XX Gonadal Dysgenesis Without Anomalies
The forms of 46,XX gonadal dysgenesis not associated with somatic anomalies is most often inherited in autosomal recessive fashion. Affected individuals are normal in stature (mean height, 165 cm)1; somatic features of Turner’s stigmata are usually absent. Presence of consanguinity pointed to autosomal recessive inheritance decades ago. Segregation analysis by the author and colleagues revealed the segregation ratio to be 0.16 for female sibs.2 Thus, two thirds of gonadal dysgenesis in 46,XX individuals is genetic. The nongenetic phenocopies could be caused by infection, infarction, infiltrative or autoimmune phenomena (see Chapter 5-88 for further discussion).
Of clinical relevance is the variable expressivity in XX gonadal dysgenesis. In some families one sib has streak gonads, whereas another had primary amenorrhea and extreme ovarian hypoplasia (presence of a few oocytes).1,2,3,4,5,6 If the mutant gene responsible for XX gonadal dysgenesis were capable of variable expression, that gene may be responsible for some sporadic cases of premature ovarian failure (POF).
The mechanism underlying failure of germ-cell persistence in XX gonadal dysgenesis without somatic anomalies is unknown, but there are many candidate genes. Any abnormality of meiosis could be manifested as ovarian failure and infertility in otherwise normal women. Other mechanisms by which mutant genes could act include interference with germ-cell migration, connective tissue abnormalities, or gonadotropin receptor abnormalities.
Identifying the specific autosomal genes responsible for the various forms of XX gonadal dysgenesis has been difficult. In many monogenic disorders efforts toward positional cloning are facilitated by the fortuitous family in which an autosomal translocation cosegregates with the disorder. Sporadic cases of XX gonadal dysgenesis have long been associated with reciprocal autosomal translocations, but there seems to be little reproducibility among autosomes involved. Genome-wide sib-pair analysis using polymorphic DNA markers (dinucleotide repeats; single nucleotide polymorphism) should theoretically identify chromosomal region(s) worthy of sequencing. Indeed, this method was used successfully in Finland to elucidate the form of XX gonadal dysgenesis caused by follicle-stimulating hormone receptor (FSHR) mutation, which we shall discuss below.5,7 Construction of cDNA libraries of ovarian specific genes represent a more omnibus approach, combined with use of gene knockout technology in the mouse and sequencing of candidate genes in the human. Many mouse genes are of potential relevance (Table 2), and perturbations in their human homologues can be sought. Many of these genes may only manifest as ovarian failure, in contrast to predictions that other organ systems would also be disturbed. An example is bone morphogenetic protein (bmp). The conclusion is that ovarian meiosis is easily perturbed, secondarily leading to germ-cell failure.
TABLE 2. Selected Mouse Models of Ovarian Failure
|
Mutant Mouse/Transgene |
Human Locus |
Function |
Mouse Phenotype |
|
Prenatal ovarian failure defects |
|
|
|
|
Zinc finger X (zfx) knockout |
Xp22.1-p21.3 |
Transcription factor |
Reduced number of oocytes, infertility, short stature [Luoh and colleagues98] |
|
Germ-cell deficient (gcd) unknown |
Unknown |
Unknown gene, generated by transgene insertion |
Lack of germ cells in early as day 11.5 of embryonic development [Pellas and colleagues99] |
|
White spotting (W) |
4p11-q12 |
Tyrosine kinase receptor |
Reduced pigmentation, anemia, lack of germ cells [Manové and colleagues100] |
|
Steel (Sl) |
12q22 |
Mast cell growth factor |
Reduced pigmentation, anemia, lack of germ cells [Matsui and colleagues101] |
|
Steroidogenic Factor 1 (SF-1) knockout |
9q33 |
Nuclear receptor factor |
Ovarian agenesis, XY sex reversal, adrenal agenesis [Luo and colleagues102] |
|
mutS (E. coli) homolog 5 (MSH 5) |
6p21.3 |
DNA mismatch repair |
Absence of ovarian structure, normal oviducts and uteri [Edelmann and colleagues103] |
|
Beta cell leukemia/lymphoma 2(Bcl-2) knockout |
18q21.3 |
Cell death repressor protein |
Accelerated atresia of primordial follicles [Ratts and colleagues104] |
|
Factor in germline α (Figα) |
Unknown |
Transcription factor |
Females lack primordial follicles, males are normal [Soyal and colleagues105] |
|
Postnatal ovarian failure defects |
|
|
|
|
Growth differentiation factor 9 (GDF9) |
5 |
Oocyte secreted growth factor |
Block in prenatal follicle development, infertility [Dong and colleagues106] |
|
Follicle-stimulating hormone β |
11p13 |
Glycoprotein hormone |
Female infertility, block of folliculogenesis before antral stage subunit ββ knockout (FSHβ) [Kumar and colleagues107] |
|
FSH receptor (FSHR) knockout |
2p21-p16 |
Hormone receptor |
Female infertility, block in folliculogenesis before antral stage [Dierich and colleagues108] |
|
Estrogen receptor α (Erα) knockout |
11q12 |
Hormone receptor |
Absent corpora lutea, arrest of preovulatory follicle maturation [Lubahn and colleagues109] |
|
Connexin 37 knockout |
1p35.1 |
Gap junction |
Lack of Graafian follicles, failure to ovulate [Simon and colleagues110] |
|
mutL (E. coli) homologue 1 (MLH1) |
3p21.3 |
DNA repair enzyme |
Failure to complete meiosis II, normal estrous cycle [Edelmann and colleagues111] |
|
Zona matrix protein 3 (mZP3) knockout |
7q11.23 |
Zona pellucida |
Infertility, oocytes lack zona pellucida [Rankin and colleagues112] |
|
Nerve growth factor-induced gene NGFI- A knockout |
2q32.3-q33 |
Transcription factor |
Lack of corpora lutea, suppressed luteinizing hormone levels [Topilko and colleagues113] |
Perrault Syndrome (XX Gonadal Dysgenesis with Neurosensory Deafness)
XX gonadal dysgenesis associated with neurosensory deafness is called Perrault syndrome.8 Perrault syndrome is inherited in autosomal recessive fashion.1,9,10,11,12 Endocrine features seem identical to XX gonadal dysgenesis without deafness.
XX Gonadal Dysgenesis Caused by Follicle Stimulating Hormone Receptor Mutation (Val566 A1a)
This disorder is found predominantly in Finland, where Aittomaki and colleagues5,7 searched hospitals and cytogenetic laboratories throughout the country to identify 75 subjects having the XX gonadal dysgenesis phenotype. Diagnostic criteria consists of 46,XX women with primary or secondary amenorrhea whose serum FSH was 40 MIU/mL or more. The 75 included 57 sporadic cases and 18 cases having an affected relative (7 different families). Most resided in north central Finland, a more sparsely populated part of the country. Prevalence was 1 per 8300 liveborn Finnish females. This relatively high incidence is attributed to a founder effect. The segregation ratio of 0.23 for female sibs was almost identical with theoretical expectations for autosomal recessive inheritance. This is consistent with a high consanguinity rate (12%).
Sib pair analysis using polymorphic DNA markers first localized the gene to chromosome 2p, a region previously known to contain the genes for both the FSHR and the luteinizing hormone receptor (LHR). One specific mutation (Val to Ala) in exon 7 of the FSHR gene was found in 6 families5,7 (Fig. 1). This cytosine-thymidine transition (called C566T) was later found in an additional 6 families.7,13
|
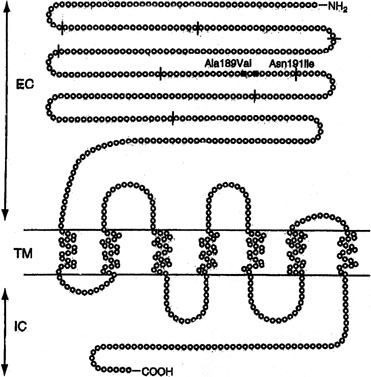
Not all Finnish XX gonadal dysgenesis cases show the C566T mutation. Thus, the C566T-negative cases in Finland could represent the previously discussed condition XX gonadal dysgenesis with no somatic anomalies. Indeed, the C566T mutation is only rarely being detected in women with 46,XX ovarian failure who reside outside Finland. In the United States, Layman and colleagues14 failed to find a mutation in FSHR gene in 35 46,XX women having hypergonadotopic hypogonadism (15 with primary amenorrhea; 20 with secondary amenorrhea). Negative findings were reported in 46,XX POF or primary amenorrhea cases from Germany,15 Brazil,16 and Mexico.17 The last report analyzed all exons of FSHR. Liu and colleagues18 found no FSHR abnormalities in one multigenerational U.S. POF family, four sporadic POF cases and two other hypergonadotopic hypogonadism cases.
Aittomaki and colleagues13 compared the phenotype of Finnish C566T XX gonadal dysgenesis (C566T) with non-C566T XX gonadal dysgenesis. The former were more likely to have ovarian follicles by ultrasound. C566T XX gonadal dysgenesis thus showed at least some of the features gynecologists have long predicted for a gonadotropin resistance disorder (so-called Savage syndrome). In general, however, the phenotype found by Aittomaki5 was unexpected, bilateral streak gonads being found. FSHR (knockout) mice similarly show failure of oogenesis; thus, the necessity of FSH for progression of oogenesis is clear.19 Interestingly, FSH is less pivotal for spermatogenesis.20
Inactivating Luteinizing Hormone Receptor Mutation (46,XX)
Another trophic hormone receptor gene, the perturbation of which causes XX gonadal dysgenesis, is the LHR. The gene is 75 kd in length and consists of 17 exons. Located on 2p near FSHR, the first 10 exons in LHR are extracellular; the last 6 are intracellular, and the 11th, transmembrane. LHR mutations have been reported predominately in 46,XY individuals, where the phenotype may extend to complete LH resistance and XY gender reversal (female). 46,XX cases are more rare than 46,XY cases, and have occurred only in sibships in which an affected 46,XY male had Leydig cell hypoplasia (XY gender reversal).
46,XX women with LHR mutations show oligomenorrhea or, less often, primary amenorrhea. Ovulation does not occur. Gametogenesis proceeds until the preovulatory stage, but not beyond. This is consistent with findings in the mouse knockout model.
Mutations in LHR are molecularly heterogeneous. Most mutations have been found in the transmembrane domain (exon). Latronico and colleagues21 reported primary amenorrhea in a 22-year-old woman whose family also included three affected males (46,XY), like the 46,XX sib homozygous for a nonsense mutation at codon 554 (Arg554ter). The resulting stop codon produced a truncated protein. The affected 46,XX female showed breast development but only a single episode of menstrual bleeding at age 20 years; LH was 37 mIU/mL, FSH 9 mIU/mL. The mutation reduced signal transduction activity of the LHR gene.
Toledo and colleagues22 studied the 46,XX female sib of two 46,XY affected males reported by Cramer and colleagues.23 The sister showed elevated gonadotropins but anatomically normal ovaries. The mutation was Ala593Pro. Two sisters reported by Laue and colleagues24 had the nonsense mutation cys545ter in exon 11; LHR function was lost. The father but not the mother had the mutation. The authors found a dominant negative effect, but more likely the mutant allele transmitted from the probably heterozygous mother has simply not yet been detected.
Cerebellar Ataxia and XX Gonadal Dysgenesis
XX gonadal dysgenesis can be found in association with a heterogeneous group of cerebellar ataxias. The hereditary ataxias are confusing nosologically, principally because of ill-defined diagnostic criteria and lack of direct access to the cerebellum. Forms of ataxia characterized by hypogonadotrophic hypogonadism also exist, but these are not considered here.
The association between hypergonadotrophic hypogonadism and ataxia was first reported by Skre and colleagues.25 In one family, a 16-year old girl was affected, whereas in the other family there were three affected sisters. In the sporadic case and in one of the three sibs, ataxia was observed shortly after birth; in the two other sibs, age of onset occurred later in childhood. Cataracts were present in all individuals described by Skre and colleagues.25
Hypergonadotropic hypogonadism and ataxia was subsequently reported by De Michele and colleagues,26 Linssen and colleagues,27 Gottschalk and colleagues,28 Fryns and colleagues,29 Nishi and colleagues,12 and Amor and colleagues.30 In these various reports the clinical features of ataxia have varied. Findings similar to those of Skre and colleagues25 were reported by De Michele and colleagues,26 Nishi and colleagues,12 and Amor and colleagues30; ataxia was usually not progressive. Mitochondrial enzymopothy was evident in one case reported by De Michele and colleagues,26 but in no other cases were mitochondrial studies conducted. Only Skre and colleagues25 observed cataracts and only Linssen and colleagues27 observed amelogenesis. Neurosensory deafness was reported by Amor and colleagues30 Mental retardation is likewise variable.30
In conclusion, a single mutant gene is unlikely to explain every single case of XX gonadal dysgenesis and cerebella ataxia. However, not every family need to be unique.
XX Gonadal Dysgenesis and Multiple Malformation Syndromes
Several other pleiotropic genes cause XX gonadal dysgenesis and various somatic features. All are rare and in perhaps even unique to a specific family. Table 3 lists these syndromes: XX gonadal dysgenesis, microcephaly, and arachnodactyly31; XX gonadal dysgenesis, cardiomyopathy, blepharoptosis, and broad nasal bridge30,32; XX gonadal dysgenesis and epibulbar dermoid33; XX gonadal dysgenesis, short stature and metabolic acidosis.34 Assuming mendelian etiology, these disorders are presumably autosomal recessive. However, subtle chromosomal rearrangements cannot be excluded.
TABLE 3. Malformation Syndromes with 46,XX Gonadal Dysgenesis
Somatic Features | Reference | Etiology |
Cerebellar ataxia, sensori-neural deafness, other somatic features | Autosomal recessive, heterogeneous | |
Microcephaly, arachnodactyly | Maximilian and colleagues31 | Autosomal recessive |
Epibulbar dermoids | Quayle and Copeland33 | Autosomal recessive |
Short stature and metabolic acidosis | Autosomal recessive | |
Blepharophimosis-ptosis-epicanthus | Autosomal dominant (FOXL2) | |
Renal parenchymal disease, ovarian failure (46,XX Frasier syndrome) Wilms tumor and genital ambiguity (Denys-Drash syndrome) different perturbations involving same gene (WT1). | Bailey and colleagues35 (Frasier syndrome) | Autosomal dominant (WT1) |
Dilated cardiomyopathy, mental retardation, bleparoptosis (Malouf syndrome) | Autosomal recessive |
In all these syndromes, an underlying biologic question is whether the ostensibly pleiotropic gene(s) causes both somatic anomalies and ovarian failure? Alternatively, could the somatic and gonadal phenotypes merely reflect closely linked genes, that is, a contiguous gene syndrome? Irrespective, do these purported genes play roles in normal ovarian differentiation, or does their perturbation merely cause ovarian failure secondary to generalized somatic disturbance?
Pleiotropic Mendelian Disorders Showing Ovarian Failure
Primary ovarian failure is observed frequently in well-established and not necessarily uncommon in mendelian disorders. All are characterized by distinct somatic features (Table 4). Pleiotrophy for these mutant genes allows clear distinction from disorders previously discussed.
Disorder | Somatic Features | Ovarian Anomalies | Etiology |
Ataxia-telangiectasia | Cerebella ataxia, multiple telangiectasias (eyes, ears, flexa surface of extremities), immunodeficiency, chromosomal breakage, malignancy, x-ray hypersensitivity | “Complete absence of ovaries”, “absence of primary follicles” [Zadik and colleagues16; Waldmann and colleagues116] | |
Carbohydrate-deficient glycoprotein syndrome, type 1 (phosphomannonutase deficiency) [Matthijs and colleagues48] | Neurologic abnormalities (e.g., unscheduled eye movements), ataxia, hypotonial/hyporeflexia strokes, joint cartractures | Ovarian failure (hypogonadism) [Kristiansson and colleagues50] | Autosomal recessive |
Cockayne Syndrome [Nance and Berry117] | Dwarfism, microcephaly, mental retardation, pigmentary retinopathy and photosensitivity, premature senility. Sensitivity to ultraviolet light | Ovarian atrophy and fibrosis [Sugarman and colleagues118] | Autosomal recessive |
Galactosemia (galactose-l-phosphate uridyl transferase deficiency) (GALT) | Hepatic failure with cirrhosis, renal failure, cataracts, cardiac failure | Ovarian failure with streak gonads [Kaufman and colleagues41; Waggoner and colleagues42; Levey and colleagues43] | Autosomal recessive |
Martsolf Syndrome [Martsolf and colleagues119] | Short stature, microbrachycephaly, cataracts, abnormal facies with relative prognathism due to maxillary hypoplasia | “primary hypogonadism” [Harbord and colleagues120] [Hennekam and colleagues121] | Autosomal recessive |
Nijmegen syndrome [Weemaes and colleagues122] | Chromosomal instability, immunodeficiency, hypersensity to ionizing radiation, malignancy | Ovarian failure (primary) [Conley and colleagues123; Chrzanowska and colleagues124] | Autosomal recessive |
Rothmund-Thompson syndrome [Hall and colleagues125] | Skin abnormalities (telangiectasia, erythrema irregular pigmentation), short statue, cataracts, sparse hair, small hands and feet, mental retardation, osteosarcoma | Ovarian failure (primary hypogonadism or delayed puberty) [Starr and colleagues126] | Autosomal recessive |
Werner syndrome [Goto and colleagues127] | Short stature, premature senility, skin changes (scleroderma) | Ovarian failure [Goto and colleagues127] | Autosomal recessive |
Of special note is Denys-Drash syndrome and Frasier syndrome, which are caused by mutations in WT-1 (Wilms’ tumor-1), a gene located on 11p. WT-1 mutations can cause either 46,XY gender reversal or 46,XY genital ambiguity. One 46,XX individual with Frasier syndrome has been reported.35 This woman manifested not only the renal parenchymal disease characteristic of Frasier syndrome, but also primary amenorrhea and ovarian failure. Gonadal failure in 46,XX Frasier syndrome could easily pass unappreciated if the primary amenorrhea is assumed to occur secondary to azotemia.
Germ-Cell Failure in Male (46,XY) and Female (46,XX) Sibs
In several sibships, male and female sibs have shown germinal cell failure. Affected 46,XX females showed streak gonads, whereas 46,XY affected males showed germ-cell aplasia (Sertoli-cell–only syndrome). In two families, parents were consanguineous, and in each there were no somatic anomalies.36,37 In three other families (see Table 1), characteristic somatic anomalies suggested distinct entities. Hamet and colleagues38 observed germ-cell failure, hypertension, and deafness; Al-Awadi and colleagues39 found germ-cell failure and alopecia; Mikati and colleagues40 reported germ-cell failure, microcephaly and short stature.
In each of these families a single autosomal gene is presumed to affect germ cell development deleteriously in both sexes. This gene(s) could act either at a specific site common to early germ cell development or exert its effect through meiotic breakdown. Elucidating such genes could help explain normal germ cell development. Indeed, a variety of attractive candidate genes exist in mouse and Drosphila (see Table 2). An example is gcd, a mouse mutant in which germ cells are deficient in both males and females.
Galactosemia
Galactosemia is caused by deficiency of galactose 1-phosphate uridyl transferase (GALT). The gene is located on 9p. Long-recognized features included renal, hepatic, and ocular defects. Kaufman and colleagues41 reported POF in 12 of 18 galactosemic women, and Waggoner and colleagues42 observed ovarian failure in 8 of 47 females. Pathogenesis presumed to involve galactose toxicity during infancy or childhood; elevated fetal levels of toxic metabolites should be cleared rapidly in utero by maternal enzymes. Consistent with this hypothesis, a neonate with galactosemia showed normal ovarian histology.43
Given the clinical severity of galactosemia and the absolute necessity for dietary treatment during childhood to prevent mental retardation, it seems highly unlikely that previously undiagnosed galactosemia would prove to be the cause of ovarian failure in women presenting solely with primary amenorrhea or POF. Of greater relevance to gynecology, therefore, was the report in 1989 by Cramer and colleagues44 that GALT heterozygotes were at increased risk for POF.44 However, the same author later failed to observe GALT abnormalities in another sample of women with early menopause,23 and Kaufman and colleagues45 likewise failed to confirm the observation. Moreover, not all homozygotes for human galactosemia are abnormal, nor are transgenic mice in which GALT is inactivated.46
Carbohydrate-Deficient Glycoprotein Intrauterine Contraceptives (Cdg) [Phosphomannomutase Deficiency [PMM2])
In type 1 carbohydrate-deficient-glycoprotein (CDG) deficiency, mannose-6-phosphate cannot be converted to mannose-1-phosphate. Thus, the lipid-linked mannose-containing oligosaccharides needed for secretory glycoproteins are lacking. The CDG gene is located on 16p13. The molecular pathogenesis is usually a missense mutation.47
The wide spectrum of neurologic abnormalities includes hypotonia, hyperreflexia, unprovoked eye movements, ataxia, joint contractions, epilepsy, and stroke-like episodes.48 Subcutaneous fat deposits, hepatomegaly, cardiomyopathy, pericardial effusion, and factor XI (clotting) deficiency develop.
Of interest here is that ovarian failure occurs; the ovaries are devoid of follicular activity.49,50
Deficiency of 17α-Hydroxylase/17,20-Desmolase (Lyase) Deficiency (Cyp17)
Sex steroid synthesis requires intact adrenal and gonadal biosynthetic pathways. Various gene products (enzymes) are necessary to convert cholesterol to testosterone and androstenedione and, hence, estrogens. The various enzyme blocks have varying but predictable consequences, depending on their site in the biosynthetic pathway (Fig. 2). The most common adrenal biosynthetic problem is involving deficiency of 21- or 11β-hydroxylase, either of which causes pseudohermaphroditism. These disorders cause genital ambiguity because of virilization, but need not be considered in the differential diagnosis of XX gonadal dysgenesis.
|
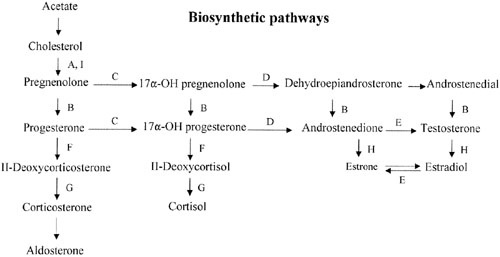
If the cytochrome P450 enzyme 17α-hydroxylase/17-20-lyase is deficient, pregnenolone cannot be converted to 17α-hydroxy-pregnenolone. If the enzyme defect were complete, cortisol, androstenedione, testosterone, and estrogens could not be synthesized; however, 11-deoxycorticosterone and corticosterone could. With compensatory increase in adrenocorticotropic hormone (ACTH secretion), 11-deoxycorticosterone and corticosterone increase to result in hypernatremia, hypokalemia, and hypervolemia. Hypertension occurs. Aldosterone is decreased, presumably because hypervolemia suppresses the renin-angiotensin system.
Females (46,XX) with 17α-hydroxylase deficiency have normal external genitalia, but at puberty fail to undergo normal secondary sexual development (primary amenorrhea). Affected males (46,XY) usually have genital ambiguity (male pseudohermaphroditism) because partial expression of the gene produced some androgens. Affected females are ordinarily encountered in differential diagnosis of XX gonadal dysgenesis. Hypertension is the major distinguishing clue. Oocytes appear incapable of spontaneously exceeding a diameter greater than 2.5 mm,51 but ovaries nonetheless respond to exogenous gonadotropins.52
17α-hydroxylase deficiency is inherited in autosomal recessive fashion. The gene (CYP17) is located on 10q24-25, and the gene product is a cytochrome P450 enzyme. More than 20 different mutations have been identified in CYP17, scattered among the 8 exons. Mutations include missense mutations, duplications, deletions, and premature protein truncation.53 Most mutations have been observed in only a single family, yet another example of molecular hetereogeneity. An exception exists in Mennonites of Dutch origin, where a 4-base duplication in exon 8 accounts for most cases.54 This founder mutation originated in Friesland.
This single gene (and enzyme) is responsible for both 17α-hydroxylase and 17,20-desmolase (lyase) actions (see Fig. 2). A few patients having deficiency of both 17α-hydroxylase and 17,20 lyase activities have been analyzed, with mutations different from those only showing deficient 17α-hydroxylase activity have been found.55,56 Transfection experiments show that only 5% of 17α-hydroxylase activity is sufficient for the estrogen production necessary for normal secondary sexual characteristics in a 46,XX individual; however, 25% of enzyme activity is necessary to virilize external genitalia in males.53,55 Targeted mutagenesis in the rat gene indicates that mutations closer to the 5′ end are more deleterious.
Aromatase Mutations (Cyp19)
Conversion of androgens (Δ4-androstenedione) to estrogens (estrone) requires cytochrome P-450 aromatase, an enzyme product of a 40-kb gene located on chromosome 15q21.1.57 Deficiency of the aromatase enzyme in 46,XX individuals is most often associated with clitoral hypertrophy or genital ambiguity, but 46,XX aromatase deficiency may present as primary amenorrhea in phenotypic females.
Ito and colleagues58 reported aromatase mutation (CYP19) in an 18-year-old 46,XX Japanese woman having primary amenorrhea and cystic ovaries. Compound heterozygosity, existed for two point mutations in exon 10. The mutant protein had no activity. Conte and colleagues59 reported aromatase deficiency in a 46,XX woman presenting with primary amenorrhea, elevated gonadatropins, and ovarian cysts. Compound heterozygositly also existed for two mutations in exon 10. One mutation was ′ C1303T (cysteine rather than arginine); the other was ′ G1310A (tyrosine rather than cysteine).
Mullis and colleagues60 reported clitoral enlargement at puberty. No breast development occurred. FSH was elevated; estrone and estradiol were decreased. Multiple ovarian follicular cysts were evident. Hormonal (estrogen and progesterone) therapy produced a growth spurt, breast development, menarche, and decreased numbers of follicular cysts. Compound heterozygosity existed in CYP19.
46,XX Agonadia
Agonadia usually occurs in 46,XY individuals but, rare 46,XX cases exist. As in 46,XY agonadia, gonads are absent, in 46,XX agonadia. This contrasts with persistence in the form of streaks (gonadal dysgenesis). External genitalia are abnormal but female-like; no more than rudimentary Müllerian or wolffian derivatives are present. External genitalia usually consist of a phallus about the size of a clitoris, underdeveloped labia majora, and nearly complete fusion of labioscrotal folds. Thus, external genitalia are nearly or somewhat female in appearance. In approximately one half of cases, somatic anomalies coexist: craniofacial anomalies, vertebral anomalies, and mental retardation.
Agonadia must be considered in the differential diagnosis of 46,XX primary amenorrhea. The 46,XX agonadia cases reported by Duck,61 and Levinson62 were sporadic. Mendonca and colleagues63 reported agonadia without somatic anomalies in phenotypic sibs having unlike chromosomal complements (46,XY and 46,XX). Kennerknecht and colleagues64 reported agonadism, hypoplasia of the pulmonary artery and lung, and dextrocardia in XX and XY sibs.