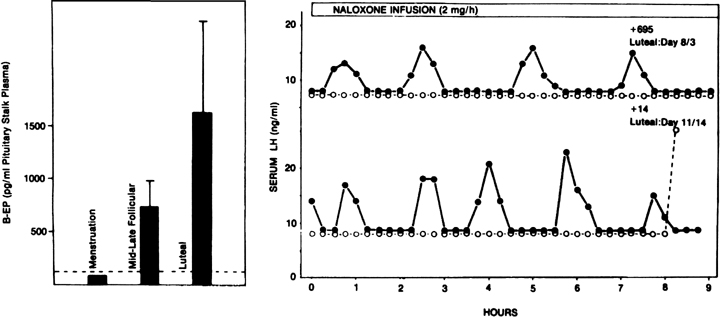 Fig. 12. The endogenous opiates and the menstrual cycle. A. Changes in hypothalamic β-endorphin activity (as determined by
its secretion into the pituitary stalk portal vasculature) during the
menstrual cycle of the rhesus monkey. In the presence of low ovarian steroids, such
as at menstruation, levels are lowest. They are highest
in the presence of progesterone during the luteal phase. B. Effects of a naloxone infusion on pulsatile luteinizing hormone (LH) secretion
in two monkeys during the luteal phase. Note the dramatic increase
in LH pulse frequency after endogenous opiate antagonism by naloxone (closed circles) compared with controls receiving saline (open circles).(A. Van Vugt DA, Lam NY, Ferin M: Reduced frequency of pulsatile luteinizing
hormone secretion in the luteal phase of the rhesus monkey: Involvement
of endogenous opiates. Endocrinology 115:1095, 1984; used by permission
of The Endocrine Society. B. Ferin M, Van Vugt D, Wardlaw S: The hypothalamic control of the menstrual
cycle and the role of endogenous opioid peptides. Rec Prog Horm Res 40:441, 1984; used
by permission of The Endocrine Society.) Fig. 12. The endogenous opiates and the menstrual cycle. A. Changes in hypothalamic β-endorphin activity (as determined by
its secretion into the pituitary stalk portal vasculature) during the
menstrual cycle of the rhesus monkey. In the presence of low ovarian steroids, such
as at menstruation, levels are lowest. They are highest
in the presence of progesterone during the luteal phase. B. Effects of a naloxone infusion on pulsatile luteinizing hormone (LH) secretion
in two monkeys during the luteal phase. Note the dramatic increase
in LH pulse frequency after endogenous opiate antagonism by naloxone (closed circles) compared with controls receiving saline (open circles).(A. Van Vugt DA, Lam NY, Ferin M: Reduced frequency of pulsatile luteinizing
hormone secretion in the luteal phase of the rhesus monkey: Involvement
of endogenous opiates. Endocrinology 115:1095, 1984; used by permission
of The Endocrine Society. B. Ferin M, Van Vugt D, Wardlaw S: The hypothalamic control of the menstrual
cycle and the role of endogenous opioid peptides. Rec Prog Horm Res 40:441, 1984; used
by permission of The Endocrine Society.)
|
Progesterone also affects the hypothalamic thermoregulatory center so that
an increase in basal body temperature accompanies increased progesterone
secretion during the luteal phase, giving rise to the typical biphasic
basal body temperature curve of the ovulatory menstrual cycle. Progesterone
dominance during this phase of the menstrual cycle modifies
the genital tract in preparation for possible implantation of a fertilized
ovum; there is increased secretory activity by the endometrial
glands and changes in the characteristics of the cervical mucus, which
is now thick and viscous. At these large luteal concentrations, progesterone
also inhibits the estradiol positive feedback loop.96 Gametogenic follicle growth also is held in abeyance during the luteal
phase in primates, presumably a local effect of progesterone.98 In primates, the life span of the corpus luteum is limited to a period
of about 14 days. According to biochemical and histologic criteria, the
corpus luteum reaches maturity 8 to 9 days after ovulation, after which
its secretory capability declines. Midway through the luteal phase, levels
of estradiol and progesterone decrease, and menstruation follows
ovulation by 13 to 15 days. Structural luteolysis is a complex process
responsible for the elimination of the corpus luteum. Steroidogenic
luteal cells undergo characteristic degenerative changes, with intense
cytoplasmic vacuolation and invasion by macrophages followed by a
perimenstrual apoptotic wave.99 The factors responsible for luteolysis in primates are unknown.80 The focus has been on three primary candidates: the reduction in LH pulse
frequency, local estradiol and prostaglandin F2α. Experimentally in primates, a decrease in pulse frequency or a temporary
cessation of LH support are not sufficient to induce luteolysis. Administration
of estrogen antagonists or aromatase inhibitors does not
substantiate a role for estrogen in this process either. Although prostaglandin
F2α seems to be an important luteolytic signal in nonprimate species, the
primate uterus is not the source of luteolytic agents because hysterectomy
does not alter cyclicity, and a role for prostaglandin in primates
remains to be substantiated. It is now believed that regression of the
corpus luteum is related to an alteration in age-dependent luteal cell
responsiveness and is dictated by various luteotropic and luteolytic
agents, the existence and dynamics of which have not been investigated.80 Only rapidly rising concentrations of chorionic gonadotropin can rescue
the corpus luteum from its regression and involution. After the dramatic decrease in estradiol and progesterone secretion at
the end of the luteal phase, there is a characteristic divergence in the
ratio of the two gonadotropins, now favoring a specific rise in FSH (see Fig. 2). The precise reason for the increase in the FSH-to-LH ratio at the end
of the cycle remains to be determined, but there are several possibilities, all
of which may play a role to a certain degree. Because FSH
seems to be slightly more sensitive to the estradiol negative feedback
loop than LH,100 a rise in FSH may be the result of the rapid decline in estradiol at that
time. The rise in FSH also may reflect differential effects of GnRH
pulse frequency on the synthesis of LH and FSH, as a lower pulse frequency
favors FSH β subunit synthesis, and a larger pool of FSH
would be available for release at the end of this period (see earlier).22 It also has been speculated that the accompanying fall in inhibin A (from
a peak in the mid luteal phase)101 may play a role in initiating the intercycle FSH rise. The increase in
the FSH-to-LH ratio heralds the new cycle with the recruitment of a new
cohort of follicles. |