46,XX/46,XY About 30 cytogenetically documented 46,XX/46,XY true hermaphrodites have
been reported. True hermaphroditism of the type 46,XX/46,XY is rarer
than 46,XX true hermaphroditism but about as common as the 46,XY type. Not
all 46,XX/46,XY individuals are true hermaphrodites; some are phenotypically
normal or have gonadal dysgenesis or male pseudohermaphroditism. In
fact, fusion of XX and XY mouse zygotes usually produces not
true hermaphroditism but, rather, clearly male or clearly female offspring6. ETIOLOGY. A 46,XX/46,XY chromosome complement could arise by 1) nondisjunction involving
a 47,XXY or 46,XY zygote, with loss of certain cell lines and
retention of others, or 2) chimerism. The etiology of 46,XX/46,XY true hermaphroditism in humans is usually suspected
to be chimerism, rather than nondisjunction. Chimerism connotes
the presence in a single individual of cells derived from different
zygotes. The phenomenon is proved by detection of two populations of
cells, usually erythrocytes with different blood types, in a single individual. At
least three types of chimerism are known: 1) blood chimerism (interchange
of blood cells between co-twins through placental anastomoses), 2) transplacental chimerism (interchange of fetal and maternal
blood cells), and 3) whole-body chimerism. Whole-body chimerism, presumably
the type of chimerism responsible for some 46,XX/46,XY true
hermaphrodites, could result from 1) fertilization of both an ovum and
its polar body, 2) fertilization of each of two ova contained within
a single binucleated follicle, 3) fertilization of ova derived from different
follicles, followed by fusion, or 4) other related phenomena. Whole-body
chimerism can be assumed if two or more genotypes are 1) present
in nonhematogenous tissues (skin or gonads) or 2) persist in hematogenous
tissues. The frequency of whole-body chimerism may be underestimated, since
chimeric individuals are usually ascertained because of
abnormal sexual development (e.g., true hermaphroditism). Thus, almost
all recognized whole-body chimeras are heterosexual, although one might
expect an equal number of isosexual chimeras. That whole-body chimerism
has been detected in phenotypically normal individuals confirms
that a 46,XX/ 46,XY karyotype is not invariably associated with true
hermaphroditism. In any case, it is assumed that presence of ovarian as well as testicular
tissue reflects products elaborated by the Y and not elaborated by
the X--probably H-Y antigen. Indeed, Winters et al7 report that the testicular but not the ovarian portion of an ovotestis
contains H-Y antigen. DIAGNOSIS. Cytogenetic Data. Ascertainment of 46,XX/46,XY true hermaphrodites has been accomplished
by studies restricted to lymphocytes as well as by studies of both lymphocytes
and other tissues (e.g., skin or gonadal fibroblasts). In 3 cases, cytogenetic
studies limited to lymphocytes would have failed to
detect more than one line. If additional tissues are studied, the minority
cell line is most likely to be detected in gonadal fibroblasts and
relatively less likely to be detected in skin fibroblasts. In addition, the
proportion of 46,XY cells in lymphocytes bears no ostensible
relationship to gonadal status. That is, a relatively high proportion
of 46,XY cells in lymphocytes does not necessarily indicate that the gonadal
tissue is predominantly testicular, nor, conversely, does a high
proportion of 46,XX cells indicate that gonadal tissue is predominantly
ovarian. The clinical significance of these observations is that analysis
of multiple tissues is the only way to detect certain 46,XX/46,XY
true hermaphrodites, although many can be detected by analyzing lymphocytes
alone. Phenotype. The external genitalia of 46,XX/46,XY true hermaphrodites are usually
either ambiguous or sufficiently masculinized to suggest to attending
physicians that the sex of rearing should be male. A female sex of rearing
was chosen in only 3 of 13 cases. Even if reared as males, affected
individuals usually show perineal or penoscrotal hypospadias and incomplete
labioscrotal fusion. The distribution of gonadal tissue is shown in Table 1. No single type predominates. Although the chromosome constitution of
gonadal tissue has been determined in only a few cases, 46,XY cells appear
more likely to be present in a testis or ovotestis than in an ovary. Additional
studies, however, are necessary to confirm this hypothesis. In
aggregate, the right gonad is much more likely to contain testicular
tissue than the left--a characteristic that 46,XX/46,XY true hermaphrodites
share with other true hermaphrodites. There is no obvious
explanation for the predilection for testicular development to take place
on the right, but it is interesting to note that in many species, the
right gonad is vestigial. Furthermore, following extirpation of the
left ovary from newborn hens, the right gonad differentiates into a
testis8. TABLE 1. Clinical Characteristics of True Hermaphrodites With Various Chromosomal
Complements
| Sex of | | | | |
Chromosomal | Rearing | | Unicornuate | | Total |
Complement | (Female/ | Uterus | or Bicornuate | Gonadal Distribution (Left:Right) | Informative |
(Total Cases)* | Total)† | Present‡ | Uterus§ | OT:OT | O:T | T:O | O:OT | OT:O | T:OT | OT:T | Other | Cases |
46,XX/46,XY | | | | | | | | | | | | |
(28) | 3/13 | 20/22 | 1/20 | 6 | 7 | 1 | 4 | 4 | 1 | 2 | 0 | 24 |
46,XX/47,XXY | | | | | | | | | | | | |
(13) | 3/10 | 8/10 | 2/8 | 4 | 2 | 3 | 1 | 0 | 0 | 0 | 2 | 12 |
45,X/46,XY | | | | | | | | | | | | |
(11) | 3/8 | 5/5 | 0/5 | 0 | 4 | 1 | 0 | 2 | 0 | 1 | 0 | 8 |
46,XY (37) | 2/17 | 17/19 | 2/17 | 0 | 12 | 4 | 4 | 0 | 4 | 2 | 2 | 28 |
Familial | | | | | | | | | | | | |
46,XX (8)|| | 1/10 | 2/10 | 0/2 | 6 | 0 | 0 | 3 | 0 | 1 | 0 | 0 | 10 |
Nonfamilial | | | | | | | | | | | | |
46,XX (131) | 34/89 | 90/108 | 18/90 | 23 | 24 | 8 | 36 | 9 | 2 | 10 | 5 | 117 |
O, ovary; T, testis; OT, ovotestis.
*Complete descriptions not available for all cases.
†Includes only those cases in which attending physicians assigned
a sex of rearing.
‡Includes cases in which uterus described as rudimentary.
§Probably an underestimate; descriptions of uteri often incomplete.
||References 20–25; cases of Kasdan et al (28) and Berger et al (29) not
included.
From Simpson JL: True hermaphroditism: Etiology and phenotypic considerations. Birth
Defects 14(6C):9, 1978.
The ovarian and testicular portions of an ovotestis are usually juxtaposed
end-to-end. By definition, both seminiferous tubules and ovarian follicles
are present. Testicular tissue, whether existing as a separate
testis or as one component of an ovotestis, is characterized by relatively
few normal germ cells. The tubules are usually hyalinized and composed
only of Sertoli cells (Fig. 2); Leydig cells may be hyperplastic. Although spermatozoa are rare, one
possible 46,XX/46,XY case9 was fertile and had a sperm count of 20 × 106/ml. By contrast, ovarian tissue often contains numerous primordial follicles
in various developmental stages. That ovarian tissue is often normal
is also evidenced by breast development, cyclic menses, and histologic
evidence of ovulation. However, histologic criteria for diagnosis
introduce some biases favoring presence of normal oocytes in true hermaphrodites. 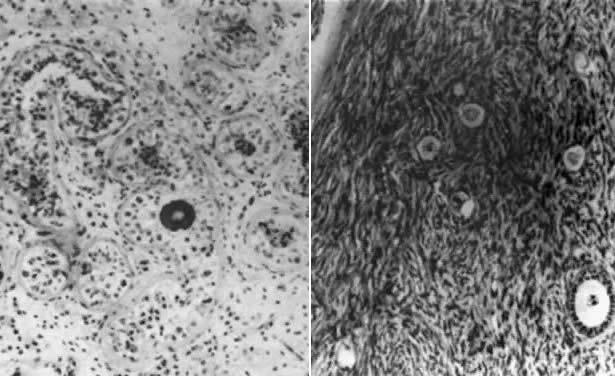 Fig. 2. Photomicrograph of testicular (left) and ovarian (right) portions of an ovotestis of 46,XX/47,XX,r(Y) true hermaphrodite. The ovarian portion contains riot only stroma but
also oocytes.(From Sarto GE, Opitz JM, Inhorn SL: Consideration of sex chromosome abnormalities
in man. in Benirschke K (ed): Comparative Mammalian Cytogenics. New
York: Springer-Verlag, 1969.) Fig. 2. Photomicrograph of testicular (left) and ovarian (right) portions of an ovotestis of 46,XX/47,XX,r(Y) true hermaphrodite. The ovarian portion contains riot only stroma but
also oocytes.(From Sarto GE, Opitz JM, Inhorn SL: Consideration of sex chromosome abnormalities
in man. in Benirschke K (ed): Comparative Mammalian Cytogenics. New
York: Springer-Verlag, 1969.)
|
True hermaphrodites with 46,XX/46,XY karyotypes usually have müllerian
derivatives, namely a uterus and one or more fallopian tubes. Rarely
is the uterus absent, although a few authors comment that uterine
development was rudimentary or “merely a remnant.” Menstruation
occurred in 3 of 7 puberal individuals with a uterus. The presence
or absence of fallopian tubes or wolffian derivatives reflects the
ipsilateral gonad. A fallopian tube is invariably present ipsilateral
to an ovary, whereas a vas deferens, epididymis, and, often, a seminal
vesicle are usually present: ipsilateral to a testis. Either fallopian
tubes or wolffian derivatives may be present on the side of an ovotestis, although
most often a fallopian tube is present. Although fallopian
tubes and wolffian derivatives are usually not both present on the
same side, even on the side of an ovotestis, this combination has been
observed occasionally, contrary to opinions stated by some authors. In 2 cases, gonadal
tumors developed. 46,XX/47,XXY True hermaphroditism associated with a 46,XX/47,XXY chromosome complement
probably results from nondisjunction involving a 46,XY or 47,XXY zygote. The
karyotype can be explained readily by postulating loss of certain
cell lines and retention of others. The possibility of chimerism
has not been excluded, however, or even, in most cases, vigorously pursued. By 1977, 13 cases had been detected, usually on the basis of cultured lymphocytes. The
sex of rearing was chosen by the attending physicians
in 10 cases. The gonadal distribution shows no obvious pattern (Table 1). A uterus was present in 8 of 10 cases, 2 being unicornuate. Prevalences
of bicornuate and unicornuate uteri seem likely to be underestimated, since
the uterus was often not completely described. The occurrence
of unicornuate or bicornuate uteri would be predicted because androgens
and the müllerian inhibitory factor (MIF) influence embryonic
ductal differentiation through local diffusion from the fetal testes. 45,X/46,XY Individuals with a 45,X/46,XY karyotype display a spectrum of phenotypes, ranging
from almost normal males to females indistinguishable from
those with 45,X gonadal dysgenesis and the Turner stigmata1. The different phenotypes presumably reflect different tissue distributions
of 45,X and 46,XY cells. This assumption, however, has not been
proved and, in fact, often cannot be demonstrated, although one should
recall that in gonadal cultures fibroblasts rather than germ cells are
cultured. The phenotype most often associated with 45,X/46,XY mosaicism is mixed
or asymmetric gonadal dysgenesis, characterized by a unilateral streak
gonad and a contralateral testis. In most 45,X/46,XY individuals, no
ovarian follicles are detectable, and hence the diagnosis of true hermaphroditism
is inappropriate. Although 11 45,X/46,XY true hermaphrodites have been reported, only 8 were
completely described. A :male sex of rearing was chosen in 5 of the 8 cases. A
uterus was found in 5 cases. Five of 8 had one ovary and
one testis, and in 4 of these 5, the testis was on the right. No case
had bilateral ovotestes (Table 1). Other Forms of Mosaicism or Chimerism True hermaphroditism has been associated with various other chromosomal
abnormalities, including 45,X/46,XX; 46,XX/47,XYY; 46,XX/46,XY/ 47,XXY; 46,XX/47,XXY/49,XXYYY; 45,X/46,XY/ 47,XYY; and 46,XX/48,XXYY. These
karyotypes probably arise by mitotic nondisjunction, although chimerism
has not been excluded. Because only a few cases have been associated
with a given karyotype, generalizations would be unwise; however, alternating
gonadal hermaphroditism occurs relatively frequently. 46,XY About 40 cases of 46,XY true hermaphroditism have been reported, although
a complete description is not .available for all of them. Over half
were Japanese; 46,XY true hermaphroditism appears to be the most common
type of true hermaphroditism in Japan, in contrast to its relative
rarity outside Asia. A sex of rearing was assigned in 19 cases, and in only 2 cases was the
female role chosen. This indicates that the external genitalia are more
masculine in appearance than in 46,XX/46,XY true hermaphroditism, a
suggestion consistent with published descriptions of external genitalia. Quantitation
of genital virilization, however, is difficult. The gonadal
distribution in Table 1 shows a high frequency of the alternating type, specifically a left ovary
and a right testis. No 46,XY true hermaphrodites definitely had bilateral
ovotestes, although 1 possible case was reported by Sandberg et
al10. A uterus was present in 17 of 19 cases for which complete descriptions
were available; 2 uteri were unicornuate and 1 was bicornuate. Development
of wolffian and müllerian derivatives was similar to that
in other true hermaphrodites. ETIOLOGY. The presence of oocytes in 46,XY true hermaphrodites could result from 1) undetected
chimerism or mosaicism, with 46,XX cells present but not
readily detectable, 2) translocation of ovarian determinants from the
X chromosome to either the Y chromosome or an autosome, or 3) a mutant
gene or genes. Certain phenotypic features in recent reviews1,2 suggest that undetected chimerism or mosaicism is often, although not
necessarily always, the cause of 46,XY true hermaphroditism. For example, in
one survey the frequency of alternating gonads was relatively high--16 of 28 cases (58%)2. Moreover, the gonadal tissue appeared more likely to be testicular than
ovarian, and the external genitalia were relatively more virilized
than in true hermaphrodites with other karyotypes. True hermaphrodites
of type 46,XY were especially likely to differ from 46,XX true hermaphrodites
with respect to gonadal distribution, extent of virilization, and
sex of rearing. The differences observed would be expected if ostensible 46,XY
cases actually had mosaicism or chimerism in which the proportion
of 46,XX cells was relatively small and hence difficult to detect. That
is, if some cells contained the factor responsible for the
true hermaphroditism and others did not, dissimilar gonads might be expected. Few
cytogenetic studies have been extensive enough to exclude
mosaicism or chimerism in 46,XY true hermaphrodites. By contrast, if 1) X-Y interchange, 2) a Y-autosome translocation, or 3) a
mutant allele were present, both gonads would theoretically seem likely
to be morphologically similar (i.e., bilateral ovotestes) because
every cell would presumably possess the factor responsible for abnormal
gonadal development. X-Y or Y-autosome translocations remain theoretic
explanations for 46,XY true hermaphroditism, but no cytogenetic data
support their existence. The relatively high frequency of 46,XY true
hermaphroditism among Japanese suggests presence of a mutant gene, since
differences in racial prevalences are often the first clue to an
underlying genetic etiology. The lack of familial aggregates and the
absence of parental consanguinity, however, fail to support the hypothesis
of recessive inheritance. On the other hand, Milner et al11 reported X-chromatin-negative siblings with true hermaphroditism; additional
cytogenetic studies were unavailable. PATHOLOGY. Two 46,XY true hermaphrodites12,13 developed gonadoblastomas, suggesting that 46,XY true hermaphrodites show
the same predilection for neoplastic transformation as do individuals
with 45,X/46,XY mosaicism or XY gonadal dysgenesis. Breast carcinoma
has also been reported in 2 true hermaphrodites; one was 46,XX14 and the karyotype of the other15 is unknown. 46,XX The karyotype most often detected among true hermaphrodites is 46,XX. About 150 cases
have been reported, including 40 among the Bantu and other
African blacks. A female sex of rearing was chosen in about one third of all 46,XY cases
in which a sex assignment was made--much more often than in the 46,XY
or 46,XX/46,XY groups. In fact, 1 true hermaphrodite16 was delivered of a male infant. Somatic anomalies were occasionally but
not often present. The gonadal distribution most commonly observed was a left ovary and a
right ovotestis. The next most common was bilateral ovotestes (Table 1). Alternating gonadal distribution occurred less frequently in 46,XX true
hermaphrodites than in 46,XY true hermaphrodites. One 46,XY true hermaphrodite
developed a dysgerminoma14, and a second developed a gonadoblastoma17. A uterus was present in 80% of the cases. Although still relatively high, this
frequency is lower than that among 46,XY or 46,XX/46,XY true hermaphrodites. The
uterus was sometimes unicornuate or bicornuate. As
in true hermaphrodites with other karyotypes, a vas deferens and an epididymis
were usually present ipsilateral to a testis, and a fallopian
tube was usually present ipsilateral to an ovotestis. Occasionally, however, only
wolffian derivatives or both müllerian and wolffian
derivatives were present. Van Niekerk14,18 observed that 1) the fimbriated end of the fallopian tube was often occluded
and 2) the cervical canal was often obliterated or characterized
by squamous metaplasia. These observations have apparently not often
been made by others19, leading one to wonder whether Van Niekerk's sample (all Bantu) indicates
genetic heterogeneity among 46,XX true hermaphrodites. In summary, 46,XX true hermaphrodites are more likely than other true hermaphrodites
to have either a left ovary and a right ovotestis, or bilateral
ovotestes. In particular, alternating hermaphroditism is less
common than in 46,XY true hermaphrodites, and a uterus is less likely
to be present. In contrast to the apparent lack of heritability in other forms of true
hermaphroditism, several familial aggregates of 46,XX true hermaphroditism
have been reported. In 4 families20,21,22,23,24,25, multiple siblings had 46,XX true hermaphroditism. In contrast to 46,XX/46,XY
or 46,XY true hermaphrodites, a uterus was present in only 2 of 10 patients, both
in the same family. A male sex of rearing was chosen
in 9 of 10 cases; puberal development was similar to that in other
true hermaphrodites. These families indicate that at least one form of 46,XX
true hermaphroditism results from an autosomal recessive gene or
a factor that acts in similar fashion. ETIOLOGY. The presence of testicular tissue in individuals who apparently lack a
Y chromosome could be explained by 1) undetected mosaicism or chimerism, 2) translocation
of the Y testicular determinants to the X chromosome, 3) translocation
of Y testicular determinants to an autosome, or 4) a
mutant allele, or alleles. Let us consider each hypothesis. First, in some cases undetected 46,XY cells are doubtless present. Cultures
of testicular fibroblasts, however, sometimes show no 46,XY cells, although
the complement in germ cells is unknown. In addition, cases
with alternating hermaphroditism are those that theoretically appear
most likely to have chimerism or mosaicism. Thus, the relatively low frequency
of alternating gonads in 46,XX true hermaphrodites suggests that
it would be unwise to assume in all cases the presence of undetected
mosaicism. Second, the presence of testicular tissue in 46,XX true hermaphrodites
could result from translocation of Y-linked testicular determinants to
an X chromosome or to an autosome (X-Y or X-autosome translocation). Supporting
this hypothesis are 1) anomalous familial distributions of
Xg alleles and 2) presence of H-Y antigen in most cases26,27. Presence of H-Y indicates Y-autosome or Y-X translocation of the Y-testicular
determinant. Translocation of testicular determinants does not
necessarily explain the true hermaphroditism phenotype; consideration
of the following shows that other phenomena must simultaneously be postulated. If 1) H-Y
antigen is the gene product of the testicular determinant
and 2) the H-Y locus is translocated to an X chromosome or to
an autosome, why is testicular differentiation abnormal in individuals
who carry the translocation? That is, why are H-Y antigen-positive 46,XX
true hermaphrodites not normal males? Wachtel27 notes that H-Y titer in these patients is lower than in 46,XY males. Thus, quantitative
decrease in H-Y could be responsible. Finally, the occurrence of familial aggregates suggests that some sporadic
cases of 46,XX true hermaphroditism result from a mutant gene. Indeed, both
dominant and recessive sex-reversal genes exist in animals1,2; lending credence to hypotheses that such genes may exist in humans. Such
sex-reversed animals have been positive for H-Y antigens27. On the other hand, an X-Y or Y-autosome translocation in a paternal gonad
could also explain sibship aggregates. Indeed, in one family13, both affected siblings were positive for H-Y antigen, suggesting either 1) X-Y
or Y-autosome translocation in a line present in the father's
testes or 2) activation of H-Y antigen through a mutation. The
postulated mutant could thus interfere with H-Y antigen, or it could repress
a male-determining locus controlled in regulatory fashion by the
Y-testicular determinant(s). Unfortunately, few authors publish adequate
genealogic data, and no H-Y data are available. It seems unlikely
that all sporadic 46,XX true hermaphrodites could be explained on the
basis of a mutant autosomal recessive allele, but some sporadic cases, especially
those with bilateral ovotestes and no uterus, might result
from such factors. In addition to a recessive gene, an autosomal dominant sex-reversal gene
might exist. Kasdan et al29 reported a kindred in which the proband was a 46,XX true hermaphrodite; both
a “male” sibling and a paternal “uncle” showed
complete sex reversal (46,XX males). In this family there thus
appeared to be segregating a dominant factor capable of causing complete
sex reversal (46,XX male), or if perhaps less completely expressed, 46,XX
true hermaphroditism. A 46,XX male proband and probably a 46,XX
true hermaphrodite sibling were also present in the family reported
by Berger et al30; the true hermaphrodite sibling, said to be 46,XX/ 46,XY, had only two 46,XY
cells studied prior to availability of banding techniques. Thus, it
is uncertain whether the complement was 46,XX or 46,XX/46,XY. In
addition, Chapelle et al reported a kindred in which 2 paternal second
cousins were 46,XX males31,32. |
