Evaluation of Cortisol Secretion The first reliable method for measuring glucocorticoid levels is known
as the Porter-Silber chromogen test. This fluorometric test measures cortisol
and cortisone levels in the plasma, or their urinary metabolites, 17-OH
corticosteroids (17-OHCS), by the intensity of a color change
that occurs when the urine or plasma reacts with phenylhydrazine. Since
the development of radioimmunoassays and competitive binding assays
in the 1950s and 1960s, more accurate tests have been developed. The
amount of hormone is quantified according to the degree that it displaces
the tracer steroid from the antibody or binding protein. Radioimmunoassays
have been developed for most of the major steroids derived from
cholesterol. Basal abnormalities of hormone levels may indicate adrenal
abnormalities. However, single determinations are affected by pulsatile
secretion and circadian rhythm. Circulating glucocorticoid levels have a diurnal rhythm. Therefore, a 24-hour
urine collection is required for a reliable measurement of 17-OHCS
level. Normal values range from 2 to 6 μg/24 hours in women and 3 to 10 μg
in men. The amount of 17-OHCS excreted is directly related
to body mass. Obese subjects may exceed the normal values despite
having normal adrenal function. Expressing urinary 17-OHCS as milliliters
per gram of urinary creatinine level will correct for obesity. The
normal value is 2 to 6.5 mg/g creatinine. The 17-ketogenic steroids (17-KGS) rarely are used as a measure of cortisol
secretion. 17-KGS constitute a greater percentage of cortisol metabolites
than do 17-OHCS, but technical problems and the lack of specificity
of 17-KGS make this measure less useful. At 8 a.m., the normal cortisol level ranges from 7 to 22 ng/dl when measured
by radioimmunoassay. This value is twice the level that is expected
when the test is performed on plasma obtained at 8 p.m. In addition, cortisol
is secreted episodically. Therefore, single estimations of
plasma cortisol level without prior suppression are considered unreliable. The 24-hour determination of urinary free cortisol (UFC) level is a reliable
test of the amount of cortisol excreted unchanged during a 24-hour
period. It reflects not only daily production rates but also the portion
that is free in the circulation. UFC can be determined by either
radioimmunoassay or competitive protein-binding radioimmunoassay. It
is particularly useful in distinguishing patients with obesity from those
with Cushing's syndrome.77,78 The determination of UFC is not useful in the diagnosis of adrenal hypofunction. Normal
values are in the range of 17 to 70 μg/24 hours. Many extra-adrenal factors can affect cortisol measurement and lead to
a false diagnosis of hyperfunction or hypofunction. Malnutrition and advanced
age are associated with decreased cortisol production, whereas
stress and obesity are associated with increased cortisol production. In pregnancy, estrogen induces an increase in CBG concentration. This increase
then causes an increase in plasma cortisol levels to maintain
the same level of functional (free) cortisol. Therefore, levels of plasma
cortisol are increased, but levels of urinary metabolites are normal. Administration
of estrogens, especially high-dose oral contraceptives, will
result in similar alterations in cortisol values. Other drugs
can affect cortisol measurements as well. Any drug that induces hepatic
enzymes, such as phenobarbital, may modify the metabolism of cortisol
and lead to low urinary 17-OHCS levels. Also, liver and renal disease
cause abnormalities in the excretion of cortisol. Evaluation of Adrenal Androgens The metabolites of adrenal androgens traditionally have been measured as
urinary 17-KS. In normal women, important precursors for 17-KS are DHEAS, DHEA, and
androstenedione; compounds with C-11 hydroxyl or ketone
groups, such as cortisol or cortisone, also contribute. Drugs that cause
fluorescent metabolites will yield falsely elevated values in a colorimetric
assay. In disorders of excess adrenal androgen production, 17-KS
excretion exceeds the normal range. However, radioimmunoassay of specific androgens generally is available, often
making 17-KS determinations unnecessary. DHEAS is exclusively an
adrenal product, and its plasma level is a more specific measure of
adrenal androgen production than is urinary 17-KS excretion. The total
production rate of DHEAS has been estimated to be greater than 10 mg/day. In
this laboratory, serum concentrations of DHEAS throughout the
menstrual cycle were 82 to 338 μg/dl. The metabolic clearance rate
of DHEAS is low, accounting in part for the long half-life of this steroid. The
combination of significant production rate, low metabolic clearance, and
long half-life results in substantial serum concentrations
of DHEAS that are subject to limited fluctuation. These features of
DHEAS and its exclusive derivation from the adrenal gland make serum DHEAS
level a valuable marker of long-term adrenal androgen activity and
raise the validity of a single serum determination of DHEAS concentration.79,80,81,82,83,84,85,86,87 Unlike testosterone and androstenedione, DHEAS cannot bind to serum sex-hormone-binding
globulin, and most of the binding is to the high-capacity, low-affinity
albumin bed.88,89 In some cases of Cushing's syndrome, chronic hyperprolactinemia, and
congenital adrenal hyperplasia (CAH), DHEAS level is elevated. Elevation
of DHEAS level to greater than 700 ng/dl, with or without elevation
of testosterone level, is strongly suggestive of an androgen-producing
adrenal tumor. Although testosterone-producing adrenal adenomas associated
with normal serum DHEAS level have been described, they are
rare and may respond to gonadotropic stimulation. A normal serum DHEAS
level essentially excludes the adrenal gland as the source of excessive
androgen production. Despite the apparent specificity of DHEAS as an adrenal marker, serum DHEAS
levels may be moderately elevated in patients who experience chronic
anovulation due to reproductive axis dysfunction.90,91,92,93 Serum DHEAS levels usually do not exceed 700 μg/dl in these patients. The
mechanisms underlying the anovulation-associated increase in serum
DHEAS levels remain uncertain. Elevation of serum DHEAS levels requires additional workup. A prolactin
level should be obtained because elevations in prolactin levels stimulate
adrenal androgen production. If the prolactin level is normal, a
short-term ACTH stimulation test should be performed to exclude an attenuated
form of CAH. Once attenuated CAH and hyperprolactinemia are excluded, and
the serum DHEAS level is less than 700 μg/μl, the options
are screening for the rare possibility of Cushing's syndrome
or empirically suppressing the reproductive axis (with gonadotropin-releasing
hormone agonists), given the likelihood that the elevated circulating
levels of DHEAS represent a functional chronic anovulatory disorder. If
the elevated DHEAS level is secondary to reproductive axis
dysfunction, suppression of the reproductive axis over a period of 3 to 6 months
should result in a reduction of serum DHEAS levels.94,95,96 However, if this decrease does not occur the patient may have an early, slowly
progressing adrenal androgen-producing tumor (e.g., adenoma). Evaluation of Hypothalamic-Pituitary-Adrenal Function In general, evaluation of adrenal function requires more provocative testing. Diagnosis
of hypercortisolism (Cushing's syndrome) is made
by the finding of failure to suppress cortisol production by dexamethasone
suppression test. The etiology of Cushing's syndrome is determined
by evaluation of baseline circulating ACTH levels, the metyrapone
test, and the CRH stimulation test. Diagnosis of an adrenal cause
of hyperandrogenism can be made by an ACTH stimulation test. An ACTH stimulation
test is required for the diagnosis of adrenal insufficiency. PLASMA ADRENOCORTICOTROPIC HORMONE RADIOIMMUNOASSAYS. Plasma levels of endogenous ACTH can be measured by radioimmunoassay. ACTH
is secreted in a pulsatile fashion, and its circadian rhythm is responsible
for the circadian rhythm of cortisol secretion. Clinically, ACTH
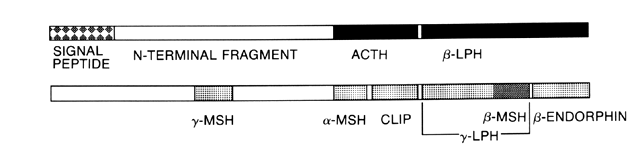
radioimmunoassays may measure not only the native 39-amino-acid
ACTH molecule, but also structurally related POMC products.97 In normal, unstressed subjects, the level of ACTH late in the evening
generally is less than 20 pg/ml, and morning values are in the range of 10 to 80 pg/ml.98 The plasma ACTH level can be helpful in determining the etiology of adrenocortical
insufficiency99 and of Cushing's syndrome. ADRENOCORTICOTROPIC HORMONE STIMULATION TESTS. The response of the adrenal gland to exogenous ACTH may be helpful from
a diagnostic standpoint. In general, the synthetic ACTH analog cosyntropin, which
consists of the 24 N-terminal amino acids of the native
peptide, is used for testing purposes. The degree of response depends
not only on the physiologic integrity of the gland but also on the degree
of prior stimulation. This test is used to diagnose adrenocortical
insufficiency by measuring cortisol levels or to diagnose congenital
adrenal hyperplasia by measuring androgen precursor levels. For the rapid ACTH test, one or two blood samples are collected to determine
basal levels of plasma cortisol, followed by intravenous injection
of 250 μg cosyntropin. Plasma may be sampled 30, 45, or 60 minutes
after ACTH administration.100 With a normal response to stimulation, plasma cortisol level will exceed 15 μg/dl
and will exhibit an incremental rise of 5 to 7 μg/dl
or more. A normal response excludes primary adrenocortical insufficiency. However, it
does not necessarily exclude partial secondary adrenocortical
insufficiency; some patients with mild or early ACTH deficiency
may have sufficient basal ACTH production to prevent adrenocortical
atrophy, yet lack the ability to activate this axis appropriately when
stressed. Prolonged ACTH stimulation also may distinguish primary from secondary
adrenocortical insufficiency. One protocol for prolonged ACTH stimulation
is collection of 24-hour urine specimens for 1 day before ACTH stimulation
and the 2 days during ACTH infusion to determine the values of 17-OHCS
and creatinine.101 A continuous intravenous infusion of 1600 μg cosyntropin, which is
equivalent to the 160 units of ACTH originally used, is administered for 48 hours. It
is not necessary to restrict diet or activity during the
infusion. Normal subjects excrete more than 27 mg/24 hours 17-OHCS
on the first day of the infusion, and more than 47 mg/24 hours on the
second day. Patients with secondary adrenocortical insufficiency generally
excrete more than 4 mg/24 hours on the first day of infusion, and
more than 10 mg/24 hours on the second day. Patients with primary adrenocortical
insufficiency usually excrete less than 3 mg/24 hours on the
first day and less than 4 mg/24 hours on the second. In patients with a high risk of primary adrenal insufficiency, glucocorticoid
therapy may be initiated at the time of a rapid ACTH stimulation
test or during a prolonged ACTH stimulation protocol. Dexamethasone
is the glucocorticoid of choice; it is 25 times more potent than cortisol, and
the amount required for treatment does not interfere with cortisol
or 17-OHCS determination. Although dexamethasone suppresses the
pituitary secretion of endogenous ACTH, it does not interfere with the
response of the adrenal to exogenous ACTH, and therefore will not alter
the results of the test. The ACTH stimulation tests for the diagnosis of congenital adrenal hyperplasia
involve the measurement of steroid precursors proximal to the
enzymatic block. Various protocols have been used. Some require overnight
dexamethasone suppression before testing, although most clinicians
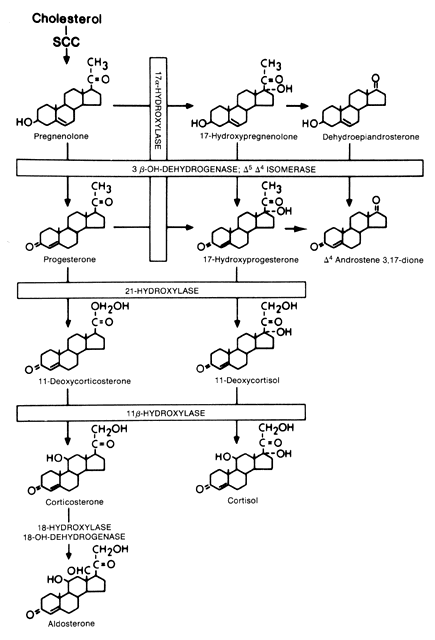
do not consider this step necessary. Levels of progesterone and 17-OH
progesterone are elevated with CAH21, the level of circulating 18-hydroxycorticosterone is elevated with CAH11, and the 17-hydroxypregnenolone:17-hydroxyprogesterone ratio is elevated
with CAH3β-HSD. DEXAMETHASONE SUPPRESSION TEST. Dexamethasone, a potent synthetic glucocorticoid, is used in suppression
tests because the small amount required to suppress ACTH does not interfere
with the assays of steroids or steroid metabolites. A small single
dose, 1 mg, is used in the overnight suppression tests to screen
for Cushing's syndrome. Liddle102 developed a higher dose dexamethasone test to confirm Cushing's syndrome
and determine its etiology. These tests and their use in the diagnosis
of Cushing's syndrome are discussed in detail in another
chapter. METYRAPONE TEST. Metyrapone decreases the adrenal secretion of cortisol, primarily by inhibiting
the enzymatic activity of 11β-hydroxylase. In the normal
person, the decline in circulating cortisol level activates the hypothalamic-pituitary
unit to increase ACTH release. ACTH stimulates adrenal
steroidogenesis, but because of the inhibition of 11β-hydroxylase, an
excess of 11-deoxycorticosteroids, primarily 11-deoxycortisol (11-DOC), is
secreted. The degree of metyrapone inhibition of cortisol
synthesis is assessed by measuring serum cortisol level. The degree
of resultant ACTH stimulation may be assessed by measuring the plasma
level of ACTH directly, the level of 11-DOC in plasma or urine, or the
urinary excretion of metabolites of 11-DOC, which are measured as 17-OHCS. The metyrapone test is appropriate for assessment of hypothalamic-pituitary
function in patients in whom there is no reason to suspect primary
adrenocortical insufficiency. Otherwise, ACTH stimulation test should
precede metyrapone testing because the administration of metyrapone
may precipitate adrenal crisis with cardiovascular collapse. If the cortisol
response to a rapid ACTH test is diminished, no additional information
will be gained from the metyrapone test. In contrast to the ACTH
stimulation test, the administration of dexamethasone or other glucocorticoids
will invalidate the metyrapone test. The patient must be hospitalized for the metyrapone test because of the
possibility of precipitating adrenal crisis. During the administration
of metyrapone, some patients with normal adrenal function will experience
side effects, most commonly vertigo, nausea, and vomiting. Gastrointestinal
symptoms are minimized by administering the metyrapone with
food. Many methods of metyrapone administration have been employed, including
single-dose tests.78,103 According to one protocol for the metyrapone test,104 beginning at 8 a.m., 750 mg metyrapone is given orally every 4 hours for
a total of six doses. Plasma is taken at 8 a.m., immediately before
the first dose; and again the next morning at 8 a.m., 4 hours after the
last dose. The cortisol level after metyrapone administration should
be less than 6 to 8 μg/dl if the test results are to be considered
valid. In normal subjects, the 11-DOC level before the administration
of metyrapone is approximately 1 ng/dl and the level after metyrapone
administration is greater than 10.5 μg/dl. Patients with inadequate
pituitary-adrenal reserve as a result of pituitary disease or long-term
exogenous glucocorticoid therapy have 11-DOC levels after metyrapone
of less than 8 μg/dl, with mean values of 2 to 3 μg/dl. INSULIN-INDUCED HYPOGLYCEMIA TEST. Insulin-induced hypoglycemia stimulates the secretion of ACTH and cortisol. Testing
with hypoglycemia provides results more rapidly than the
metyrapone test, and it also provides an assessment of growth hormone
secretion. Additionally, it is preferred by some authors over the metyrapone
test as an evaluation of ACTH reserve.100,105 Hypoglycemia is contraindicated in patients with cardiovascular disease, who
are at risk for the development of arrhythmia, and in patients
with epilepsy, who are at risk for the development of seizure. As is true
of the metyrapone test, no useful information about the hypothalamic-pituitary-adrenal
axis will be gained with insulin testing if the cortisol
response to ACTH is subnormal. For the test, the patient fasts overnight. A physician must be in attendance
throughout the test to assess the mental status of the patient and
administer glucose as necessary. A plasma sample is drawn at 8 a.m. for
cortisol and glucose determination. A bolus of regular insulin 0.1 U/kg
is given intravenously, and blood is sampled 30, 60, 90, and perhaps 120 minutes
after the insulin dose. If pituitary disease is strongly
suspected, the insulin dosage is decreased to 0.05 U/kg because severe
hypoglycemia may result from the larger dose of insulin. Virtually
all patients who achieve suppression of plasma glucose to the degree
required for an adequate test will have tachycardia and diaphoresis. If
the patient becomes confused, the test should be terminated immediately
by infusing glucose. Hypoglycemic stimulus is adequate if the glucose level falls to a level
that is less than 50% of the baseline value. If the stimulus has been
adequate, normal subjects exhibit a 5-μg/dl increment in plasma cortisol
level and a concentration of greater than 15 μg/dl in the 60- or 90-minute
sample.100 The best objective measure of the response of the hypothalamic-pituitary-adrenal
axis to hypoglycemic stress may be the cortisol/glucose slope
value, which is obtained by plotting serial cortisol values against
the corresponding glucose values.103 In addition to its value in assessing ACTH reserve, the cortisol response
to insulin-induced hypoglycemia may be useful for distinguishing patients
with Cushing's syndrome from those with false-positive dexamethasone
suppression test findings. Patients with Cushing's syndrome
generally have blunted responses.78 CORTICOTROPIN-RELEASING HORMONE TESTING. Corticotropin-releasing hormone testing is replacing metyrapone and insulin-induced
hypoglycemia testing to assess the hypothalamic-pituitary-adrenal
access. The test is performed by intravenous injection of 1 μg/kg
ovine or human CRH followed by measurement of baseline and stimulated
serum ACTH and plasma cortisol levels. This test induces maximal
ACTH and cortisol levels. Human CRH has a more rapid clearance than
ovine CRH. The increment of cortisol is greater if the test is performed
at 8:00 p.m., when the circulating cortisol levels are lowest, because
the peak cortisol levels induced by the stimulation test are unchanged
regardless of the time that the test is performed. Side effects
of flushing, metallic taste, and increased respiratory rate have been
reported. However, unlike the metyrapone test, it is considered safe
to perform on an outpatient basis. The test has been useful in distinguishing patients with Cushing's
disease, who have normal or exaggerated hormonal increases, from patients
with ectopic ACTH secretion or non-ACTH-dependent Cushing's
syndrome, who do not respond to stimulation.106,107,108,109 The test also has been used in conjunction with inferior petrosal vein
sampling to lateralize ACTH-secreting pituitary adenomas. ACTH response
also may be blunted in patients with anorexia nervosa and primary affective
disorders.110,111 Its usefulness in adrenal insufficiency is limited. Patients with adrenal
insufficiency due to hypothalamic abnormalities usually have greater
increases in the ACTH level after stimulation compared with patients
with pituitary abnormalities.112 |