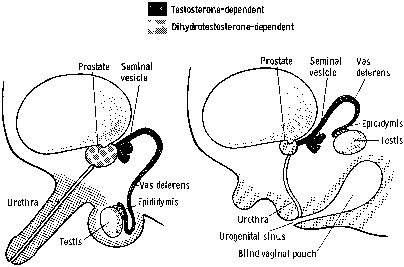
Steroid 5α-reductase isozymes located in the microsomes of the cell are nicotinamide-adenine dinucleotide phosphate (NADPH)-dependent proteins that reduce the double bond at the 4-5 position of a variety of c19 and c21 steroids. These isozymes convert testosterone to the more potent androgen, dihydrotestosterone (DHT). Testosterone and DHT bind to the intracellular androgen receptor (a member of the nuclear steroid/thyroid hormone receptor superfamily) and interact with a cognate androgen DNA response element to regulate target gene expression.1,2 Although testosterone and DHT interact with the same androgen receptor, they produce distinct biological responses in certain conditions.3 The molecular mechanism for this is unclear even though DHT has been reported to bind to the androgen receptor more avidly than testosterone,4 and the DHT-receptor complex is more efficiently transformed to the DNA-binding state than is the testosterone-receptor complex.5
In the early 1960s, it was theorized that multiple 5α-reductase isozymes existed.6 In 1975 and 1976, Moore and Wilson7,8 detected different pH optima for 5α-reductase activity in genital and nongenital skin. In the genital skin, the major enzymatic activity had a narrow, acidic pH optimum of 5.5,8 which was found to be low in the genital skin of male pseudohermaphrodites with 5α-reductase deficiency.7,9 Another enzymatic activity had a neutral to alkaline pH (pH 7 to 9), which was present in both normal genital and nongenital skin and was found to be normal in the genital skin of male pseudohermaphrodites with 5α-reductase deficiency. Kinetic analysis of 5α-reductase activity in the epithelium and stroma of the prostate also suggested that there were different 5α-reductase activities.10,11 Pharmacologic studies of specific 5α-reductase inhibitors provided further evidence of multiple 5α-reductase isozymes.12 Although many attempts were made to purify 5α-reductases, they were not successful because of the extreme insolubility of the protein.
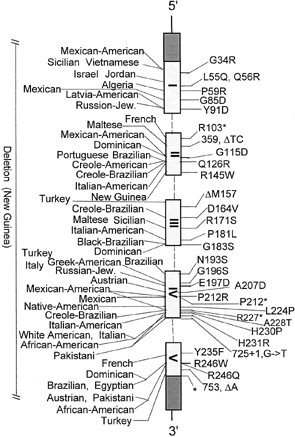
In the early 1990s, two genes encoding two isozymes13,14,15,16 were cloned using expression cloning technology, steroid 5α-reductase type 1 (gene symbol:SRD5A1) and steroid 5α-reductase type 2 (gene symbol:SRD5A2). Mutations in the 5α-reductase-2 gene are responsible for male pseudohermaphrodi-tism from 5α-reductase deficiency.14,17 The characteristics of the two 5α-reductase isozymes are summarized in Table 1.
Table 1. Comparison of Human 5α-Reductase Isozymes
| Type 1 | Type 2 |
Gene structure | 5 exons, 4 introns | 5 exons, 4 introns |
Gene, chromosome location | SRD5A1, 5p15 | SRD5A2, 2p23 |
Size | 259 amino acids | 254 amino acids |
| Mr = 29,462 | Mr = 28,398 |
Tissue distribution | Liver, nongenital skin, | Prostate, epididymis, |
| prostate, brain, ovary, testis | seminal vesicle, genital |
|
| skin, liver, uterus, breast, |
|
| hair follicle, placenta, testis |
PH optima | Neutral to basic | Acidic or neutral |
Prostate level | Low | High |
Activity in 5α-reductase deficiency | Normal | Mutated |
Finasteride inhibition | Ki | Ki = 3–5 nM |
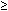 300 nM
300 nMThe human 5α-reductase-2 gene has five exons and four introns, encodes a highly hydrophobic 254 amino acid protein with the molecular weight of approximately 28.4 kilodaltons (kd), and maps to the short arm of chromosome 2 band 23.14,16 The type 2 isozyme has a much higher affinity for testosterone (apparent Km = 4 to 50 nM) than type 1 isozyme (Km = 1 to 5 μM); however, the apparent Km (3 to 10 μM)for NADPH cofactor is similar in both isozymes. The type-2 isozyme is sensitive to finasteride, a 5α-reductase inhibitor. The type 2 isozyme has an acidic pH optimum in enzymatic assays7,14,16; however, it may work in a neutral pH optimum in its native state.18 The type 2 isozyme is expressed in the external genital tissues early in gestation.19 In adulthood, its expression in prostate, genital skin, epididymis, seminal vesicle, and liver is relatively high, whereas it is quite low in other tissues. Recently, it has been reported that this isozyme may also be expressed in the ovary and hair follicles.20,21
The 5α-reductase-1 gene is normal in male pseudohermaphrodites with 5α-reductase deficiency.14 It also has 5 exons and 4 introns and is located on the short arm of chromosome 5 band 15. This isozyme has 259 highly hydrophobic amino acids with a molecular weight approximately 29.5 kd.16 There is approximately 50% homology between human type-1 and type-2 isozymes in amino acid compositions. 5α-reductase-1 has a broad alkaline pH optimum, a lower substrate affinity, and a lower sensitivity to finasteride inhibition.16,17 This isozyme is expressed in nongenital skin, liver, and certain brain regions; however, its presence in the prostate, genital skin, epididymis, seminal vesicle, testis, adrenals, and kidney is low. Its expression is detected at birth in the liver and nongenital skin and is present throughout life, although its expression in embryonic tissues is low. The physiologic function of 5α-reductase-1 is still obscure. It may play a significant role in parturition.22