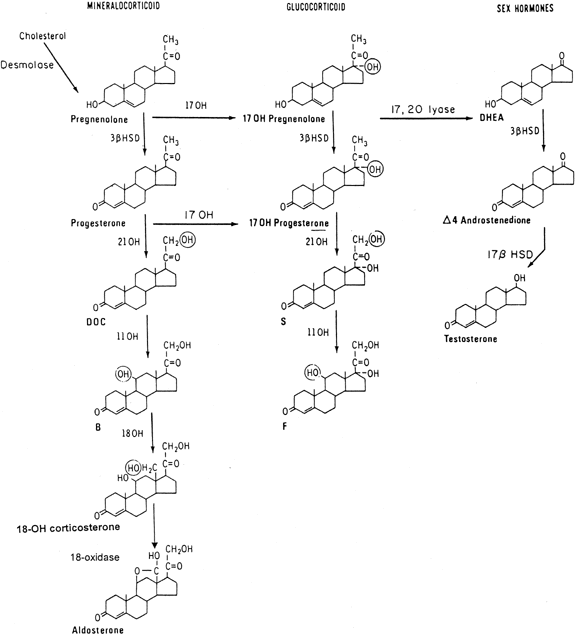
The following enzymatic defects of steroidogenesis may cause female pseudohermaphroditism
or virilization4,5,6: - 3β-Hydroxysteroid dehydrogenase deficiency (classic and nonclassic CAH)
- 21-Hydroxylase deficiency (salt-wasting, simple virilizing, and nonclassic
CAH)
- 11β-Hydroxylase (hypertensive classic and nonclassic CAH)
These syndromes are discussed in detail later in this chapter. 21-Hydroxylase Deficiency CAH is the most common cause of female pseudohermaphroditism and virilization, and
decreased cortisol synthesis owing to reduction or loss of 21-hydroxylase
enzyme function is the most common biochemical cause
of CAH.7,8,9 The decreased plasma cortisol elevates adrenocorticotropic hormone (ACTH) secretion,10,11,12 stimulating increased adrenal production of cortisol and of the androgen
precursors and androgens, which do not require 21-hydroxylase for their
biosynthesis. Early clinical studies13 showed increased urinary levels of pregnanetriol, the principal metabolite
of 17-OHP, and also of the 17-ketosteroids, metabolites of the androgens
DHEA and Δ4-A, and testosterone, in patients with 21-hydroxylase deficiency. Determinations
of serum levels of 17-OHP and Δ4 by radioimmunoassay allow more accurate diagnosis of CAH than could be
provided formerly by the assessment of 24-hour urinary levels of hormonal
metabolites.14,15 CLASSIC 21-HYDROXYLASE DEFICIENCY. The prominent feature of 21-hydroxylase deficiency is progressive virilization
with advanced somatic development. The classic disorder produces
ambiguous or masculine external genital formation in female infants
at birth. It occurs in two major forms, simple virilizing and salt-wasting. Because
a salt-wasting crisis within the first few weeks of life
can have profound effects on the infants, leading even to death, every
infant born with ambiguous genitalia must have a prompt, thorough evaluation
to exclude the salt-wasting forms of CAH. Simple Virilizing Form Developmental genital anomalies are manifest in females as varying degrees
of genital ambiguity, which should alert the physician to the presence
of the condition. CAH caused by 21-hydroxylase deficiency is the
most common cause of ambiguous genitalia in the newborn female, and because
affected females have the capacity for an entirely normal female
sex role, including childbearing, it is very important to recognize this
disorder in newborns with ambiguous genitalia. Without treatment, there
is progressive virilization and early fusion of the epiphyses with
resulting short stature. Salt-Wasting Form In 75% of patients with classic 21-hydroxylase deficiency, salt wasting
occurs in infancy and is life-threatening. It is characterized by hyponatremia, hyperkalemia, inappropriate natriuresis, and low serum and
urinary aldosterone with concomitantly high plasma renin activity (PRA). The
increase in the proportion of salt-wasting cases in recent years
may be attributed in part to advances in diagnostic techniques as well
as to increased survival because of the availability of exogenous mineralocorticoid
supplements. Salt wasting is caused by an enzyme deficiency that significantly impairs
aldosterone synthesis, and low levels of this essential mineralocorticoid
result in inadequate sodium retention by the renal distal tubule. It
may also result from the effect of certain hormonal precursors, thought
to be mineralocorticoid antagonists, found in increased levels
in 21-hydroxylase deficiency. Salt wasting may be particularly prominent
in infancy, given the marginally competent sodium-conserving mechanism
of the immature newborn renal tubule, and may lessen with age.16,17,18 Careful monitoring of PRA in patients will aid in determining their changing
dietary sodium, glucocorticoid, and mineralocorticoid requirements. The extent of virilization may be the same in simple virilizing and salt-wasting
CAH. Thus, even mildly virilized infants with 21-hydroxylase
deficiency should be observed carefully for signs of a potentially life-threatening
crisis within the first few weeks after birth. Glucocorticoids are essential for the normal development and functioning
of the adrenal medulla. A recent study has shown that CAH compromises
both the development and functioning of the adrenomedullary system.19 A 40%to 80% reduction of plasma epinephrine and metaepinephrine concentrations
was found in patients with CAH. This reduction is probably caused
by a combination of the lack of intraadrenal cortisol secretion and
abnormal adrenomedullary formation. Until recently, the literature has indicated that with few exceptions,20 presence or absence of salt wasting is consistent within a family, and
it has been predicted that subsequent affected offspring will have the
same form of the disease as the index case. However, several families
have been reported in which concordance for salt wasting was lacking
among human leukocyte antigen (HLA)-identical siblings16 (see later discussion on HLA and 21-hydroxylase). In some cases, differences
in aldosterone-synthesizing capacity could also be demonstrated
between affected siblings as well as in unrelated individuals carrying
identical mutations. NONCLASSIC 21-HYDROXYLASE DEFICIENCY. An attenuated form of adrenal hyperplasia was first suspected in the early 1950s
by gynecologists in clinical practice who used glucocorticoids
to treat women with physical signs of hyperandrogenism, including infertility.21,22 The first documentation of suppression of 21-hydroxylase precursors in
the urine of such women after glucocorticoid therapy was by Decourt and
co-workers23 in 1957. During the next two decades, the empiric use of glucocorticoids
for the treatment of virilized women became commonplace, as adrenal
androgens were often assumed to be elevated in those patients. Development
of a radioimmunoassay for 17-OHP in the 1970s made it possible to
diagnose 21-hydroxylase defects by measuring serum elevations of this
index compound (the principal substrate for 21-hydroxylase in the adrenal
zona fasciculata).24 The autosomal-recessive mode of genetic transmission of the nonclassic
form of 21-hydroxylase deficiency became apparent through family studies
of classic 21-hydroxylase deficiency.25,26,27 Individual 21-hydroxylase genotypes are revealed by the 17-OHP response
to ACTH stimulation testing. Serum concentration samples are obtained
at 0 (baseline) and 60 minutes after ACTH administration, with concentrations
plotted on a reference nomogram (Fig. 3). The establishment of linkage to HLA28,29 confirmed the existence of this disorder as an allele of classic 21-hydroxylase
deficiency.26,30 The HLA associations for nonclassic 21-hydroxylase deficiency28,29,31,32 are distinct from those found in classic 21-hydroxylase deficiency and
differ according to ethnicity.29,33 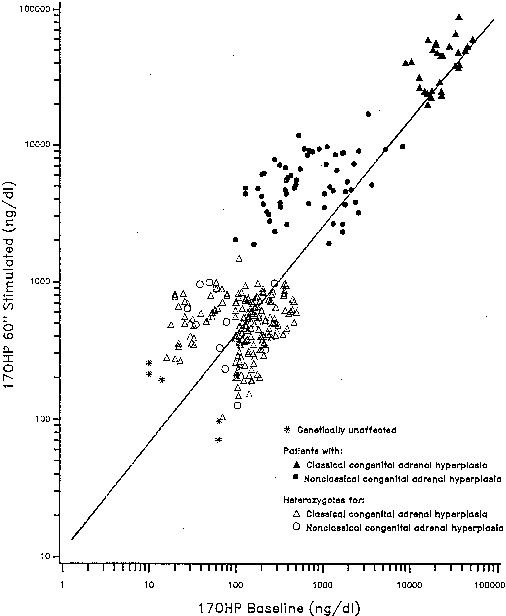 Fig 3. Diagnosis 21-hydroxylase deficiency. ACTH 0.25 mg is given IV bolus at 8:00 a.m.. Serum
samples are obtained at 0 and 60 minutes. The 0-minute
concentration is plotted on the abscissa, the 60-minute on the ordinate. Patients
segregate into groups on the regression line, as indicated, allowing
clearcut hormonal diagnosis of classic patients and nonclassic
patients from heterozygote and unaffected subjects. Fig 3. Diagnosis 21-hydroxylase deficiency. ACTH 0.25 mg is given IV bolus at 8:00 a.m.. Serum
samples are obtained at 0 and 60 minutes. The 0-minute
concentration is plotted on the abscissa, the 60-minute on the ordinate. Patients
segregate into groups on the regression line, as indicated, allowing
clearcut hormonal diagnosis of classic patients and nonclassic
patients from heterozygote and unaffected subjects.
|
Clinical Features Clinical symptomatology of nonclassic 21-hydroxylase deficiency is variable;symptoms
may appear at any age (Fig. 4), and longitudinal follow-up shows they may wax and wane with time. Certain
patients hormonally identified as affected with nonclassic 21-hydroxylase
deficiency are entirely asymptomatic up until the time of detection (usually
as part of a family study). It is now thought, however, that
emergence of overt hyperandrogenism is to be expected at some
point in almost all cases. 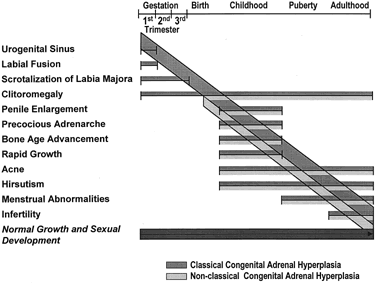 Fig 4. Clinical spectrum of steroid 21-hydroxylase deficiency.(New MI, Dupont B, Grumbach K et al: Congenital adrenal hyperplasia and
related conditions. In Stansbury JB, Wyngaarden JB, Fredrickson DS et
al [eds]: The Metabolic Basis of Inherited Disease, 5th ed. New
York, McGraw-Hill, 1983) Fig 4. Clinical spectrum of steroid 21-hydroxylase deficiency.(New MI, Dupont B, Grumbach K et al: Congenital adrenal hyperplasia and
related conditions. In Stansbury JB, Wyngaarden JB, Fredrickson DS et
al [eds]: The Metabolic Basis of Inherited Disease, 5th ed. New
York, McGraw-Hill, 1983)
|
Nonclassic 21-hydroxylase deficiency can result in premature development
of pubic hair in children; to our knowledge, the youngest such patient
was noted to have pubic hair at 6 months of age.27 Elevated adrenal androgens promote the early fusion of epiphyseal growth
plates, and it is commonly found that children with the disorder have
advanced bone age and accelerated linear growth velocity and are ultimately
shorter than the height that might be predicted on the basis
of mid-parental height.34 Severe cystic acne refractory to oral antibiotics and retinoic acid has
been attributed to nonclassic 21-hydroxylase deficiency. In addition, male-pattern
baldness in young women with this disorder has been noted
as the sole presenting symptom. Menarche may be normal or delayed, and secondary amenorrhea is a frequent
occurrence. Of patients with polycystic ovary syndrome, there is a
subgroup of women with nonclassic 21-hydroxylase deficiency. The pathophysiology
of this phenomenon probably relates to adrenal sex-steroid
excess disrupting the usual cyclicity of gonadotropin release or the direct
effects of adrenal androgens on the ovary, leading ultimately to
the formation of ovarian cysts, which may then autonomously produce androgens. Chronic hypersecretion of androgen precursors can induce a reduction in
insulin sensitivity in female patients with nonclassic 21-hydroxylase
deficiency.35 Although the androgen profile in serum and urine in both the basal and
ACTH-stimulated states in this syndrome may not be markedly different
from that demonstrated by women with polycystic ovary syndrome, the response
of 17-OHP to ACTH clearly identifies those patients who have an
adrenal 21-hydroxylase defect.36 Figure 5 presents nomograms for establishing the diagnosis of 21-hydroxylase deficiency (see
later discussion on nonclassic 3β-HSD deficiency). Even ultrasonograms of the ovary do not distinguish between
women with excess androgens from polycystic ovary syndrome and
women with nonclassic 21-hydroxylase deficiency, and ACTH tests are required
for the differential diagnosis. The response of the hypothalamic-pituitary-gonadal
axis to luteinizing hormone-releasing hormone is variably
abnormal in virilized women with nonclassic 21-hydroxylase deficiency.37
|
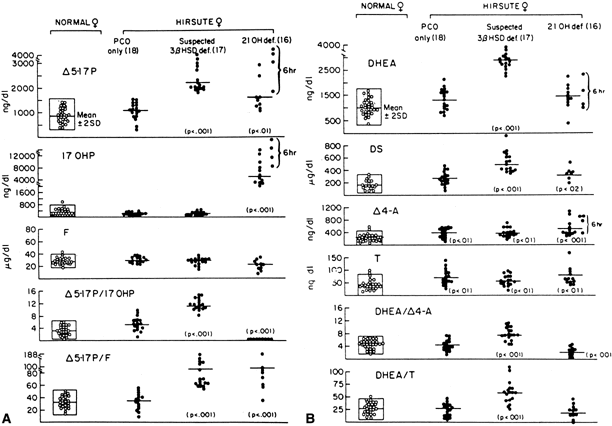 Fig 5. Hormonal responses to ACTH stimulation at 0 minutes (A) and 60 minutes ( B ). 3 β -HSD deficiency is indicated when all the following criteria are two standard
deviations above the mean:D5-17-hydroxypregnenolone (Δ5 -17P) response, DHEA response, Δ5 -17P:17-0HP, and D5-17P:cortisol ( F ). (Pang S, Herner AJ, Stoner E et al: Late-onset adrenal steroid 3 β -hydroxysteroid dehydrogenase deficiency: I. A cause of hirsutism in pubertal
and postpubertal women. J Clin Endocrinol Metab 60:428, 1985; © The
Endocrine Society)
Fig 5. Hormonal responses to ACTH stimulation at 0 minutes (A) and 60 minutes ( B ). 3 β -HSD deficiency is indicated when all the following criteria are two standard
deviations above the mean:D5-17-hydroxypregnenolone (Δ5 -17P) response, DHEA response, Δ5 -17P:17-0HP, and D5-17P:cortisol ( F ). (Pang S, Herner AJ, Stoner E et al: Late-onset adrenal steroid 3 β -hydroxysteroid dehydrogenase deficiency: I. A cause of hirsutism in pubertal
and postpubertal women. J Clin Endocrinol Metab 60:428, 1985; © The
Endocrine Society)
|
Treatment with glucocorticoids is effective in suppressing adrenal androgen
production, and with time, clinical signs of androgen excess show
improvement. Given the 9-month life expectancy of established hair follicles, remission
of hirsutism generally takes at least 1 to 2 years. Because
of the presumptive identification of the first nonclassic patients
approximately 30 years ago, it has been recognized that infertility
in women may be reversed during glucocorticoid therapy.21,22,23,38 Variability of Symptoms The virilizing signs of nonclassic 21-hydroxylase deficiency are extremely
variable among patients despite similar androgen levels. Thus, some
patients with nonclassic 21-hydroxylase deficiency develop acne; others
develop hirsutism, oligomenorrhea, or even reduced fertility; and
some remain asymptomatic. The basis for the specific and idiosyncratic
organ response to excess androgen remains unexplained. There may be interindividual
variability in receptor sensitivity; alternatively, there
may be both interindividual and intraindividual variation over time
in the peripheral metabolism of skin or hair follicles. The disorder
may be progressive, as demonstrated by the increasing prevalence of hirsutism
with age that is observed among female patients with nonclassic
CAH.39 Ultrasonography frequently demonstrates cystic ovaries, similar to those
found in polycystic ovary syndrome, in patients with CAH caused by defects
in 21-hydroxylase and other enzymes, the loss of which leads to
a low level of cortisol. Will the suppression of adrenal androgens, readily
accomplished in the treatment of CAH, cause reversal of the cystic
changes in the ovary?Preliminary evidence indicates that this is likely, suggesting
that cystic changes of the ovary in humans can result
from excess androgens from adrenal or other extraovarian sources. Further, it
may be valuable to test all women with cystic ovaries seen on
ultrasonography for inborn errors of steroidogenesis, especially those
women from ethnic groups at high risk for nonclassic 21-hydroxylase
deficiency. High Prevalence of 21-Hydroxylase Deficiency The most common cause of female pseudohermaphrodi tism is CAH caused by 21-hydroxylase
deficiency; both the classic and nonclassic forms of the
disorder occur with high frequency in the populations that have been
studied. Neonatal screening tests suggest an incidence of 1 in 15,000 in most white
populations for classic 21-hydroxylase deficiency.40 This disorder is thus relatively common for an inborn error of metabolism. Phenylketonuria, the
most common of the inherited diseases for which
neonatal screening is mandated, occurs in approximately 1 in 15,000 births
in white populations. The nonclassic form of 21-hydroxylase deficiency is even more common and
is probably the most frequent autosomal-recessive genetic disorder in
humans. Its incidence is especially high in Ashkenazi Jews (5%), Hispanics (2%), Yugoslavians (1.4%), and Italians (0.3%).33 In the diverse white population, the incidence is at least 0.1%.33,41,42 HUMAN LEUKOCYTE ANTIGENS AND 21HYDROXYLASE DEFICIENCY. The genetic region called human leukocyte antigen (HLA), which is a part
of the major histocompatibility complex (MHC), is a cluster of genes
located on the short arm of chromosome 6. The class I antigens (HLA-A, -B, and -C), expressed
on all nucleated cells, are the major barriers
for allogenic transplantation. The class II antigens (principally HLA-D
and -DR), expressed primarily on activated T-lymphocytes, participate
in immune responses. The HLA complex also contains a linkage group
of genes expressing products with immediate functions outside histocompatibility
and immune response. The major components of class III antigens
are the genes for C2 and C4 (C4a and C4b) of serum complement, factor
B of the alternate complement pathway, and the 21-hydroxylase enzyme, cytochrome
P450c21. 21-Hydroxylase deficiency is inherited as a monogenic autosomal-recessive
trait closely linked to the HLA complex.43,44 Thus, with few exceptions,45 a sibling sharing both HLA haplotypes with the proband is predicted to
be affected; one who shares a single haplotype is predicted to be heterozygote; and
one who shares no HLA haplotype is predicted to be unaffected. De
novo pathologic mutations are identified in those rare cases
in which an HLA-identical sibling is not affected.46,47,48,49 HLA/21-Hydroxylase Linkage The linkage between HLA and the 21-hydroxylase gene was first shown by
Dupont and associates43 in a study on six families. A study of persons with intra-HLA recombinations
suggested that the locus for 21-hydroxylase is situated between
HLA-B and HLA-D, within the class III region.50,51 Linkage Disequilibrium In addition to being linked to the adjacent HLA loci in general, 21-hydroxylase
deficiency is found more frequently than expected with certain
specific HLA antigens. Haplotypic associations (occurrence of genes
linked on the same chromosome) in the HLA complex often include specific
alleles of the other class III genes, complement components Bf, C2, and
C4(C4A/B).52,53,54,55,56 Salt-wasting 21-hydroxylase deficiency is associated with HLA-Bw60 and
with the extended haplotype HLA-A3;Bw47;DR7. This haplotype carries a
null allele at locus C4B (thus expressing only one isotype of complement
C4). Simple virilizing disease is associated with antigen HLA-Bw51 in
selected ethnic groups, and the partial haplotype HLA-B14;DR1 is found
to be associated with nonclassic disease in all ethnic groups examined
except the Yugoslavian population. The B14;DR1 haplotype includes
a duplication of one of the C4 loci.57 Another extended haplotype, HLA-A1,B8,DR3, is negatively associated with 21-hydroxylase
deficiency; this hormonally normal haplotype, like the
haplotype B47;DR7 (severe 21-hydroxylase deficiency), also expresses
only one C4 isotype, in this case carrying a null allele at locus C4A. Molecular Genetics The structural gene encoding the adrenal cytochrome P450 specific for steroid 21-hydroxylation (P450c21) is named CYP21 or CYP21B and contains 10 exons.58,59,60 This gene and a 98% identical pseudogene (CYP21P or CYP21A) are located
in close proximity (30 kb) in the HLA complex adjacent to and alternating
with the C4B and C4A genes encoding the fourth component of the
serum complement.61,62 The pseudogene CYP21P does not produce a detectable mRNA or a protein, owing
to several deleterious mutations. Mutations in CYP21 appear to be
generated by either of two types of recombination mechanisms. Misalignment
of the tandem C4A-CYP21P-C4B-CYP21 arrangement during meiosis leads
to unequal crossing over, resulting in a complete deletion of a DNA
segment, including C4B and CYP21. Alternatively, small deleterious
mutations appear to be transferred from CYP21P to CYP21 in gene conversion
events.63,64 The frequency of gene deletions in different ethnic groups ranges from 11%to 35%, and
many of these are found in association with the haplotype
HLA-B47;DR7.65 CORRELATION OF GENOTYPE WITH PHENOTYPE. In general, mutant P450c21 enzymes carrying specific amino acid substitutions
identified in patients with 21-hydroxylase deficiency display
activities that correspond roughly to the clinical severity of the disease
and to the associated biochemical abnormalities (Table 1). Table 1.
| | Mutation | | | |
Gene | Exon/Intron | Name/Type | AA | Comments | Reference |
CYP21 | E1 | Insertion (conversion) | +L9 | Normal polymorphism | 66 |
| E1 | Nonsense mutation | W22X | | 67 |
| E1 | Frameshift | W22 +1nt | Insertion of 1 nucleotide | 68 |
| E1 | Missense mutation (conversion) | P30L | NC phenotype | 69 |
| E1 | Missense mutation | P30Q | SW allele | 70 |
| E1 | Frameshift mutation | Y47Δ1nt | Deletion of thymidine at nt 141 leads to L51X | 71 |
| I1 | Aberrant splicing of intron 1 | W23X nt 295 A G G | | 67 |
| E2 | Missense mutation | G90V | Spanish patient | 72 |
| I2 | Aberrant splicing of intron 2 | nt 387 GA | Intron 2 splice donor site Chinese patient | 73 |
| I2 | Aberrant splicing of intron 2 (conversion) | nt 656 A/CG | Part of intron (end 19 bases) retained in mRNA processing. Most frequent nondeleted
allele | 74 |
| E3 | Nonsense mutation | Y97X | | 75 |
| E3 | Missense mutation | P106L | NC allele | 76 |
| E3 | Eight-base deletion (conversion) | G110Δ8nt | Frameshift: 20-AA + stop | 66 |
| E4 | One-base deletion | C169Δ1nt | Frameshift | 77 |
| E4 | Missense mutation (conversion) | I172N | Affects anchoring in membrane | 78 |
| E5 | Missense mutation | G178A | SW allele | 72 |
| E5 | Three-base deletion | ΔE196 | Deletion of nucleotides 1158–1160 | 79 |
| E6 | Cluster (conversion) | I236N V237E* M239K* | *2 more charges added in region with multiple charged residues | 74 |
| E7 | Missense mutation (conversion) | V281L | Major NC mutation HLA-B14;DR1 associated | 80 |
| E7 | Missense mutation | V281G | | 81 |
| E7 | Missense mutation | G291S | AA substitution | 76 |
| | | | CT at conserved | |
| | | | position | |
| | | b398 | At position +9 of | |
| | | | intron (secondary | |
| | | | effect?) | |
| E7 | Missense mutation | G291C | | 72 |
| E7 | Missense mutation | L300F | | 81 |
| E7 | Nonsense mutation | W302X | Finnish patient | 82 |
| E7 | Single base insertion (conversion) | F306 +1nt | Frameshift: +T at codon 305–7 | 83 |
| I7 | Loss of splice donor site at Intron 7 | nt 1784 GC | Aberrant splicing Found in one SW patient | 84 |
| I7 | Loss of splice donor site at Intron 7 | nt 1785 TG | Aberrant splicing Found in one NC patient | 85 |
| E8 | Nonsense mutation | R316X | Chinese patient | 73 |
| E8 | Nonsense mutation (conversion) | Q318X | | 86 |
| E8 | Frameshift | S330 Δ10 nt | Chinese patient | 73 |
| E8 | Missense mutation | R339H | NC allele | 87 |
| E8 | Missense mutation | R354H | 0% activity in transfected cells | 72 |
| E8 | Missense mutation | R354C | | 81 |
| E8 | Missense mutation | R356W | Radical AA | 88 |
| | (conversion) | | substitution | |
| | | | May impair redox interactions | |
| E8 | Missense mutation | R356P | May impair redox | 89 |
| | | | interactions | |
| E8 | Missense mutation | R356Q | May impair redox | 89 |
| | | | interactions | |
| E9 | Missense mutation | E380D | | 90 |
| E9 | Duplication | V397 +16nt | Frameshift Chinese patient | 73 |
| E9 | Nonsense mutation | W405X | | 76 |
| E10 | Missense mutation | G424S | Brazilian patient | 91 |
| E10 | Missense mutation | P453S+ plus P105L | NC allele AA substitution of conserved (P453) and nonconserved (P105) residue | 76, 92 |
| E10 | Frameshift mutation | P475 Δ1nt | | 85 |
| E10 | Missense mutation | R483P | Possible first step of 2-step mechanism generating no. 39 | 93 |
| E10 | Compound frameshift mutation | R483 Δ1nt | Replaces last 11 AA and extends protein by a further 45 AA | 76 |
NC, Nonclassical.
*Nucleotide number.
Like a homozygous deletion that precludes the expression of any enzyme, deletion
in conjunction with a stop mutation or with a cluster of mutations
at exon 6, which confers zero enzyme activity in vitro, would be
predicted to result in 0% overall 21-hydroxylase activity in vivo and
the severe salt-wasting phenotype. Homozygosity for the mutation Ile-172-Asn, which
confers approximately 2% of normal activity on the gene
product, usually results in the simple virilizing phenotype. However, the
distinction between the two forms of classic 21-hydroxylase deficiency
is not absolute. Speiser and associates94 classified 90 patients into three mutation groups based on the degree
of predicted enzymatic compromise. Mutation group A (no enzymatic activity) consisted
primarily of salt-wasting patients, group B (2% activity) of
simple virilizing patients, and group C (10% to 20% activity) of
nonclassic patients. Mutation groups were correlated with clinical diagnosis, but
each group contained patients with phenotypes either more
or less severe than predicted. The phenotype was accurately predicted
in 87% (54/62) of group A, in 72% (16/22) of group B, and in 62.5% of
group C. A recent study by Wilson and colleagues found that the 10 most
common mutations observed in the 21-hydroxylase gene result in phenotypes
that are not always concordant with genotype.95 Genetic heterogeneity has been found in all populations studied thus far.81,96,97,98,99,100,101,102,103,104,105 Thus, the phenotype of a patient cannot be predicted from the genotype
with complete certainty. Patients with phenotypes that are more severe
than predicted from the genotype and who are discordant with siblings
may have additional, as yet unidentified, mutations within the CYP21 gene. It
is also plausible that at least some differences in clinical
disease expression are governed by factors remote from the CYP21 locus. One
could postulate that phenotypic severity is influenced by parental
imprinting or by negative allelic complementation (i.e., exaggerated
gene dosage effect).106 Activity of other gene-encoding proteins other than P450c21 that have
steroid 21-hydroxylase activity is another possibility to explain phenotypic
heterogeneity.102 Finally, patients with CAH lacking mutations in the entire 21-hydroxylase
gene have also been described.107 3 β -Hydroxysteroid Dehydrogenase Deficiency In 3β-HSD deficiency, as in the other two common forms of CAH known to produce
ambiguous genitalia in the newborn, there is a spectrum of clinical
phenotypes, including both salt-wasting and non-salt-wasting forms.108 The degree of severity of the enzyme defect cannot be determined from
the appearance of the external genitalia at birth. A defect in 3β-HSD was first described by Bongiovanni109 in 1962. On the basis of pedigree analysis, a monogenic autosomal-recessive
mode of inheritance seemed most likely.108,109,110 This disorder affects the synthesis of all classes of adrenocortical steroids. Deficiency
of the 3β-HSD enzyme may be diagnosed by measuring elevated levels of the Δ5-steroids: pregnenolone, 17α-hydroxypregnenolone, and DHEA in serum, and
pregnanetriol and 16-pregnanetriol in urine. An elevated ratio
of Δ5 to Δ4 steroids characterizes the biochemical findings in patients with 3β-HSD deficiency. Unlike 21-hydroxylase, this enzyme is active in the gonads as well as in
the adrenal glands, and a deficiency of 3β-HSD may cause both male and female pseudohermaphroditism. The resulting
androgen deficiency in affected males will usually cause some degree
of hypospadias (often the severe perineal-scrotal form) and palpable
testes. Affected females, although frequently normal, may have clitoromegaly
from very high levels of the weak androgen DHEA, which may undergo
peripheral conversion to more potent androgens. The deficiency of
aldosterone in classic cases of 3β-HSD deficiency results in salt wasting.109,111,112,113,114 Many cases, however, have been described in which the ability to conserve
sodium was intact.108,110,111,113,115,116,117,118,119,120,121,122,123 It is claimed that 3β-HSD deficiency is the second most common steroidogenic defect,124 but as yet there have been no epidemiologic studies. There have been no
reports of geographic or ethnic predominance of the disorder. As is
the case for 21-hydroxylase deficiency, an attenuated or nonclassic form
of the disease appears more common than the severe deficiency.
Nonclassic 3 β -HSD Deficiency Nonclassic 3β-HSD deficiency is usually identified in girls with premature adrenarche
or in adolescent and young adult women with hirsutism, acne, and oligomenorrhea.36,125 Until recently, these young women were usually diagnosed as having polycystic
ovary syndrome, but careful scrutiny of ACTH-stimulated adrenal
hormone responses has allowed differentiation of this subgroup.36 Little is known about symptomatic effects in males; cases presenting clinically
at a later age have all had some degree of hypospadias, indicating
presence of an enzyme defect already in prenatal life. Mild 3β-HSD deficiency appears to be a relatively common cause of hirsutism in
women. Fifteen percent of women with hirsutism in one study had the disorder, compared
with 14% with late-onset 21-hydroxylase deficiency;the
rest had no diagnosable adrenal defect.36 The mean age at diagnosis was 20 years, and although the patients tended
to be of normal stature, there was some indication that the more severely
affected women experienced an early growth acceleration in childhood, but
as adults were short for mid-parental height. They were not
obese. They tended to have a somewhat early pubarche and thelarche; 13% of
patients with premature pubarche in one study had mild 3β-HSD deficiency,126 but the age at menarche was normal (11.8 years). Hirsutism or acne had
its onset at the time of menarche or shortly thereafter, and about half
of the women had onset of oligomenorrhea at the time of, or within
the next 2 years after, onset of hirsutism or acne. A recent study has
excluded partial 3β-HSD deficiency among 78 hirsute women.127 The diagnosis of mild 3β-HSD deficiency requires the demonstration of abnormally elevated Δ5-17α-hydroxypregnenolone (Δ5-17P) and DHEA levels in response to a standard ACTH stimulation test.36 Especially helpful in the diagnosis are the ratios of Δ5-17P:17-OHP and Δ5-17P:cortisol, which are characteristically elevated in 3β-HSD deficiency, thus differentiating it from 21-hydroxylase deficiency (see Fig. 5A). Baseline gonadotropin levels tend to be in the normal early- to late-follicular
range, regardless of menstrual disturbance or presence of
ovarian cysts, although quite a few patients may manifest an elevated
base line luteinizing hormone. Adrenal computed tomography (CT) and abdominal
ultrasound, when adequately performed, unequivocally show mild
to marked bilateral adrenal hyperplasia. Molecular Genetics The enzyme 3β-HSD has the compound function of 3β-hydroxysteroid dehydrogenation and 3-oxosteroid isomerization. Two genes
encoding 3β-HSD mapped to the 1p13 chromosome have been cloned to date: a skin-placental
form (type I) and an adrenal-gonadal form (type II). Studies on
the type II 3&gr;-HSD gene from index cases have revealed a number
of mutations associated with different phenotypic forms of 3β-HSD deficiency.128,129,130,131,132,133 Molecular analysis to date has not shown conclusively that DNA abnormality
is present in patients with the nonclassic form of the disease,134 although a heterozygous mutation of the type II gene in one case135 demonstrated that it is likely. 11 β -HYDROXYLASE DEFICIENCY. Eberlein and Bongiovanni136,137 were the first to describe a congenital disorder of steroid biosynthesis
causing virilization and hypertension with a pattern of steroid secretion
indicating defective 11β-hydroxylation. This form accounts for approximately 5% of all cases of
CAH.7,8,9,138 Classic 11 β -Hydroxylase Deficiency As in 21-hydroxylase deficiency, excess fetal androgen production causes
prenatal virilization of females, resulting in ambiguous external genitalia
with normal female internal reproductive organs. In newborn males
with 11β-hydroxylase deficiency, the external genitalia may be normal, but in either
sex, virilization ensues postnatally if the disorder is untreated. Deficiency of 11β-hydroxylase results in rising levels of the 11-deoxysteroids, 11-deoxycortisol (compound
S), and 11-deoxycorticosterone (DOC), a moderately
potent salt-retaining steroid. The elevated DOC causes sodium retention, plasma
volume expansion, and suppression of PRA. Indeed, suppressed
PRA is considered a hallmark of this defect, but exceptions have been
noted.139 If present, hypertension is the single clinical feature of this disorder
that distinguishes it from 21-hydroxylase deficiency. Hypokalemia and
alkalosis resulting from mineralocorticoid hormone excess are inconstant
features of this form of CAH. It is not entirely clear whether DOC
is the agent causing elevation of blood pressure.89 The presence of mineralocorticoid excess and hypertension is not necessarily
proportional to the degree of hypokalemia, nor is there a direct
correlation between the degree of virilization and hypertension.140 Deficiency of 11β-hydroxylase accounts for approximately 5% of the cases of CAH worldwide. Molecular Genetics Deficiency of 11β-hydroxylase activity is inherited in an autosomal recessive manner, but
unlike 21-hydroxylase deficiency, it is not HLA linked.141,142,143 Distinct isoenzymes of the mitochondrial P450 enzyme P450c11 participate
in cortisol and aldosterone synthesis in humans. These isoenzymes, encoded
by two genes CYP11B1 and CYP11B2, are 93%identical and located
on the long arm of chromosome 8.144,145 In the zona fasciculata, 11β-hydroxylation of 11-deoxycortisol yields cortisol in a single step. In
the zona glomerulosa, P450c11 hydroxylates DOC to corticosterone and
has 18-hydroxylase and 18-oxidase activities, converting corticosterone
to 18-hydroxycorticosterone and then aldosterone.146 Errors in these steps can lead to aldosterone synthase deficiency, types
I and II. In the zona fasciculata, there is some 18-hydroxylation of
DOC and corticosterone, but no aldosterone synthesis. Studies of patients
with defective cortisol or aldosterone synthesis caused by respective
deficiencies in 11β-hydroxylase and aldosterone synthase type II activities have confirmed
the hypothesis that the isoenzyme encoded by CYP11B1 synthesizes cortisol
in the zona fasciculata, whereas the isoenzyme encoded by CYP11B2 synthesizes
aldosterone in the zona glomerulosa.147,148 Mutations in the zona fasciculata gene result in defective cortisol synthesis
and hypertension caused by elevated DOC levels, whereas mutations
in the zona glomerulosa gene result in defective aldosterone synthesis
and salt wasting. Many patients in the Moroccan-Israeli population
share the same mutation, Arg-448-His, in CYP11B1, which encodes a defective
enzyme.149 Among the number of known mutations,150,151,152,153,154,155,156 most are clustered in exons 6 to 8. This clustering may reflect the location
of functionally important amino acid residues within the enzyme
or an increased tendency to develop mutations within this region of the
gene. Nonclassic 11 β -Hydroxylase Deficiency Mild, nonclassic, and even asymptomatic forms of 11β-hydroxylase deficiency have been reported157,158,159,160,161,162 and may represent allelic variants. Elevated serum 17-OHP:cortisol or 11-deoxycortisol:cortisol
ratios are seen in some heterozygotes, but in
one study of obligate heterozygote parents, no consistent biochemical
defect could be established in the base line state or after ACTH stimulation.163 It appears that hormonal values for carriers of (presumed) mild-deficiency
alleles overlap with the normal range. If this disorder follows the model of 21-hydroxylase deficiency in which
structural gene mutations have been correlated with disease, then it
is anticipated that mutations in the 11β-hydroxylase structural gene can produce the spectrum of clinical symptoms
seen for 11β-hydroxylase deficiency (hypertension and virilization) and for 18-hydroxylase
deficiency and 18-dehydrogenase deficiency (salt wasting without
virilization). Further work is required, however, to determine whether
the clinical polymorphism results from genetic allelism. |