The observation of dimorphic sex chromosomes in 1905 led to the idea that
a gene on the Y chromosome was testis determining.18 In humans, this was not confirmed until methods for cytogenetic analysis
improved in the 1950s. In that era, the presence of a Y chromosome
was shown to be male sex-determining, regardless of the number of X chromosomes
present, and the absence of a Y chromosome was shown to be female
sex-determining.19,20 Amid these observations, a number of individuals were observed who had
genetic sex reversal, genetic sex of one type and a phenotypic sex of
the opposite type. These individuals were classified either as 46,XX
males or 46,XY females with gonadal dysgenesis; the latter occurred because
their gonads regressed after an initial period of differentiation. A
model for explaining genetic sex reversal was proposed in 1965 in
which nonhomologous recombination between X and Y chromosomes translocated
the testis-determining factor from the Y onto the X chromosome.21 This hypothesis was proved correct for many cases of 46,XX maleness and
was instrumental for the cloning of the Y-linked testis-determining
factor (Fig. 2A). Still, not all cases of sex reversal could be explained by this observation, and
the study of individuals with other forms of genetic sex
reversal has been important for identifying autosomal- and X-linked sex-determining
genes (see Fig. 2B). |
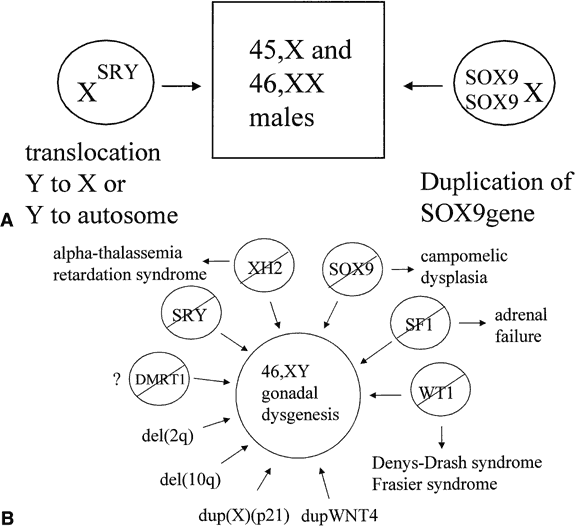 Fig. 2. A. Genetic mechanisms known to cause 46,XX maleness. B. Genetic mechanisms known to cause 46,XY gonadal dysgenesis. A slash means
a loss of function mutation.
Fig. 2. A. Genetic mechanisms known to cause 46,XX maleness. B. Genetic mechanisms known to cause 46,XY gonadal dysgenesis. A slash means
a loss of function mutation.
|
From the 1970s on, a number of candidate genes for the testis-determining
factor, including HY antigen, bkm, and ZFY, were proposed and then subsequently excluded.22,23,24 In 1990, the gene called sex-determining region Y (SRY in humans, Sry in mice) was shown to the testis-determining factor on the Y chromosome.5,6 This gene was found on the Y chromosomes of most other eutherian mammals
in which it was tested (e.g., chimp, rabbit, pig, horse, cow, and tiger). It
was also found in all human and mouse XX males who have this
phenotype on the basis of nonhomologous recombination between the X and
Y chromosomes. Deletion of the SRY/Sry gene prevents testes differentiation
from occurring.25 The timing of SRY/Sry expression in the gonadal ridges of the humans and mice was as expected (i.e., at
the induction of testicular differentiation).26,27 The human and mouse SRY/Sry genes are not functionally equivalent. XX mice transgenic for an intact Sry gene have normal testis differentiation, whereas XX mice transgenic for
an intact human SRY gene do not.28 The SRY proteins from all mammalian species contain an HMG box with a nuclear
localization signal, suggesting that the proteins might act as
nuclear transcription factors.29 The HMG boxes of the SRY proteins from various species all bind DNA at
a similar consensus sequence (AATAAC) and bend DNA to a varying degree (Fig. 3).30 The structures of the SRY proteins, even among closely related species
of mice and primates, are very different outside the HMG box.31,32 The mouse(M. musculus) Sry gene encodes glutamine/histidine-rich repeat 223 amino acid residues in
length at the N-terminus of the protein. This is highly polymorphic
in different mouse species and shortened in nonmusculus species. The human SRY gene does not contain a glutamine/histidine-rich region. The mouse Sry
protein with the long glutamine-rich region functions as a transcriptional
activator.29 Shortening of the glutamine-rich domain prevents transcriptional activation
from occurring both in vitro and in vivo in transgenic mice.33,34 There is no evidence that SRY functions as a transcriptional activator
in other species. Rather, it might function as an architectural factor
that binds other transcriptional regulators. The identification of SIP1, a
human protein with two PDZ-protein-binding domains that can interact
with the C-terminal seven amino acids of SRY, suggests a mechanism
by which this can happen.35 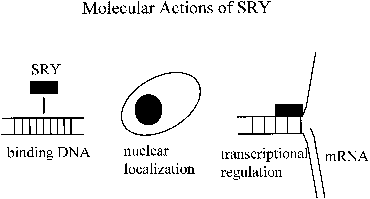 Fig. 3. Molecular actions of SRY. Fig. 3. Molecular actions of SRY.
|
A number of autosomal genes have been cloned that play a role in testis
determination (see Fig. 2B). The first to be identified was the Wilms' tumor suppressor, WT1.36,37 This gene encodes a transcription factor with zinc-finger domains that
bind to specific DNA sequences. The gene is expressed both in the undifferentiated
gonadal ridge and in the sex cords after testicular determination
in human embryos, suggesting that it has roles both in early
gonadal development and auxiliary effects in the testis pathway.38 Many different isoforms are produced through alternate splicing of the WT1 RNA. Deletion of WT1 from the germline is associated with Wilms' and other tumors, especially
gonadoblastoma, and with renal and gonadal anomalies.39,40 Germline mutation varies the efficiency of splicing WT1 RNA, thus yielding different ratios of the WT1 isoform that lead to sex reversal rather than normal gonadal differentiation.41,42 SOX9 (SRY-like HMG-box protein 9) was the second autosomal, sex-determining
gene to be identified.43 Mutations in this gene are associated with campomelic dysplasia, a skeletal
malformation syndrome in which the 46,XY individuals commonly have
sex reversal.44SOX9 encodes a protein with two activation domains, suggesting that it acts
by upregulation of other genes.45 The expression of SOX9 in the gonads of 46,XY human embryos follows a pattern similar to that
of SRY.27 The expression commences with testicular induction and increases over
the next several days with maximal detection observed over the sex cords, most
likely in Sertoli cells. No expression in the gonads of early 46,XX
female embryos, is consistent with a primary role for SOX9 in testicular determination; however, low levels of SOX9 transcripts in the embryonic rete ovarii are detected at a later time. Several
bits of genetic evidence suggest that all of the effects of SRY in causing male sexual differentiation may channel through SOX9. A 46,XX male with a chromosomal duplication encompassing the SOX9 gene suggests that enhanced expression of SOX9, even in the absence of SRY, is sufficient to cause testis determination (see Fig. 2A).46 Similarly, derepression of SOX9 expression in XX gonads leads to male development in Odsex mice.47 Ordinarily, this derepression might be mediated by Sry. Steroidogenic factor-1 (SF1), a transcription factor that regulates the
expression of a number of genes involved in steroid hormone production
and male sexual differentiation, falls into the class of so-called orphan
nuclear receptors.48,49 In addition to a zinc-finger DNA binding domain, the gene encodes a receptor
domain for an as-yet-unidentified ligand. Human SF1 is expressed
in the adrenal gland and the gonadal ridge before testicular differentiation. The
expression in both tissues continues after the period of
testicular differentiation.38 Testicular expression is observed both in Leydig and Sertoli cells, suggesting
a role both in testis differentiation and in hormone production. A de novo hemizygous mutation that disrupts DNA binding by the SF1 protein was associated
with adrenal insufficiency and with gonadal dysgenesis in an
individual with a 46,XY karyotype.50 This confirms a role for SF1 as a testis-determining factor. At least two X chromosomal loci have been implicated to play a role in
gonadal development. When mutated, XH2 results in the alpha-thalassemia, mental retardation syndrome.51,52 Some 46,XY individuals have gonadal dysgenesis as part of their phenotype, suggesting
that XH2 plays a role in testicular development. This gene encodes a helicase that
unwinds DNA, making it accessible to transcription factors. The dosage-sensitive
site has apparent anti-testis properties, when overexpressed, but
is not essential for testis determination. This region was
identified in individuals with 46,XY gonadal dysgenesis who had duplications
of the X chromosome in the region of band Xp21 and is not subject
to X chromosome inactivation in these individuals with partial duplication.53,54 Deletion of a gene within this region has been associated with adrenal
hypoplasia congenita. As a result, the gene has been called DAX1 (dosage-sensitive
sex reversal, adrenal hypoplasia congenita, and X chromosome).55 DAX1 encodes an orphan nuclear receptor with zinc fingers for DNA binding and
a domain for binding an unknown ligand.49 Low levels of DAX1 expression are observed before and after testicular induction in humans, most
notably over the sex cords after the Sertoli cells have differentiated.38 As a result, the anti-testis properties of DAX1 overexpression might act
before or after the expression of SRY. WNT4 may also function as a dosage-sensitive, sex-determining gene. Targeted, homozygous
deletion of this gene results in masculinization of
XX mice. These mice have a marked reduction in the number of oocytes in
their ovaries at birth,56 with the remaining occytes being observed in the process of degeneration. A
few sex-cordlike structures have been observed in these ovaries
and the supporting cells in these sex cords express the Sertoli cell markers, Mis
and Dhh. In addition, Leydig cell differentiation is not suppressed
and testosterone biosynthesis occurs. Mutations in Wnt4 have extragonadal effects on sexual differentiation. Homozygous mutant
mice have regress of their Mullerian ducts and masculinization of their
Wolffian ducts. Overexpression of WNT4 was observed in the lymphocytes of a 46,XY female with gonadal dysgenesis
whose chromosomal constitution included a 1p31-p35 duplication.57 This duplicated segment included the WNT4 gene. In transfected cells, WNT4 upregulated DAX1, suggesting a common mechanism for the two forms of dosage-sensitive
sex reversal. Several testis-determining genes have been identified in 46,XY knockout
mice with gonadal dysgenesis. These include fibroblast growth factor 9 (Fgf9) and Dmrt1. Homozygous knockout of the Fgf9 gene results in lung hypoplasia and, in XY embryos, a variety of gonadal
phenotypes, ranging from gonadal dysgenesis to testicular hypoplasia.58 To date, mutations in the Fgf9 gene have not been described in humans with sex reversal. The Dmrt family of genes contains a zinc finger-like DNA binding domain homologous
to the sex-determining genes in C. elegans (Mab-3) and Drosophila (dsx). Both invertebrate proteins are functionally related because they
are required for differentiation of male-specific sense organs, regulation
of yolk protein transcription, and normal male mating behavior.59 They show expression exclusively in gonadal ridge and early ridge that
is enhanced in males.60,61 Homozygous knockout of the mouse Dmrt1 gene causes severe testicular hypoplasia in XY mice, a phenotype similar
to that observed in humans with a deletion of the short arm of chromosome 9.62,63,64 However, chromosome 9 contains three DMRT genes. Deletion mapping and mutation analysis have been unable to distinguish
which of these genes is essential for the testicular development.65 The products of several testis-determining genes interact to regulate MIS, a downstream target of sexual differentiation (Fig. 4 ). SOX9 binds to a high-affinity site in the MIS promoter and initiates transcription.66 If the high-affinity site is eliminated, no transcription is initiated. Other
factors binding to one another and to the MIS promoter act as quantitative regulators of MIS transcription. SF1 binds to the promoter and to SOX9 to increase transcription.67 Elimination of the SF1 binding site leads reduces the levels of Mis transcripts in mice, but to a level of expression that is still sufficient
to cause complete Mullerian duct regression.66 An isoform of WT1 associates with SF1 to form a heterodimer that increases MIS expression.68 WT1 missense mutations, associated with 46,XY gonadal dysgenesis, do not
synergize with SF1. DAX1 antagonizes the synergy between SF1 and WT1, most
likely through a direct interaction with SF1.68,69,70,71 These findings suggest that WT1 and DAX1 may also functionally oppose
each other during testis development by modulating SF1-mediated transactivation. 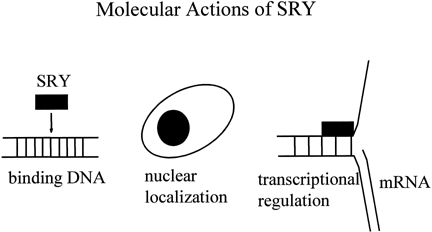 Fig. 4. Interactions of sex-determining transcriptional factor to regulate the MIS promoter. SOX9 is a transactivator. SF1 increases SOX9 tarnsactivation. WT1 synergizes
with SF1. DAX1 antagonizes synergy. Fig. 4. Interactions of sex-determining transcriptional factor to regulate the MIS promoter. SOX9 is a transactivator. SF1 increases SOX9 tarnsactivation. WT1 synergizes
with SF1. DAX1 antagonizes synergy.
|
WT1 also plays a role in regulating the expression of SRY.72 WT1 is known to be temporally expressed in the gonadal ridge before as
the expression of SRY—as might be expected for a transcriptional
regulator. This induction of SRY expression occurs through a proximal
EGR1-like DNA-binding sequence in the core promoter of the SRY gene. |