Physical Changes of Puberty
The detailed longitudinal studies of British children by Tanner and Marshall1,2,3 provide information about the sequence and timing of the physical and sexual changes that occur throughout puberty. For convenience, breast and pubic hair development in girls and genital and pubic hair development in boys were arbitrarily divided into five stages (Tables 1 and 2), referred to as Tanner breast, pubic hair, and genital stages.
TABLE 1. Tanner Staging of Female Puberty
Stage | Breasts | Pubic Hair |
1—Prepubertal | Elevation of papilla only | Any vellus over pubes no different from abdominal hair |
2 | Breast bud with elevation of breast and papilla and enlargement of areola | Slightly pigmented, downy hair along the labia |
3 | Further enlargement of breast and papilla with no separation of their contours | Darker, coarser, more curled hair over pubes |
4 | Projection of areola and papilla to form a secondary mound | Adult pubic hair that does not reach thighs (axillary hair) |
5—Adult | Mature breast, projection of papilla only as areola conforms to breast contour | Adult hair now on thighs |
TABLE 2. Tanner Staging of Male Puberty
Stage | Genitalia | Pubic Hair |
1—Prepubertal | Testes, penis, and scrotum same size and proportion as early childhood | Any vellus over pubes no different from abdominal hair |
2 | Early enlargement of testes >2 cm3; scrotal skin reddens and changes in texture | Slightly pigmented, downy hair at base of penis |
3 | Penis lengthens; testes enlarge 3–6 cm3; growth of scrotum | Darker, coarser, more curled, over pubes |
4 | Further penile and scrotal growth; testes 8–12 cm3 | Adult pubic hair that does not reach thighs (axillary hair) |
5—Adult | Genitalia adult in size and shape, testes 15–25 cm3 | Adult hair now on thighs |
Several factors seem to influence the time of initiation of puberty, including genetic factors, geographic location, exposure to light, and general health and nutrition. In family studies, the onset of the mother’s puberty seems more important than the father’s puberty; the daughters and sons of early-maturing mothers are early to mature.4 Children closer to the equator, children at lower altitudes, children in urban areas, and obese children start earlier than children in northern latitudes, children at higher elevation above sea level, rural children, and normal-weight children. British girls begin menarche at a mean age of 13.5 years, whereas a 1970 study showed that American girls begin menarche at a mean age of 12.8 years.5
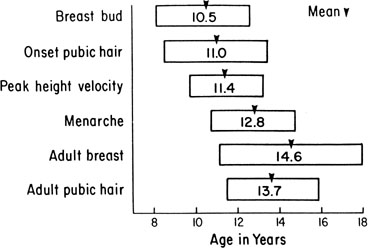
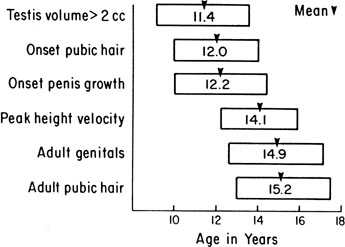
The sequence and timing of pubertal changes in girls and boys are shown in Figures 1 and 2. Figure 1 has been modified from the data of Marshall and Tanner2 to take into account the fact that menarche in American girls is 0.7 years earlier than in British girls. The first sign of puberty in a girl is breast budding, which occurs at a mean age of 10.5 years; the first sign of puberty in boys is testicular enlargement, which occurs a year later at a mean age of 11.5. The size of the testes can be measured accurately by comparison with a set of elliptical beads of increasing volume (the Prader orchidometer). Normal values for testicular volume are presented in Figure 3.6 The normal range (mean ± 2.5 SD) is 8 to 13 years for breast budding and 9 to 14 years for testicular enlargement. In about 20% of children, pubic hair growth is the first sign of puberty. A cross-sectional study based on pubertal staging of more than 17,000 girls from age 3 to 12 years was carried out in pediatric office practices across the United States to evaluate the age of onset of puberty in African-American and white girls. This study reported that white girls had onset of pubic hair growth at a mean of 10.51 years, breast development at 9.96 years, and menarche at 12.88 years. In comparison, African-American girls had the onset of pubic hair growth at a mean of 8.78 years (range, 4.8 to 12.8 years), breast development at 8.87 years (range, 5 to 12.7 years), and menarche at 12.16 years (range, 9.7 to 14.6 years).7 The maximal growth rate occurs 1 year after onset of breast budding in girls at a mean age of 11.5 years and 2.5 years after testicular enlargement in boys at a mean age of 14.1 years. A criticism of this cross-sectional study is the fact that pubertal staging was rated by inspection only and not palpation. It is possible that occasional erroneous classification of a Tanner 1 girl as a Tanner 2 girl may have occurred. This cross-sectional study is supported, however, by data from the National Health Examination Surveys (NHES) from 1963 to 1970 and the National Health Examination Survey (NHANES III) from 1988 to 1994. The NHANES data indicate that 12% to 14% of girls have Tanner 2 breast development by age 8 years. The median age of Tanner 2 breast development is 9.5 years in the aforementioned cross-sectional study and 9.7 years in NHANES. The onset of menses is 12 years in all three studies, supporting the idea that although the onset of puberty is occurring earlier, it is not being completed earlier.8 Based on these studies, it has been proposed that the lower limit of normal onset of puberty be redefined to 7 years for white girls and 6 years for African-American girls. An organic cause for precocious puberty should be sought in white girls less than 7 years old and African-American girls less than 6 years old. No change has been made in guidelines for the assessment of precocious puberty in boys, who still require investigation for organic causes if the onset of puberty occurs at less than 9 years of age.9 Girls and boys who experience rapid progression of pubertal development, even if it occurs after the above-mentioned age cutoffs, require diagnostic studies to determine the cause of their accelerated puberty.
|
|
|

The pattern of growth velocity from birth to adult life, including the timing of the maximal pubertal growth spurt, is illustrated in Figure 4. The maximal growth rate always precedes menarche. The mean age of menarche is 12.8 years, with a normal range of 10.3 to 15.3 years (mean ± 2.5 SD). Menarche occurs 2.3 ± 2.1 years after breast budding. Although there is not a corresponding landmark in boys, a Scandinavian study reported the average age of spermarche (as judged by appearance of spermatozoa in the urine) to be 13.4 years (range, 11.7 to 15.3 years).10 The age of menarche has been decreasing at a rate of about 1 year for every 25 years from 1830 to 1960. This change probably is related to improved general health and nutrition in particular. Over the last generation, this trend has leveled off.
|

All these figures may vary depending on the individual’s genetic and environmental background, and there is a wide variation of normal for the onset and sequence of each stage. The pubertal growth spurt coincides with increasing serum insulin-like growth factor I (IGF-I) concentrations concomitant with the rise in sex steroid levels.11 Evidence supports the conclusion that estrogens and androgens augment human pituitary growth hormone (hGH) secretion. Studies using 24-hour sampling show higher mean hGH concentrations, with more hGH peaks and higher peak amplitude.12 Children with precocious puberty and hGH deficiency still undergo a growth spurt, however, although less than normal, and manifest serum IGF-I concentrations intermediate between prepubertal children with hGH deficiency and children with sexual precocity and intact hGH secretion.13 These studies support a smaller but direct effect of sex steroids on IGF-I production.
Other important somatic changes accompany the pubertal growth spurt. In girls, total body fat increases from 16% to 23.5% of total body weight; in boys, total body fat decreases in early to mid puberty. In boys, muscle mass, as measured by radiologic studies of muscle cross-sectional area, increases fourfold; in girls, muscle mass increases twofold.
It has been proposed that when girls reach a critical weight of 47.8 kg, menarche is triggered (the so-called Frische-Revelle hypothesis).14 Other studies show that there is a large variation in total body weight at menarche, and menarche seems better correlated with the increase in total body fat to 23.5%. There are exceptions to this hypothesis, however: Girls with idiopathic central precocious puberty may undergo menarche with a total body fat of 19%, children with sexual precocity secondary to hypothyroidism have a total body fat of 29%, and obese girls with no signs of puberty may have a total body fat of 27%.15 It seems more reasonable to hypothesize that central mechanisms activate the hypothalamic-pituitary axis (HPA), leading to gonadotropin stimulation of ovarian sex steroids that in turn stimulate growth to the critical weight and an increase in body fat composition to 23.5%.
The cross-sectional study cited earlier documented a trend toward initiation of puberty at earlier ages in white and African-American girls. This trend may be linked to an increase in the prevalence of obesity in young girls. Significantly higher body mass index z scores have been shown in white pubertal versus prepubertal 6- to 9-year-old girls. In African-American girls, a smaller difference in body mass index z score was detected, suggesting that other genetic and environmental influences may play a role in their earlier pubertal onset. The practical implications of these findings for clinicians are that white and African-American girls who are overweight may be more likely to display signs of early puberty. Physicians should consider the contribution of obesity to the incidence of early puberty when decisions regarding searching for pathologic causes or consideration of treatment of early puberty is made.16
Leptin produced by adipocytes may play a role in modulating reproduction. Leptin-deficient Ob/Ob mice are infertile, with fertility restored with leptin replacement. In rats, minimal leptin levels seem to be required to initiate puberty. Serum leptin concentrations are correlated positively with Tanner stages, and adolescent girls have higher levels than boys. Obese adolescent girls have higher serum leptin concentrations, suggesting that these girls overcome leptin resistance to achieve reproductive capacity.17
Endocrine Changes at Puberty
Although several pieces of the puzzle of the mechanism of onset of puberty have been put into place, the complete picture of the factors responsible for activation of the hypothalamic-pituitary-gonadal axis is not clear. During fetal life, pituitary follicle-stimulating hormone (FSH) and luteinizing hormone (LH) concentrations rise and peak near midgestation; gonadotropin levels then fall, perhaps under the inhibition of placental steroids. Early in infancy, again there is a rise in gonadotropins, perhaps resulting from loss of placental steroid inhibition, with FSH predominating in girls and LH in boys. Serum gonadotropin concentrations reach levels seen in adults (FSH, 1 to 31 mIU/mL in women; LH, 3 to 25 mIU/mL in men), peak around 2 to 4 weeks of life, then fall to prepubertal low levels over the next several months.18,19 These pituitary gonadotropins stimulate temporary rises in estradiol in girls (10 to 60 pg/mL) and testosterone in boys (60 to 400 ng/dL).20,21 Even during the quiescent prepubertal period, sensitive immunofluorometric assays show pulsatile secretion, albeit at low amplitude, of LH and FSH, with nocturnal levels higher than daytime levels.22 The prepubertal pituitary can respond to exogenous gonadotropin-releasing hormone (GnRH) stimulation, with an FSH peak predominating in girls and a slight LH peak predominating in boys.23 The prepubertal gonad also shows a rise in sex steroids in response to exogenous gonadotropin stimulation.24 Immaturity of the prepubertal pituitary-gonadal axis does not seem to prevent the onset of puberty. These studies indirectly support the hypothesis that an inhibitor from higher brain centers restrains the HPA. Although the nature of this inhibitor is unknown, opioid peptides are a potential candidate. In a study of pubertal boys, administration of the opioid receptor antagonist naltrexone resulted in significantly higher mean LH concentration, LH pulse frequency, and area under LH time curve.25 Alternatively, animal studies support a neuronal-to-glial signaling pathway, perhaps via increased production of growth factors, such as transforming growth factor α, directly stimulating GnRH.26
The onset of puberty may result from diminished brain inhibition or positive glial-neuronal stimulation of GnRH secretion (Fig. 5). The maturation process that triggers this pubertal alarm clock is unknown, but it seems to be tied to normal development of the HPA during fetal life and infancy, physical growth and body composition changes, and skeletal maturation.
|
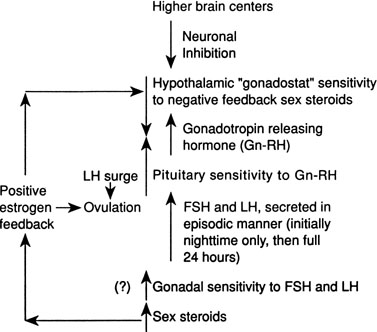
GnRH is secreted in a pulsatile pattern about once every 90 minutes. The frequency and amplitude of this GnRH pulse generator determine the pattern of FSH and LH secretion. During infancy, GnRH secretion is elevated followed by a long period of relative quiescence that occurs until late childhood, when pubertal maturation begins. The transition period from childhood quiescence to an adolescent pattern of GnRH secretion occurs gradually rather than abruptly. Animal models and human studies indicate that hypothalamic neurons are capable of secreting GnRH throughout childhood, and small pulses of GnRH-induced LH and FSH secretion are found in 4-year-olds.27 In girls in early puberty, FSH and LH are secreted at higher frequency and amplitude at night, and there is an equal FSH and LH response to GnRH stimulation. In mid to late puberty, FSH and LH are secreted during the day and night, with higher LH peak amplitudes, both spontaneous and in response to exogenous GnRH stimulation.23 In boys in early puberty, nocturnal LH secretion predominates; in late puberty, daytime LH secretion predominates. LH secretion, spontaneous and in response to exogenous GnRH stimulation, is greater than FSH secretion throughout puberty in boys.23
Increased pituitary gonadotropin secretion results in growth and development of the gonads and a gradual increase in sex steroid production. The normal ranges of serum gonadotropins and sex steroids through the Tanner stages are presented for girls in Table 3 and for boys in Table 4. Before the pubertal rise in gonadotropins, an increase in adrenal sex steroids, such as dehydroepiandrosterone (DHEA) and androstenedione, has been documented.28 Evidence supports separate control mechanisms regulating adrenarche and gonadarche, although some investigators have speculated that the prepubertal rise in these adrenal steroids may play a role in the activation of the hypothalamic “gonadostat.”29 A disproportionate number of girls with premature adrenarche, resulting, for example, from late-onset congenital adrenal hyperplasia (CAH), experience an early onset of menarche.30
TABLE 3. Blood Hormone Concentrations During Female Puberty*
|
Tanner Stage |
FSH† (mIU/ml) |
LH† (mIU/ml) |
Estradiol (pg/ml) |
DHEA (ng/dL) |
Androstenedione (ng/dL) |
17-OH-Progesterone (ng/dL) |
Progesterone (ng/dL) |
|
1—Prepubertal |
2.16 ± 1.14 |
0.03 ± 0.03 |
1–20 |
31–345 |
8–50 |
3–82 |
<10–33 |
|
2 |
3.44 ± 1.58 |
0.71 ± 1.04 |
10–24 |
150–570 |
42–100 |
11–98 |
<10–55 |
|
3 |
4.88 ± 2.11 |
2.10 ± 2.33 |
7–60 |
200–600 |
80–190 |
11–155 |
10–450 |
|
4 |
6.19 ± 2.55 |
3.67 ± 2.22 |
21–85 |
200–780 |
77–225 |
18–230 |
10–1300 |
|
5—Adult: Follicular |
6.63 ± 2.19 |
5.76 ± 3.46 |
3–100 |
215–850 |
82–240 |
15–70 |
15–70 |
|
Luteal |
70–300 |
35–290 |
200–2500 |
To convert to International SI units, multiply:
FSH × 1 = IU/L LH × 1 = IU/L Estradiol × 3.67 = pmol/L DHEA × 34.67 = pmol/L Androstenedione × 34.92 = pmol/L 17-OH-progesterone × 30.26 = pmol/L Progesterone × 31.80 = pmol/L
* Taken from normal ranges reported by Endocrine Sciences, Tarzana, CA, 1990, except as noted below.
† Mean ± 1 SD taken from Neely EK, Hintz RL, Wilson DM et al: Normal ranges for immunochemiluminometric gonadotropin assays. J Pediatr 127:40, 1995
TABLE 4. Blood Hormone Concentrations During Male Puberty*
|
Tanner Stage |
FSH† (mIU/ml) |
LH† (mIU/ml) |
Testosterone (ng/dL) |
DHT (ng/dL) |
DHEA (ng/dL) |
Androstenedione (ng/dL) |
17-OH-Progesterone (ng/dL) |
|
1—Prepubertal |
0.97 ± 0.59 |
0.05 ± 0.05 |
<3–10 |
<3 |
31–345 |
8–50 |
3–90 |
|
2 |
2.75 ± 1.84 |
1.80 ± 1.30 |
18–150 |
3–17 |
110–495 |
31–65 |
5–115 |
|
3 |
2.94 ± 1.55 |
1.86 ± 1.41 |
100–320 |
8–33 |
170–585 |
50–100 |
10–138 |
|
4 |
4.47 ± 1.88 |
2.65 ± 1.81 |
200–620 |
22–52 |
160–640 |
48–140 |
24–180 |
|
5—Adult |
4.91 ± 2.02 |
4.51 ± 1.99 |
350–970 |
24–65 |
250–900 |
65–210 |
24–175 |
To convert to International SI units, multiply:
FSH × 1 = IU/L; LH × 1 = IU/L; Testosterone × 34.67 = pmol/L; Dihydrotestosterone × 34.44 = pmol/L; DHEA × 34.67 = pmol/L; Androstenedione × 34.92 = pmol/L; 17-OH-progesterone × 30.26 = pmol/L
* Taken from normal ranges reported by Endocrine Sciences, Tarzana, CA, 1990, except as noted below.
† Mean ± 1 SD taken from Neely EK, Hintz RL, Wilson DM et al: Normal ranges for immunochemiluminometric gonadotropin assays. J Pediatr 127:40, 1995
In mid to late puberty in girls, there is development of positive estrogen feedback on the hypothalamus.31 This feedback seems to be important in the maturation of the hypothalamic cyclic center in girls, and it is this positive estrogen feedback that stimulates the midcycle LH surge, which is important for ovulation.