The differential diagnoses of hirsutism can be divided into those causes
that are independent of excessive androgen action (i.e. nonandrogenic
causes), those that are related to excessive production or ingestion
of androgens (androgen causes), and idiopathic hirsutism (IH), which
is presumed to occur in response to the excessive peripheral utilization
of androgens. Nonandrogenic Causes ACROMEGALY. Between 0% and 50% of acromegalic women have been reported to present
with hirsutism.53 Accompanying the hirsutism is an enlargement of the hands and feet and
a coarsening of the facial features. This cause of hirsutism is extremely
rare, and the diagnosis is based on a determination of excessive
growth hormone secretion. The mechanisms underlying the association between
acromegaly and hirsutism, if any exist, remain to be confirmed. CHRONIC SKIN IRRITATION. Teleologically, hairs protect the skin, and chronic dermatologic irritation
or injury, particularly of the face, can stimulate hair growth.18 Excessive waxing or plucking,54 abuse of depilating agents, and chronic use of retinoic acid for the treatment
of age-associated wrinkles55 have all been reported to increase the length and diameter of facial hairs
and possibly convert to vellus to terminal hairs. DRUGS. Certain medications, such as phenytoin, cyclosporine, minoxidil, calcium
channel blockers, diazoxide, and erythropoietin, can cause a generalized
growth of body and facial hair.56 However, these drugs generally produce vellus hypertrichosis (i.e. overgrowth
of vellus hairs) and not hirsutism. Androgenic Causes ANDROGENIC DRUGS. On occasion the use or abuse of androgenic drugs such as danazol, methyltestosterone, stanazolol, and oxandrolone lead to the development of
acne and hirsutism. This problem is most frequently seen in women being
treated with long-term danazol for hereditary angioneurotic angioedema, young
women engaged in body building, and menopausal women who are
receiving androgens as part of the hormonal replacement. MALE PSEUDOHERMAPHRODITISM. The peripubertal onset of hirsutism, breast development, and clitoromegaly, accompanied
by primary amenorrhea in association with an absent
uterus, suggests the presence of either incomplete androgen insensitivity (i.e. partial
testicular feminization) or a Y-bearing dysgenetic gonad.57,58 If undiagnosed, progressive masculinization may result. On examination, these
persons demonstrate a blind vaginal pouch and varying degrees
of clitoromegaly and labial fusion. These patients may also demonstrate
bilateral or unilateral vulvar swelling secondary to the descent of
one or both gonads. In some patients with XY gonadal dysgenesis, a uterine
remnant is present, invariably accompanied by some degree of genital
asymmetry. Overall, this cause of hirsutism is extremely rare. NONCLASSIC ADRENAL HYPERPLASIA. Adrenal enzyme deficiencies that lead to the appearance of hyperandrogenic
symptoms at some time after birth have been called late-onset, postpubertal, attenuated, mild, or acquired, although the term nonclassic
adrenal hyperplasia (NCAH) is preferred. By definition, NCAH is an autosomal
recessive disorder that causes symptomatic hyperandrogenemia (e.g. hirsutism, acne, oligomenorrhea) peripubertally or postpubertally, in
the presence of normal female external genitalia. If a girl presents
with evidence of an adrenal enzyme deficiency and demonstrates virilization of the genitalia, the disorder should be classified
as congenital or classic adrenal hyperplasia (CAH), because the
adrenal hyperandrogenemia was present in utero during genital development. A defect in 21-hydroxylase (21-OH) accounts for more than 95% of all cases
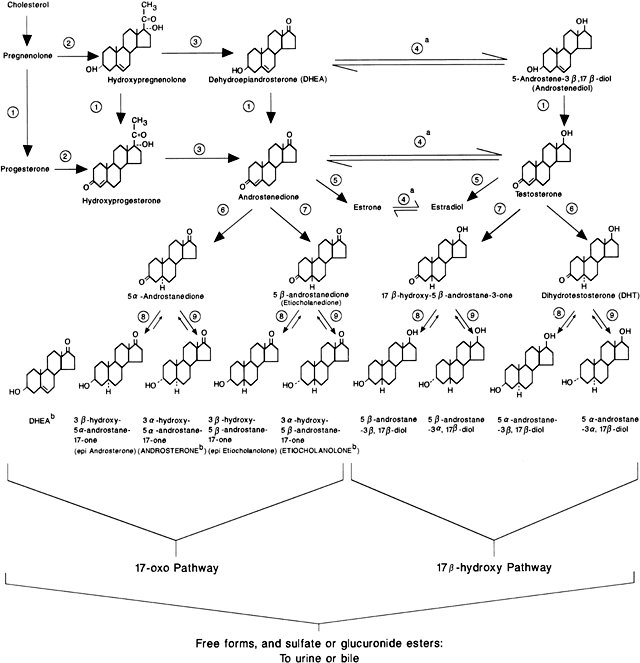
of NCAH (and CAH), although defects of 3β-hydroxysteroid dehydrogenase (3β-HSD) and 11β-hydroxylase (11-OH) have also been
reported to cause the disorder59 (Fig. 4). These patients have a genetic defect of one of these enzymes that decreases
its catalyzing efficiency. Consequently, the production of appropriate
amounts of the enzymatic product (e.g. 11-deoxycortisol in the
case of 21-OH deficiency) requires greater concentrations of precursor
owing to the changes in the enzymatic kinetic constant.60 The excess amounts of precursor (e.g. 17 α-hydroxyprogesterone [17-HP] in
the case of 21-OH deficiency) are then also converted
to greater amounts of androgens. Overall, and in contrast to CAH, patients
with NCAH do not generally demonstrate a decrease in cortisol production
nor an elevation in circulating adrenocorticotropic hormone (ACTH) levels.61 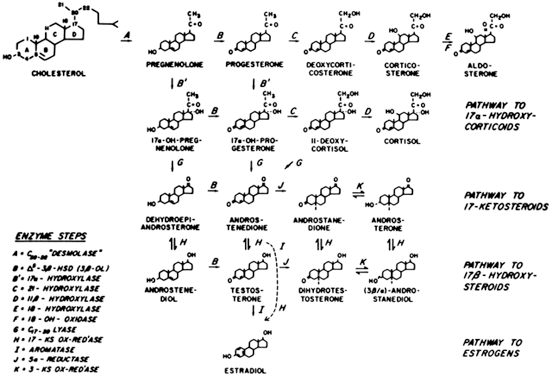 Fig. 4. Major pathways of steroid biosynthesis from cholesterol. The flow of hormonogenesis
is usually to the right and downward. C20–22 desmolase (enzyme step A) is also termed the cholesterol side-chain cleavage
system (i.e. P450scc ).(Hatch R, Rosenfield, RL, Kim MH et al: Hirsutism: implications, etiology, and
management. Am J Obstet Gynecol 140:815, 1981.) Fig. 4. Major pathways of steroid biosynthesis from cholesterol. The flow of hormonogenesis
is usually to the right and downward. C20–22 desmolase (enzyme step A) is also termed the cholesterol side-chain cleavage
system (i.e. P450scc ).(Hatch R, Rosenfield, RL, Kim MH et al: Hirsutism: implications, etiology, and
management. Am J Obstet Gynecol 140:815, 1981.)
|
21-Hydroxylase-Deficient NCAH. A deficiency in 21-OH activity constitutes the most common cause of NCAH, affecting
between 1% and 10% of hyperandrogenic women, depending on
ethnicity.59 Overall, only 1% to 2% of hirsute individuals of Anglo-Saxon descent are
affected, and 5% to 6% of hirsute patients of Latin descent (e.g. Spanish, Italian, French) have the disorder. The prevalence of NCAH appears
to be particularly high among Ashkenazi Jews and patients from the
Middle East.
In contrast to CAH, NCAH patients do not demonstrate virilization of
the external genitalia, although occasional patients may have mild clitoromegaly.
Hyperandrogenic symptoms most commonly appear peripubertally or postpubertally,
although some children may present with premature adrenarche. Women with
NCAH generally demonstrate relatively mild symptoms of hyperandrogenism,
if any at all.62 Biochemically it is difficult
to distinguish women with NCAH from other hyperandrogenic patients, and
circulating T and DHEAS levels are not different from those observed in
patients with PCOS.62,63,64
In fact, the level of DHEAS, an exclusive adrenal androgen, is often normal.
The measurement of a morning 17-HP level in the follicular phase of the
menstrual cycle can be used to screen for 21-OH deficiency in NCAH.63,65 The majority of women with untreated NCAH demonstrate basal 17-HP levels
higher than 2 ng/ml (Fig. 5). If used to screen for 21-OH deficient NCAH, the basal 17-HP should be
obtained in the follicular phase of the menstrual cycle, because this
progestogen increases in the luteal phase.65 Furthermore, dexamethasone should not be administrated before sampling, because
it will suppress the circulating 17-HP level. 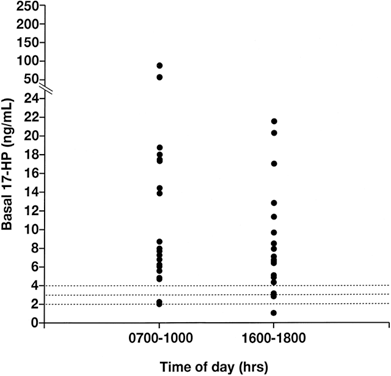 Fig. 5. The basal 17-hydroxyprogesterone (17-HP) level was determined in 20 patients
with 21-hydroxylase-deficient nonclassic adrenal hyperplasia, in
the morning (7 to 10 a.m.) and in the afternoon (4 to 6 p.m.). All 20 samples
obtained in the morning were found to exceed 2 ng/ml, and 18 exceeded
both 3 ng/ml and 4 ng/ml. Of samples obtained in the afternoon, 19, 18, and 17 exceeded 2, 3, and 4 ng/ml, respectively.(Azziz R, Hincapie LC, Knochenhauer ES et al: Screening for 21-hydroxylase
deficient nonclassic adrenal hyperplasia: A prospective study. Fertil
Steril 72:915, 1999.) Fig. 5. The basal 17-hydroxyprogesterone (17-HP) level was determined in 20 patients
with 21-hydroxylase-deficient nonclassic adrenal hyperplasia, in
the morning (7 to 10 a.m.) and in the afternoon (4 to 6 p.m.). All 20 samples
obtained in the morning were found to exceed 2 ng/ml, and 18 exceeded
both 3 ng/ml and 4 ng/ml. Of samples obtained in the afternoon, 19, 18, and 17 exceeded 2, 3, and 4 ng/ml, respectively.(Azziz R, Hincapie LC, Knochenhauer ES et al: Screening for 21-hydroxylase
deficient nonclassic adrenal hyperplasia: A prospective study. Fertil
Steril 72:915, 1999.)
|
Hyperandrogenic women who demonstrate a basal follicular phase 17-HP
level higher than 2 ng/ml merit an ACTH stimulation test to rule out NCAH
(see later discussion). If the 17-HP level 30 or 60 minutes after intravenous
administration of one vial of ACTH-(1-24) (Cortrosyn) is greater than
10 ng/ml, the diagnosis of 21-OH-deficient NCAH is established, although
most patients have ACTH-stimulated 17-HP values in excess of 15 ng/ml
(Fig. 6).62,63,64,65,66,67
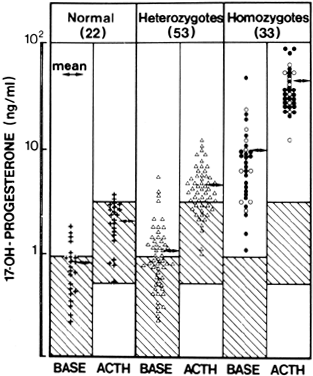 Fig. 6. Plasma concentrations of 17-hydroxyprogesterone before and after acute
stimulation with adrenocorticotropic hormone (ACTH) in patients with late-onset 21-hydroxylase
deficiency and members of their families. Closed
circle, proband; open circle, homozygotes; open triangle, heterozygotes; plus
sign, normals.(Kuttenn F, Couillin P, Girard F et al: Late-onset adrenal hyperplasia
and hirsutism. N Engl J Med 313:2224, 1985.) Fig. 6. Plasma concentrations of 17-hydroxyprogesterone before and after acute
stimulation with adrenocorticotropic hormone (ACTH) in patients with late-onset 21-hydroxylase
deficiency and members of their families. Closed
circle, proband; open circle, homozygotes; open triangle, heterozygotes; plus
sign, normals.(Kuttenn F, Couillin P, Girard F et al: Late-onset adrenal hyperplasia
and hirsutism. N Engl J Med 313:2224, 1985.)
|
The genetics of 21-OH-deficient NCAH have been extensively characterized. The 21-OH
gene (i.e. CYP21) is located on the short arm of chromosome 6, in
the midst of the human leukocyte antigen region. Molecular genetic
studies have been able to establish the majority of the mutations
resulting in 21-OH-deficient NCAH.68 In close proximity to CYP21 lies a pseudogene (CYP21P) that is not active
but is very similar in DNA sequence. Its proximity probably accounts
for the high frequency of inherited abnormalities of the 21-OH gene, due
to gene conversion or deletion. Patients with NCAH may carry mutations
in each of the homologous CYP21 alleles, which encodes for a mild
defect (but usually they are different). Alternatively, approximately
one half to two thirds of patients with NCAH are “compound heterozygotes,” carrying a mild mutation of one CYP21 and a more severe
mutation (which would result in CAH if present in homozygous form) on
the homologous allele. 11 β -Hydroxylase-Deficient NCAH. Patients with 11-OH deficient NCAH, like those with the 21-OH variant, have
been reported to be clinically indistinguishable from other hyperandrogenic
women.69 The blood pressure in patients with 11-OH-deficient NCAH ranges from normal
to moderately elevated. The basal hormonal profile is also not distinctive. However, the
prevalence of 11-OH-deficient NCAH and its diagnostic
criteria remain unclear. Using a level of 11-deoxycortisol greater
than three times the upper limit of controls as a criterion, we69 and others70 have suggested that the prevalence of presumed 11-OH-deficient NCAH among
hirsute patients is 1% or less. Nonetheless, the diagnosis of 11-OH-deficient
NCAH has yet to be confirmed by molecular analysis of the
responsible gene, CYP11a.71 3 β -Hydroxysteroid Dehydrogenase-Deficient NCAH.
The diagnosis of 3β-HSD-deficient NCAH has been assumed to be
based on abnormally high levels of 17-hydroxypregnenolone (17-HPREG) or
DHA, or both, after ACTH stimulation.72
However, molecular defects of the 3β-HSD-II gene (the enzyme form
predominant in the adrenal cortex) have yet to be demonstrated in adult
women with suspected 3β-HSD-deficient NCAH.73,74,75,76,77,78
The few cases of molecularly confirmed 3β-HSD deficiency that have
been diagnosed in children73,74,75
suggest that the level of 17-HPREG diagnostic of NCAH is more than 50
times the upper normal value.75
Furthermore, even with the much less strict diagnostic criterion of an
ACTH-stimulated 17-HPREG level three times greater than control values (similar
to the difference in 17-HP levels found in 21-OH-deficient NCAH), we
were unable to diagnose any affected patients among 86 consecutive
hyperandrogenic women79 or among 30 women with increased DHEAS levels.80 Therefore, it appears that 3β-HSD deficient NCAH, if it exists, is, like
its 11-OH-deficient counterpart, extremely rare. THE HYPERANDROGENIC-INSULIN RESISTANT-ACANTHOSIS NIGRICANS (HAIRAN) SYNDROME.
Between 1% to 5% of hyperandrogenic women have this disorder. These
patients demonstrate extreme insulin resistance with secondary hyperinsulinemia
and hyperandrogenism. Although exact diagnostic guidelines have yet to
elucidated, it appears that the disorder can be diagnosed by the presence
of extremely high circulating levels of insulin, usually more than 80
μU/ml in the fasting state and/or more than 300 μU/ml after
a 2- or 3-hour oral glucose tolerance test.81,82,83,84
In the early stages of the disorder, particularly in children or adolescents,
glucose levels are relatively normal. Nonetheless, over time many of these
patients experience progressive islet cell failure with the development
of type 2 diabetes.
The high levels of insulin are generally caused by a genetic defect in
post-insulin receptor action.84 Rarely,
some of these women demonstrate abnormal insulin receptors85,86,87
or circulating anti-insulin receptor antibodies.88,89
The elevated circulating insulin concentrations act synergistically with
luteinizing hormone (LH) to augment ovarian androgen (but not adrenal)
production. This syndrome should not be confused with PCOS (discussed
later), a disorder that is also associated with insulin resistance, although
of a much lesser degree than that of the HAIRAN syndrome.
Patients with HAIRAN syndrome often demonstrate ovarian hyperthecosis, a
pathologic finding characterized by islands of hyperplastic luteinized
theca cells located throughout the stroma and the presence of relatively
few and small atretic follicles.90 In fact, the finding of hyperthecosis usually suggests the presence of
the HAIRAN syndrome,91 although a few patients with PCOS may also demonstrate this pathologic
finding. In HAIRAN patients with hyperthecosis, circulating levels of
LH and follicle-stimulating hormone (FSH) may be normal to low (4 to 8 mIU/ml) due
to the extremely high circulating levels of T. Consequent to the presence of ovarian hyperthecosis, many patients with
HAIRAN are severely hyperandrogenic, and even present with a moderate
degree of virilization. Patients with HAIRAN syndrome also exhibit acanthosis
nigricans, a velvety, hyperpigmented change of the crease areas
of the skin (Fig. 7). Some of these patients may also demonstrate forms of lipodystrophy. Many
also have, or will have, glucose intolerance or type 2 diabetes mellitus (DM), hypertension, and dyslipidemia, particularly suppressed
high-density lipoproteins and hypertrigliceridemia. Overall, both morbidity
and mortality in these patients are quite significant, and these
women require intensive counseling, follow-up, and treatment of both
hyperandrogenic and metabolic abnormalities. 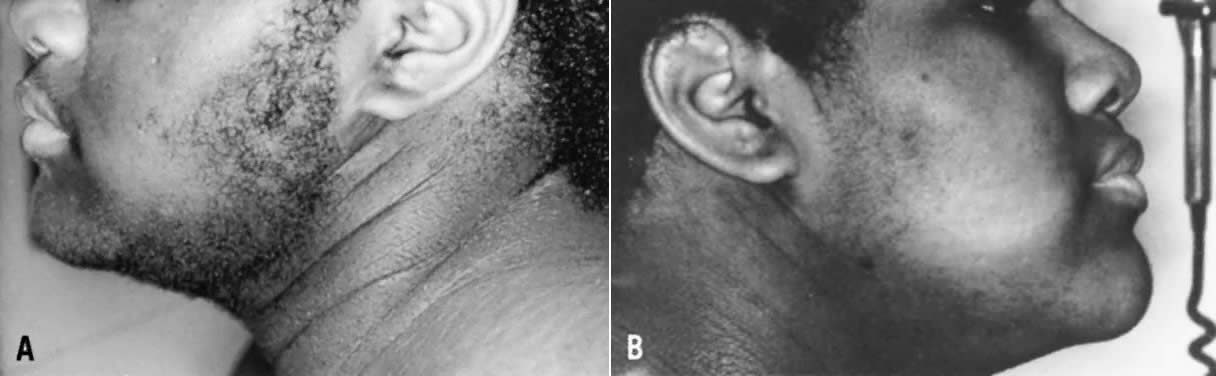 Fig. 7. Thirteen-year-old patient with HAIRAN syndrome before (left) and after 6 months of treatment with leuprolide (right). Note acanthosis nigricans, and marked improvement in hirsutism after
treatment with the long-acting growth hormone-releasing hormone (GnRH) agonist.(Azziz R: Hirsutism in the pediatric or adolescent patient. In Carpenter
SE, Rock JA, [eds]: Pediatric and Adolescent Gynecology, p 239. New
York: Raven Press, 1992.) Fig. 7. Thirteen-year-old patient with HAIRAN syndrome before (left) and after 6 months of treatment with leuprolide (right). Note acanthosis nigricans, and marked improvement in hirsutism after
treatment with the long-acting growth hormone-releasing hormone (GnRH) agonist.(Azziz R: Hirsutism in the pediatric or adolescent patient. In Carpenter
SE, Rock JA, [eds]: Pediatric and Adolescent Gynecology, p 239. New
York: Raven Press, 1992.)
|
When undergoing treatment for hyperandrogenism, most patients with HAIRAN
syndrome respond to standard therapy with antiandrogens and oral contraceptives, although
some require more aggressive treatment.92 Ketoconazole93 or longacting gonadotropin-releasing hormone (GnRH) analogues (GnRH-a)92,94 have been used in nonresponsive cases, with good results (Fig. 7). Long-acting GnRH-a inhibits LH-dependent androgen production by reducing
circulating gonadotropins to castrate levels. At least 3 to 6 months
of gonadotropin suppression is required to achieve full androgen suppression
in HAIRAN syndrome, and treatment can be continued for years
when combined with add-back estrogen/progestin replacement. If other
treatments fail, a bilateral ovarian wedge resection or even oophorectomy
may be considered. CUSHING'S DISEASE.
Hirsutism is present in 60% to 70% of women with Cushing's syndrome.
It is generally diffuse, mild, and accompanied by varying degrees of vellus
hypertrichosis. Hirsutism, with or without menstrual irregularity, was
among the presenting symptoms in 15 of 45 such women, and was the only
presenting complaint in 11.95 Overall menstrual
irregularities are seen in 80% to 100% of women with Cushing's syndrome,
and acne is present in 40% to 50%.96,97,98
Excessive ACTH-dependent adrenocortical function, owing to either an ectopic
ACTH-producing tumor or a pituitary tumor (Cushing's disease), can
lead to excessive adrenal androgen secretion and hyperandrogenemia. Imura
and colleagues did not note virilization in women with ectopic
ACTH-producing neoplasms.99 However, all of their patients were Asian, a population that is notably
resistant to the development of hirsutism. In contrast, elevated circulating
T levels have been documented in women with pituitary Cushing's
syndrome.100 An adrenal origin was ascribed to the increased androgen level, because
dexamethasone caused a parallel decrease in cortisol and T, ACTH stimulation
induced an increase in T that was more profound in women with
Cushing's disease compared with normal subjects, and bilateral adrenalectomy
dramatically decreased circulating T, correcting the oligomenorrhea
and hirsutism. An increase in the adrenal secretion of androgens may not be the only factor
accounting for the hyperandrogenic symptoms in Cushing's disease. Patients
with adrenal adenoma generally demonstrate low levels
of adrenal androgens, particularly DHEAS.101 Nevertheless, these women demonstrate frequencies of menstrual irregularity, hirsutism, and
acne similar to those of patients with generalized
adrenal hyperplasia (reflecting increased ACTH stimulation).97 Therefore, it is possible that an increase in ovarian androgen secretion
may, in part, account for the hyperandrogenism of these patients. The
role played by the increased insulin resistance and gonadotropin abnormalities
often noted in patients with Cushing's syndrome in promoting
ovarian hyperandrogenemia is unknown. Finally, a direct effect
of long-term hypercortisolemia on hair growth, particularly on vellus
hairs, cannot be ruled out. Despite the high frequency of hirsutism reported among women with Cushing's
syndrome, hirsute women are rarely diagnosed as cushingoid. As
illustration, among approximately 1000 hyperandrogenic women with whose
care the author has been involved, only 1 patient has been diagnosed
as having Cushing's disease. ANDROGENIC TUMORS. Androgen-producing tumors, either ovarian or adrenal, are relatively rare. They
should be suspected when the onset of androgenic symptoms is
rapid and sudden, and when virilization and masculinization ensue. Steroid-secreting
neoplasms are occasionally associated with other systemic
symptoms, including weight loss, anorexia, abdominal bloating, and
back pain. Suppression and stimulation tests (including corticosteroid, oral
contraceptive, human chorionic gonadotropin, and ACTH administration) can
be misleading and are not encouraged for the screening or
diagnosis of these neoplasms.102 Furthermore, because computed tomographic (CT) scanning for the diagnosis
of an adrenal neoplasm, or transvaginal sonography for an ovarian
tumor, are relatively sensitive and noninvasive, there is little need
today for adrenal or ovarian vein catheterization in the diagnosis of
androgen-secreting neoplasms.
Ovarian androgen-secreting neoplasms occur in approximately 1 of 300
to 1 of 1,000 hyperandrogenic patients.103,104,105,106
They are usually palpable on pelvic examination or associated with a unilateral
ovarian enlargement seen on ultrasound or CT. Functional ovarian neoplasms
generally are not malignant, and they include Sertoli-Leydig cell tumors
and lipoid cell tumors. Ovarian tumors should be suspected when hirsutism
or virilization is of sudden onset and rapidly progressive. The diagnosis
can be further suspected when transvaginal sonography demonstrates an
asymmetric ovarian enlargement.
Androgen-producing tumors of the adrenal are less common than ovarian neoplasms
and include adenomas and carcinomas. Adrenal carcinomas usually
are associated with the development of cushingoid features and can
be diagnosed as a large (greater than 6 cm) irregular adrenal mass on
CT scanning. The prognosis for patients with adrenocortical carcinoma
is poor. The frequency with which these patients present with oligoovulation
and hirsutism depends to a significant degree on tumor histopathology. More
differentiated tumors tend to produce higher level of androgens, whereas
less differentiated tumors produce only minimal amounts
of androgens.107 Overall, adrenocortical neoplasias demonstrate less efficient steroidogenesis
than normal adrenocortical tissue. However, because these tumors
are frequently very large, their overall steroid secretion can be significant. Because adrenal adenomas secreting solely androgens are exceedingly rare, most
patients with an androgen-secreting adrenal neoplasm present with
cushingoid features. Furthermore, physicians should keep in mind that
signs and symptoms of virilization are encountered with equal frequency
in patients with androgen-secreting adrenal adenomas and those with
adrenal carcinomas.108 POLYCYSTIC OVARY SYNDROME. PCOS affects approximately 4% of reproductive-aged women in the United
States.4 The disorder is one of the most common causes of oligo-ovulatory infertility109 and hirsutism,110 and it is associated with increased risks of cardiovascular disease (CVD), type 2 DM, endometrial carcinoma, and psychosocial dysfunction. Patients with PCOS comprise between 60% and 80% of all hyperandrogenic
women seen. Although there is no widespread agreement on the definition
of PCOS, useful criteria arose from a conference sponsored by the National
Institute of Child Health and Human Development in April of 1990.111 Most conference participants indicated that the major criteria for PCOS “should
include (in order of importance): (i) hyperandrogenism
and/or hyperandrogenemia, (ii) oligo-ovulation, [and] (iii) exclusion
of other known disorders, such as Cushing's syndrome, hyperprolactinemia, or
congenital [nonclassic] adrenal hyperplasia.” Hence, the
diagnosis of PCOS is established by the presence
of hyperandrogenic oligo-ovulation, after exclusion of other causes
of androgen excess or oligo-ovulation, such as NCAH, hyperprolactinemia, and
thyroid dysfunction. The disorder may be evident as early as
the age of 12 years.112
Pathologically, the ovarian cortex contains multiple intermediate and
atretic follicles measuring 2 to 5 mm in diameter, which give the ovary
its “polycystic” appearance. However, not all patients with
PCOS have “polycystic” ovaries.113,114
In fact, the sole appearance of “polycystic” ovaries, either
on laparoscopy or more commonly at sonography, is not diagnostic for PCOS.113,114,115,116
Excessive levels of circulating androgens from any source will
lead to the disruption of follicular development and the accumulation
of many small, atretic follicles in the ovarian cortex, producing a “polycystic”
picture51,52
(Fig. 8). Phenotypically,
40% to 60% of PCOS patients are obese, predominantly demonstrating abdominal/visceral
fat deposition. Although most, if not all, patients with PCOS have ovulatory
dysfunction, up to 30% claim to have regular menses.110
In addition, PCOS may manifest as primary amenorrhea.117,118
Although a majority of patients with PCOS demonstrate hirsutism, this
clinical finding is by no means uniform.22,119,120,121,122
In one study, 19% of 32 consecutive oligo-ovulatory patients without hirsutism
demonstrated increased levels of at least one androgen, suggesting that
one fifth of these nonhirsute oligo-ovulatory patients have PCOS.122
Hyperandrogenemia was primarily observed among patients with menstrual
cycles greater than 45 days in length, of which 38% had elevated androgens.
Hirsutism is also relatively infrequent among Asian women with PCOS.123
Patients with PCOS usually do not present with virilization.
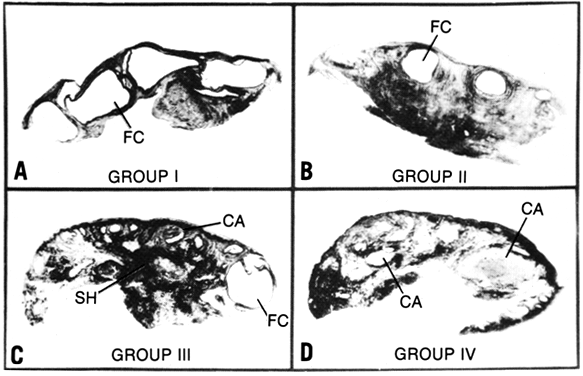 Fig. 8. The spectrum of histologic findings in polycystic ovary syndrome. f.c., follicular
cysts; s.h., stromal hyperplasia; c.a., corpora albicantia.(Givens JR: Polycystic ovaries: a sign, not a diagnosis. Semin Reprod Endocrinol 2:271, 1984) Fig. 8. The spectrum of histologic findings in polycystic ovary syndrome. f.c., follicular
cysts; s.h., stromal hyperplasia; c.a., corpora albicantia.(Givens JR: Polycystic ovaries: a sign, not a diagnosis. Semin Reprod Endocrinol 2:271, 1984)
|
Biochemically, patients with PCOS frequently demonstrate increased circulating
levels of free T and a reduction in SHBG, accompanied by variable increases
in total T.11,124,125
Approximately 30% demonstrate an abnormally high DHEAS level, although
rarely higher than 6000 ng/ml.126,127
The prolactin concentration is usually normal, although as many as 17%
to 23% of such patients demonstrate mild intermittent elevations (usually
less than 80 ng/ml, depending on the assay) without any other apparent
cause.128 An increased LH/FSH ratio, exceeding
a 2:1, is observed in approximately 60% of these patients.129,130,131
Most patients with PCOS also demonstrate a variety of metabolic abnormalities.
Most significant among these is the presence of insulin resistance, accompanied
by compensatory hyperinsulinemia, which affects approximately 30% to 80%
of these patients.123,132
The “resistance” to the action of insulin in PCOS refers
to the impaired action of this hormone on glucose transport and antilipolysis
in adipocytes, in the presence of normal insulin binding.133,134,135
The insulin resistance of PCOS appears to be associated with postreceptor
signaling aberrations, although these remain to be confirmed. It should
be noted that the insulin resistance of PCOS differs from that observed
in type 2 DM and simple obesity. However, although the unique insulin
resistance of PCOS occurs even in normal-weight individuals, it is further
aggravated by the concomitant presence of obesity.
This resistance to the cellular actions of insulin leads to a compensatory
hyperinsulinemia in PCOS, with the excess insulin causing an exaggerated
effect in other, less traditionally responsive, tissues. These
effects include androgen secretion by the ovarian theca, excess growth
of the basal cells of the skin resulting in varying degrees of acanthosis
nigricans, increased vascular and endothelial reactivity, and abnormal
hepatic and peripheral lipid metabolism. The increased insulin levels found in patients with PCOS appear to directly
enhance LH-stimulated androgen secretion from the ovary.136,137 The persistence of hyperinsulinemia in women who have been both surgically
and medically castrated refutes hyperandrogenism as the cause of
hyperinsulinemia.138,139 More obviously, normal men have free androgen levels some 30-fold higher
than those in women, yet they do not exhibit a greater prevalence of
insulin resistance or hyperinsulinemia. Increased levels of insulin
also serve to decrease circulating SHBG levels, thus resulting in higher
concentrations of free androgens.140
The polycystic ovary syndrome has a strong familial component, appearing
to demonstrate a multifactorial form of inheritance with a single dominant
gene effect.141,142
For example, if a patient has PCOS, her sister and her daughters have
an approximately 50% risk of also having the disorder.143
Therefore, patients should be counseled appropriately. Multiple attempts
have been made to detect a molecular genetic abnormality. Although some
promising associations have been reported,144,145,146
no single defect has yet been found to account for the disorder in even
a subgroup of patients. Finally, patients with PCOS should be counseled
regarding their increased risks of developing gestational diabetes, type
2 diabetes, hypertension, endometrial cancer, and, possibly, CVD.
Idiopathic Hirsutism Although IH is often referred to as “familial hirsutism,” this
represents a gross misnomer. It is now well established that other
forms of hyperandrogenism (e.g. PCOS) also demonstrate a strong familial
predisposition. Therefore, it is misleading to regard IH as being
solely familial, because this often translates to “untreatable” in
the mind of the patient and the clinician. The diagnosis of IH
is reached when an obviously hirsute patient demonstrates normal ovulatory
function and, frequently, normal circulating androgen levels. Approximately 15% to 25% of
hirsute women are diagnosed as having IH, depending
on the extent of endocrine evaluation.110,147 It is critical, however, to confirm the presence of normal ovulatory function (e.g. by
using a basal body temperature chart and luteal progesterone
measurements) in those hirsute patients who claim to have regular
menstrual cycles. Up to 40% of these women actually are oligo-ovulatory.110
Some women with IH demonstrate excessive 5α-reductase activity of
the hair follicle, which results in hirsutism despite “normal”
circulating androgen levels.148 The measurement
of serum 3α-androstanediol glucuronide, a metabolite of DHT, may
reflect peripheral 5α-reductase activity and help to confirm “hyperfunction”
of this enzyme.148,149,150
Nevertheless, the diagnosis of IH is usually one of exclusion, made when
repeated measurements of free and total circulating androgen levels are
found to be normal in a patient with hirsutism and regular ovulatory function.
|