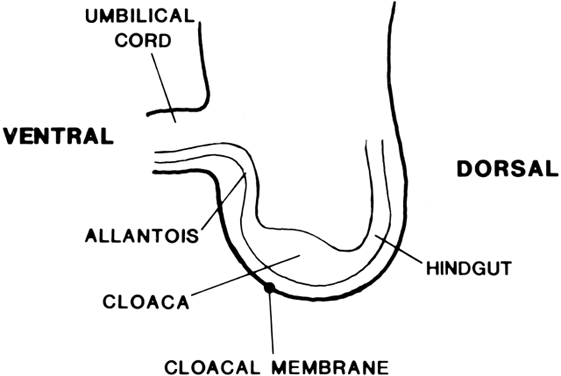
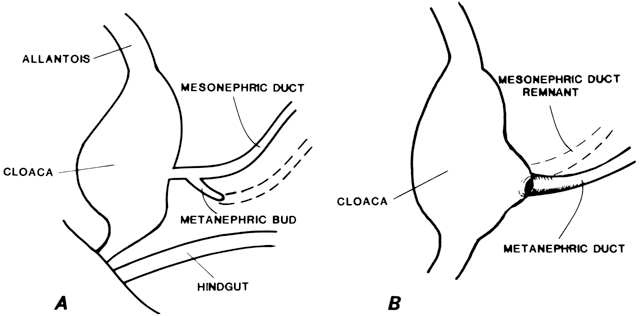
Vaginal Atresia The vagina is shortened or absent in many females whose external genitalia
are ambiguous (pseudohermaphrodites), but in the present context, we
consider only those females who lack a vagina and whose external genitalia
are otherwise normal. Two groups of individuals fulfill these
characteristics: those with absence of most of the vagina and all or almost
all of the uterus ( müllerian aplasia) and those with absence
of a portion of the vagina but presence of a normal uterus (vaginal
atresia). The two conditions are embryologically and anatomically distinct (Fig. 4). Of individuals with an absent vagina, 80% to 90% have müllerian
aplasia; the remainder have vaginal atresia.5,6,7,8,9,10 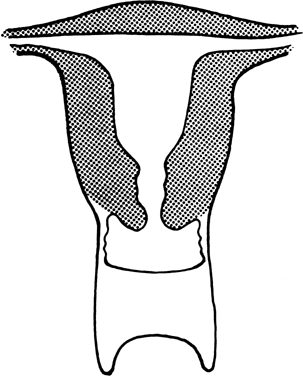 Fig. 4. Vaginal atresia.(From Sarto GE, Simpson JL: Abnormalities of müllerian and wolffian
duct systems. Birth Defects: Original Article Series 14(6a):37, 1978.) Fig. 4. Vaginal atresia.(From Sarto GE, Simpson JL: Abnormalities of müllerian and wolffian
duct systems. Birth Defects: Original Article Series 14(6a):37, 1978.)
|
In vaginal atresia the urogenital sinus fails to contribute the caudal
portion of the vagina. The lower fifth to third of the vagina is replaced
by 2 to 3 cm of fibrous tissue, above which lie a well-differentiated
upper vagina, cervix, uterine corpus, and fallopian tubes (Fig. 4). Ultrasound, magnetic resonance imaging, or rectal examination may verify
presence of müllerian derivatives. Hydrometrocolpos can develop. Familial aggregates of isolated vaginal atresia are rare if not nonexistent. However, vaginal
atresia is often reported as part of a large series
of patients with absence of vagina. Analysis of a heterogeneous sample
of patients might obscure findings that would be evident if the
two disorders were analyzed separately. Vaginal Atresia in the Multiple Malformation Syndromes Etiologically distinct from vaginal atresia in otherwise normal women is
vaginal atresia present as one component of a multiple malformation
complex. Table 1 summarizes several syndromes. Winter and coworkers described four siblings
with a previously unrecognized autosomal recessive syndrome characterized
by vaginal atresia, renal hypoplasia or agenesis, and middle ear
anomalies (malformed incus, fixation of the malleus and incus).11 A second malformation syndrome in which vaginal atresia occurs is the
Fraser syndrome, characterized by vaginal atresia and by cryptophthalmus
with its resultant blindness.12 Other syndromes include Antley-Bixler and Bardet-Biedl (Table 1). TABLE 1. Multiple Malformation Syndromes Associated With Vaginal Atresia
Syndrome | Somatic Anomalies | Etiology |
Antley-Bixler | Craniosynostosis, choanal atresia, humeroradial | Autosomal |
| synostosis, gracile ribs, bowed femora, | recessive |
| camptodactyly, renal anomalies | |
Bardet-Biedl | Degeneration of retinal pigment (retinitis pigmen- | Autosomal |
| tosa), polydactyly, obesity, mental retardation | recessive |
Fraser | Cryptophthalmia, nose and ear anomalies, stenotic | Autosomal |
| larynx, skeletal defects, syndactyly, renal agenesis, | recessive |
| large clitoris and labia majora, mental retardation | |
Winter | Lacrimal duct stenosis, external and middle ear | Autosomal |
| anomalies, renal agenesis | recessive | Transverse Vaginal Septa and the McKusick-Kaufman Syndrome Transverse vaginal septa occur at several locations and may be complete
or incomplete. These septa are usually about 2 cm thick and located near
the junction of the upper third and lower two thirds of the vagina;1,2,13 however, septa may be present in the middle or lower third of the vagina13 (Fig. 5). Perforations are usually central but may be eccentric in location.14,15,16,17 If no perforation exists, mucus and menstrual fluid cannot egress; hydrocolpos
or hydrometrocolpos may develop. Other pelvic organs are usually
normal, although occasionally the uterus is bicornuate. 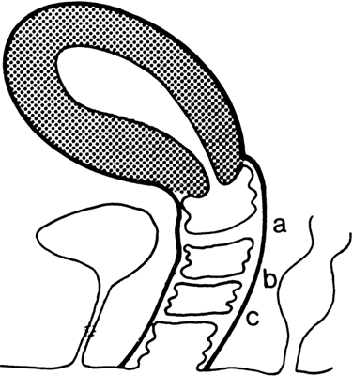 Fig. 5. Potential sites of transverse vaginal septa. A. High septum. B. Midvaginal septum. C. Low septum.(From Simpson JL, Verp MS, Plouffe L Jr: Female genital system. In Stevenson
RE, Hall JG, Goodman RM [eds]: Human Malformations and
Related Anomalies, vol 11, pp 563–588. New York: Oxford University
Press, 1993.) Fig. 5. Potential sites of transverse vaginal septa. A. High septum. B. Midvaginal septum. C. Low septum.(From Simpson JL, Verp MS, Plouffe L Jr: Female genital system. In Stevenson
RE, Hall JG, Goodman RM [eds]: Human Malformations and
Related Anomalies, vol 11, pp 563–588. New York: Oxford University
Press, 1993.)
|
Vaginal septa presumably result from failure of urogenital sinus derivatives
and the müllerian duct derivatives to fuse or canalize. This
explanation is deduced from the location of the septa, which is usually
at the predicted sites of urogenital sinus müllerian fusion, and
the histologic nature of the septa. The cranial surfaces of septa
are usually lined by columnar ( müllerian) epithelium, whereas caudal
surfaces are lined by squamous epithelium (i.e. urogenital sinus invagination). Many patients have transverse vaginal septa, polydactyly, and cardiac defects.18 The original description and most subsequent cases have been in the Amish. The
eponym McKusick-Kaufman syndrome is applied to such subjects.18 The latter cases could indicate that the postulated mutant is pleiotropic, a
suggestion to which Pinsky subscribes.19 Alternatively, presence of multiple abnormalities may indicate a different
mutant gene. Familial aggregates are rarely observed in non-Amish
kindreds; it is difficult to distinguish between these possibilities. In support of the thesis of a single pleiotropic gene is the analysis of 54 cases
by Chitayat and colleagues.20 Hydrometrocolpos was estimated to be present in 95% of Amish cases, polydactyly
in 93%, and cardiovascular malformations in 9%. Amish individuals
may show all three anomalies, various pairwise combinations of two, or
only one.21 Sonte and colleagues estimated penetrance to be 70% for hydrometrocolpos
in females, 60% for polydactyly in both sexes, and 15% for cardiovascular
defects.22 Given these probabilities, 9% of males and 3% of females could be expected
to have the gene in completely nonpenetrant state. The gene has been localized to chromosome 20p12. Homozygosity mapping for
short tandem repeat polymorphism (STRP) was performed in two large
Amish pedigrees; 385 markers were analyzed.22 The peak two-point logarithm of odds (LOD) score was 3.33, and the peak
three-point LOD score was 5.21. Region 20p12 includes the locus for
the Alagille syndrome, an autosomal recessive multiple malformation syndrome
characterized by cardiac anomalies, hepatic ductal hypoplasia, and
abnormal (“butterfly”) vertebrae. Alagille syndrome is
thought to be caused by a perturbation of jagged1 (JAG-1).23 However, no sequence abnormalities in jagged1 were found in two Amish cases of transverse vaginal septum (i.e. McKusick-Kaufman).22 Two mouse mutants show urogenital and skeletal anomalies: dominant hemimelia (dh) and
loop tail (lp). Both loci map to mouse chromosome 1, a
region not syntenic to human 20p.24,25,26 These mouse mutants are probably not good models for transverse vaginal
septa or at least the McKusick-Kaufman syndrome variant if distinct. Another
possible mouse model is ivp (imperforate vagina), an autosomal
recessive mutant not yet mapped.27 Vaginal Longitudinal Septa Vaginal septa may be longitudinal (sagittal or coronal) (Fig. 6) or transverse. Longitudinal septa, which rarely produce clinical problems, probably
result from abnormal mesodermal proliferation or persisting
epithelium. Occasionally, these septa impede the second stage of
labor. Heritable tendencies are not obvious, although no systematic studies
have been reported. 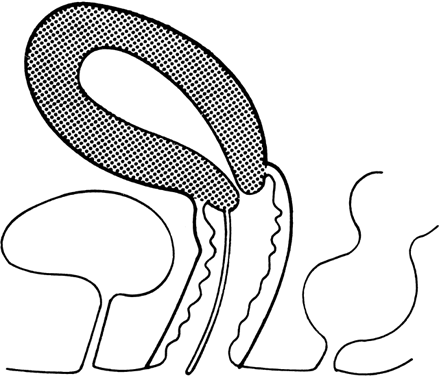 Fig. 6. Longitudinal vaginal septum.(From Simpson JL, Verp MS, Plouffe L Jr: Female genital system. In Stevenson
RE, Hall JG, Goodman Rm [eds]: Human Malformations and
Related Anomalies, vol 11, pp 563–588. New York: Oxford University
Press, 1993.) Fig. 6. Longitudinal vaginal septum.(From Simpson JL, Verp MS, Plouffe L Jr: Female genital system. In Stevenson
RE, Hall JG, Goodman Rm [eds]: Human Malformations and
Related Anomalies, vol 11, pp 563–588. New York: Oxford University
Press, 1993.)
|
Edwards and Gale reported an autosomal dominant syndrome characterized
by longitudinal vaginal septum, hand anomalies, and urinary incontinence
possibly because of a bladder neck anomaly.28 Longitudinal vaginal septa also occurs in the Johanson-Blizzard syndrome, which
is probably an autosomal recessive disorder29 (Table 2). TABLE 2. Syndromes Associated With Longitudinal Vaginal Septa
Syndrome | Somatic Anomalies | Etiology |
Edwards-Gale | Flexion contractures of distal inter- | Autosomal |
(camptobrachydactyly)28 | phalangeal joints, brachydactyly, poly- | dominant |
| dactyly, syndactyly, urinary incontinence | |
Johanson-Blizzard29 | Scalp defects, deafness, hypoplastic alae | Autosomal |
| nasi, microdontia, primary hypothyroidism, | recessive |
| malabsorption, mental retardation, | |
| hypotonia, short stature | | Absence or Atresia of the Uterine Cervix Isolated absence or hypoplasia of the cervix associated with a normal uterine
corpus and a normal vagina is rare. Relatively few cases have been
described, and there have been no reports of multiple affected family
members.30,31 The disorder presumably results from failure of müllerian duct canalization
or increased local epithelial proliferation after canalization. Hydrometrocolpos
should be anticipated. The cervical canal may also
be absent in true hermaphrodites.32 In 1997, Fujimoto and colleagues reported 7 new cases and reviewed the 51 previously
reported cases.33 They concluded that one half of all cases with cervical absence or atresia
had normal vaginas; one half had complete or partial vaginal atresia (Fig. 7). Surgically created uterovaginal canalization led to menstruation in 60% of
cases overall but more often if concomitant vaginoplasty was not
concurrently needed (68% versus 43%). After surgical correction, pregnancies
have occurred only exceptionally.31,32,33,34 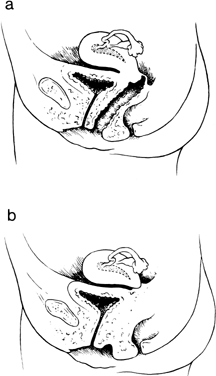 Fig. 7. A. Isolated congenital cervical atresia with normal vaginal development. B. Congenital cervical atresia with complete vaginal agenesis.(From Fujimoto VY, Miller JH, Klein NA et al: Congenital cervical atresia: Report
of seven cases and review of the literature. Am J Obstet Gynecol 177, 1419, 1977.) Fig. 7. A. Isolated congenital cervical atresia with normal vaginal development. B. Congenital cervical atresia with complete vaginal agenesis.(From Fujimoto VY, Miller JH, Klein NA et al: Congenital cervical atresia: Report
of seven cases and review of the literature. Am J Obstet Gynecol 177, 1419, 1977.)
|
Müllerian Aplasia Aplasia of the müllerian ducts leads to absence of the uterine corpus, the
uterine cervix, and the upper portion of the vagina (Fig. 4). The foreshortened 1 to 2 cm vagina is presumably derived exclusively
from invagination of the urogenital sinus. Individuals with müllerian
aplasia usually consult physicians because of primary amenorrhea. Secondary
sexual development is normal, but no uterine structures are
palpable. Uterine remnants may exist in the form of bilateral cords. The
term Rokitansky-Küster-Hauser syndrome, is often applied, sometimes if remnants persists and sometimes synonymously
with müllerian aplasia. The only disorder that ordinarily needs to be considered in the differential
diagnosis is complete androgen insensitivity. Androgen insensitivity
can be excluded on the basis of chromosomal studies and gonadal composition. Puberal
patients with müllerian aplasia invariably have
pubic hair, whereas those with androgen insensitivity usually do not. Renal anomalies are associated with müllerian aplasia more frequently
than expected by chance9,35,36,37,38 (Table 3). The most frequent renal anomalies are pelvic kidney, renal ectopia, and
unilateral aplasia. Skeletal anomalies, especially vertebral anomalies, are
not uncommon. Excretory urography and vertebral roentgenograms
are obligatory in the clinical evaluation of müllerian aplasia. TABLE 3. Urologic Anomalies in Müllerian Aplasia
| | No. | No. With |
| Total | Undergoing | Renal |
Study | Sample | Urography | Anomaly |
Phelan et al.35 | 129 | 72 | 26/72 (8 “minor”) |
Thompson et al.36 | 17 | 17 | 8/17 |
Leduc et al.9 | 10 | 10 | 4/10 |
Fore et al.37 | 32 | 32 | 20/32 |
Carson et al.38 | 23 | 22 | 4/22 (18.2%) |
Total | 211 | 153 | 62/153 (40.5%) | Familial aggregates of müllerian aplasia have been reported, namely
affected siblings.39,40,1,42,43 However, Lischke and associates observed three sets of discordant monozygotic
twins, and autosomal recessive inheritance therefore is an unlikely
explanation for all cases.44 Autosomal dominant inheritance was considered by Shokeir to exist in Saskatchewan
families in which the proband had müllerian aplasia.43 In 13 of 16 families, the proband showed complete absence of the uterine
cervix and corpus; in the remaining 3, uterine remnants ( Rokitansky-Küster-Hauser) were
present. None of the 3 individuals with uterine
remnants had an affected relative, but 10 of the 13 with complete
absence of the uterine cervix and corpus did. Two of these 10 had affected
siblings, whereas the other 8 had other affected paternal relatives (i.e. aunts, first cousins, second cousins, or great-aunts). Such observations
suggest sex-limited (female) autosomal dominant inheritance, although
other genetic mechanisms cannot be excluded. Females with the postulated
mutant would manifest müllerian abnormalities, whereas males
would show no deleterious effect. In contrast to the conclusions of Shokeir were 23 U.S. families reported
by Carson and coworkers.38 Not a single relative was affected. Absence of affected relatives among 30 postpubertal
sisters, 31 paternal aunts, and 41 maternal aunts makes
sex-limited autosomal dominant inheritance at least uncommon; however, dominant
genes could be restricted to certain populations, and fresh
dominant mutations can never be excluded. However, absence of affected
siblings and lack of paternal consanguinity speaks against autosomal
recessive inheritance. Women with müllerian aplasia have normal ovaries. A current strategy
is to obtain oocytes from affected women, perform fertilization in vitro with their husband's sperm, and transfer fertilized embryos to surrogate
uteri of another woman in hormonal synchrony. Resulting offspring
would genetically reflect the affected woman. Petrozza and colleagues45 surveyed U.S. assisted reproductive technology (ART) programs to accumulate 34 pregnancies
in women with müllerian aplasia. Of the 34 offspring, 17 were
female, and none was affected; one male child had a
middle ear defect and hearing loss. These data suggest that the most logical explanation for müllerian
aplasia is polygenic/multifactorial inheritance. This is the usual mode
of inheritance for malformations affecting a single organ system or
embryologically related systems. Müllerian aplasia clearly fulfills
these characteristics. Polygenic/multifactorial inheritance could
explain the occasional reports of multiple affected siblings. After the
birth of one child with a polygenic/multifactorial disorder, the recurrence
risk for first-degree relatives of affected probands approximates
the square root of the incidence of the trait in the population. Because
müllerian aplasia is rare, the recurrence risk for siblings
should be low. A theoretical recurrence risk could be calculated if
accurate incidence data existed. (Risk equals square root of incidence
of the trait.) Failure to detect affected sibs in a relatively small
sample is consistent with polygenic/multifactorial inheritance and a low (1% to 2%) recurrence
risk for first-degree relatives. Another plausible explanation is genetic (etiologic) heterogeneity. A dominant
or recessive gene could explain a minority of cases, perhaps those
in certain populations; nongenetic factors or polygenic/multifactorial
inheritance could explain the remainder. Genetic heterogeneity could
account for the discordant results between the study of Carson and
associates38 and that of Shokeir.43 The latter sample was derived from a different population in which a different
gene could have been segregating. Genetic heterogeneity could be deduced if some individuals affected with
a given malformation show a distinctive somatic anomaly that others
lack. Renal and vertebral differentiation and therefore anomalies are
apparently embryologically related, but a few patients display fusion
of cervical vertebrae (Klippel-Feil anomalad). However, coexistence of
müllerian aplasia and Klippel-Feil anomalad is sometimes associated
with middle ear anomalies.9,46,47,48,49,50 This triad could indicate an entity distinct from more common forms of
müllerian aplasia, especially given that renal anomalies were not
present in individuals with Klippel-Feil anomalad. Neurosensory hearing
loss in the high-frequency range has been observed.51 In several multiple malformation syndromes, müllerian aplasia is one
component (Table 4). The mechanisms presumably reflect perturbation of genes different from
those responsible for müllerian aplasia in otherwise normal individuals. TABLE 4. Syndromes Associated With Müllerian Aplasia
Syndrome | Somatic Anomalies | Etiology |
Fraser | Cryptophthalmia, nose and external ear anomalies, stenotic larynx, skeletal
defects, syndactyly, renal agenesis, large clitoris and labia majora, mental
retardation | Autosomal recessive |
Meckel-Gruber | Microcephaly, posterior encephalocele, eye anomalies, cleft palate, polydactyly, polycystic
kidneys | Autosomal recessive |
MURCS association | Renal aplasia, cervicothoracic somite dysplasia, Klippel-Feil anomaly, deafness, short
stature | Unknown |
Thalidomide teratogenicity | Nasal hemangioma, neurosensory hearing loss, ear anomalies, limb reduction
defects, visceral anomalies | Teratogen |
Urogenital adysplasia, hereditary | Oligohydramnios, flattened (Potter) facies, pulmonary | Autosomal dominant |
(hereditary renal adysplasia; bilateral renal agenesis) | hypoplasia, unilateral or bilateral absent kidneys, limb deformities | |
Winter | Lacrimal duct stenosis, external and middle ear anomalies, renal agenesis | Autosomal recessive | True Duplication of the Müllerian Ducts True duplication of the uterus is a rare anomaly that probably results
from division of one or both müllerian ducts early in embryogenesis. Affected
individuals have two separate uteri, each of which may have
two fallopian tubes.2 One or both uteri may be rudimentary or bicornuate. True duplication should
be distinguished from incomplete müllerian fusion, the much
more frequent condition in which each of two hemiuteri is associated
with only a single fallopian tube. Understandable because hemiuteri are
so much more common than true duplications, the frequent practice of
referring to bicornuate uteri as a “double uterus” actually
constitutes a misnomer. No familial aggregates have been reported. Incomplete Müllerian Fusion The müllerian ducts are originally paired organs that fuse and canalize
to form the upper vagina, uterus, and fallopian tubes. Failure of
fusion results in two hemiuteri, each associated with no more than one
fallopian tube. Sometimes one müllerian duct fails to contribute
to the definitive uterus, leading to a rudimentary horn. Figure 8 shows the different varieties of incomplete müllerian fusion. Renal
anomalies coexist with all. If one uterine horn is atretic, ipsilateral
renal agenesis is especially common. 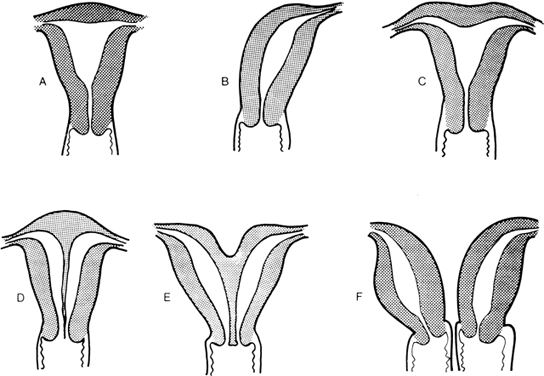 Fig. 8. Uterine fusion anomalies. A. Normal uterus. B. Unicornuate uterus. C. Arcuate uterus. D. Septate uterus. E. Bicornuate uterus. F. Didelphic uterus with a septate vagina.(From Simpson JL: Disorders of Sexual Differentiation: Etiology and Clinical
Delineation. New York; Academic Press, 1976.) Fig. 8. Uterine fusion anomalies. A. Normal uterus. B. Unicornuate uterus. C. Arcuate uterus. D. Septate uterus. E. Bicornuate uterus. F. Didelphic uterus with a septate vagina.(From Simpson JL: Disorders of Sexual Differentiation: Etiology and Clinical
Delineation. New York; Academic Press, 1976.)
|
Familial aggregates of incomplete müllerian fusion include multiple
affected siblings and affected mothers and daughters.52,53,54,55,56,57,58,59,60 Individuals in the same kindred may show different forms of incomplete
müllerian fusion.60 In the only formal genetic study reported,61 only 1 (2.7%) of 37 sisters had a clinically symptomatic uterine anomaly. There
were no affected mothers (0 of 24), maternal aunts (0 of 44), or
paternal aunts (0 of 50). The 2.7% prevalence in siblings constitutes
a minimum frequency because relatives could have been a minor uterine
anomaly in asymptomatic form. Ideally, hysteroscopy, hysterosalpingography, or
surgical exploration could be performed on relatives. Female
relatives in some families had not yet attempted pregnancy, limiting
the opportunity to manifest symptoms that would suggest an anomaly. Even
with such inherent limitations, the likelihood of first-degree
female relatives being similarly affected with müllerian fusion anomalies
would seem to be too low to be compatible with an autosomal dominant
or autosomal recessive origin. That approximately 3% of female
siblings were affected in the one formal study is consistent with predictions
based on polygenic/multifactorial cause, assuming further studies
confirm the previously given data. Hand-Foot-Genital Syndrome The hand-foot-genital (HFG) syndrome is an autosomal dominant disorder
in which incomplete müllerian fusion is a major component. First
reported by Stern and associates, multiple kindreds have now been recognized.62,63,64 A family first identified by our group55 was updated by Donnenfeld and colleagues.65 The syndrome is characterized by skeletal (hand and foot) malformations
and incomplete müllerian fusion in females or hypospadias in males (with “hand-foot-genital” replacing the original appellation
of “hand-foot-uterus”).65,66 Limb abnormalities include short first metacarpals, small distal phalanges
on the thumbs, short middle phalanges on the small finger and fusion
of the wrist bones. Analogously, the great toe is short because of
a shortened metatarsal, and the phalanx is small and pointed. Urinary system anomalies include urinary incontinence (female), ventral
displaced urethral meatus (male and female), and malposition of the ureteral
orifices in the bladder wall (female).67 These urologic anomalies differ from those usually associated with incomplete
müllerian fusion. Vertebral anomalies do not seem characteristic
of the HFG syndrome. HFG syndrome should be sought in all females with uterine anomalies, because
offspring of affected women have a 50% likelihood of inheriting
the mutant gene. Inquiry should be made concerning the presence or absence
of skeletal anomalies or genital anomalies in male and female relatives. Even
in the absence of a positive family history, HFG syndrome
may be considered to be present if an individual displays characteristic
skeletal and genital anomalies. Such an individual would probably
represent a new mutation. It is also relevant to recall that varied expressivity
is characteristic of all autosomal dominant disorders. It is
possible that some females with the HFG gene may manifest only uterine
anomalies or only skeletal anomalies, whereas others in the same kindred
show both. That the skeletal anomalies in HFG syndrome were reminiscent of the hypodactyly (Hd) mutant
in the mouse was recognized by Mortlock and colleagues,68 who had earlier detected a deletion in murine exon 1 of HOXA13.69 HOXA13 was a good candidate gene for human HFG. A HOXA13 nonsense mutant was observed in a member of the original HFG family reported
by Stern and coworkers.63 The manner by which perturbation of HOXA13 produces HFG is still uncertain, but HOXA13 is integral for differentiation or fusion/canalization of müllerian
derivatives. Incomplete Müllerian Fusion in Other Multiple Malformation Syndromes HFG is not the only one multiple malformation syndrome associated with
incomplete müllerian fusion. Table 5 shows a more complete list. Many different genes and nonmendelian factors
must remain intact for normal uterine development. Whether wild-type
genes for these syndromes are integral for normal müllerian differentiation
is unclear. In some of these syndromes uterine anomalies
may arise secondary to connective tissue or vascular perturbations. The
wild-type genes are not part of the normal müllerian differentiation
cascade. TABLE 5. Syndromes Associated With Incomplete Müllerian Fusion
Syndrome | Somatic Anomalies | Etiology |
Bardet-Biedl | Retinal pigmentary degeneration (retinitis pigmentosa), polydactyly, obesity, mental
deficiency polydactyly, obesity, mental deficiency | Autosomal recessive |
Beckwith-Wiedemann | Macroglossia, omphalocele, macrosomia | Autosomal dominant, after uniparental disomy |
Donohue (leprechaunism) | Elfin facies with thick lips; large, low-set ears; prominent breasts and
external genitalia; hirsutism; abnormal carbohydrate metabolsim; failure
to thrive; motor and mental retardation | Autosomal recessive |
Fraser | Cryptophthalmia, external ear and nose anomalies, laryngeal stenosis, syndactyly, skeletal
defects, renal agenesis, large clitoris and labia
majora, mental retardation | Autosomal recessive |
Hand-foot-genital (HFG) | Metacarpal and metatarsal anomalies, malformed thumbs, displaced urethral
meatus, urinary incontinence | Autosomal dominant |
Johanson-Blizzard | Deafness, hypoplastic alae nasi, primary hypothyroidism, mental retardation | Autosomal recessive |
Laryngeal atresia | Hydrocephaly, complete or partial laryngeal obstruction, tracheoesophageal
fistula or atresia, renal hypoplasia, varus deformity of feet | Unknown |
Meckel-Gruber | Microcephaly, posterior encephalocele, eye anomalies, cleft palate, polycystic
kidneys, polydactyly | Autosomal recessive |
Roberts | Sparse, silvery blond hair; midfacial hemangioma; cleft lip with or without
cleft palate; limb reduction defect; intrauterine growth retardation | Autosomal recessive |
Rudiger | Bifid uvula, coarse facies, absent ear cartilage, hydronephrosis secondary
to ureterovesical stenosis, short digits | Autosomal recessive |
Thalidomide teratogenicity | Nasal hemangioma, neurosensory hearing loss, ear anomalies, limb reduction
defects, visceral anomalies | Teratogen |
Trisomy 18 | Prominent occiput, malformed ears, micrognathia, short sternum, cardiac
defects, horseshoe kidney, overlapping fingers, intrauterine growth retardation, severe
developmental retardation | Chromosomal aneuploidy |
Trisomy 13 | Microcephaly, microphthalmia, malformed ears, cleft lip and palate, cardiac
anomalies, polydactyly, intrauterine growth retardation, severe developmental
retardation | Chromosomal aneuploidy |
Urogenital adysplasia, hereditary (hereditary renal agenesis) | Oligohydramnios, flattened (Potter) facies, pulmonary hypoplasia, unilateral
or bilateral absent kidneys, limb deformities | Autosomal dominant | Imperforate Hymen Ordinarily, the central portion of the hymen is patent (perforate), allowing
outflow of mucus and blood. If the hymen is imperforate, mucus and
blood accumulate in the vagina or uterus (i.e. hydrocolpos or hydrometrocolpos). An imperforate hymen is not uncommon. Fortunately, the
anomaly is easily corrected by surgical incisions, preferably
cruciform. McIlroy and Ward reported siblings who possibly
had the disorder, but no other familial aggregates have been described.70 Isolated Absence of Fallopian Tubes Absence of a fallopian tube in an otherwise normal female is rare.2,71 Fallopian tubes usually persist despite regression of all other müllerian
derivatives (i.e. uterus, cervix, and upper vagina). Unilateral absence of the ovary may
accompany ipsilateral absence of the fallopian tube.56 This implies pathogenesis involves a vascular accident or torsion after
completion of gonadal and ductal differentiation, perhaps analogous
to the cause of anorchia.72 No familial aggregates have been reported. Persistence of Müllerian Derivatives in Otherwise Normal Males The uterus and fallopian tubes ( müllerian derivatives) may persist
in ostensibly normal males (PMD males). External genitalia, wolffian (mesonephric) derivatives, and testes develop as expected; pubertal virilization
occurs. Infertility is common, and about 5% of reported individuals
develop a seminoma or other germ cell tumor. The disorder is
sometimes ascertained because the uterus and fallopian tubes are found
in inguinal hernias; the appellation hernia uteri inguinale has been applied. Multiple affected siblings or monozygotic twins have
been recognized.73 In one family, maternal half-siblings were affected, and in another maternal
first cousins.74,75 Two genes are integral to pathogenesis. One codes for anti müllerian
hormone (AMH) (i.e. müllerian inhibitory substance); the other codes for the AMH receptor. The
AMH gene is located on 19p, is 2800 bp long, and consists of
five exons.76 AMH can be measured by enzyme-linked immunosorbent assay, but the assay
is informative only before sexual maturation, because AMH production
is suppressed thereafter. When AMH is absent in PMD males, a mutation
is the structural gene can usually be demonstrated. Imbeaud and colleagues77 studied 38 PMD cases. Mutations in the AMH structural gene were found
in 16, all of whom had low or nondetectable AMH levels. Fifteen different
mutations were found, involving every exon except number 4. The PMD
cases due to the AMH gene mutations were found by the French investigators
to be predominately their Arab or Mediterranean patients. The rate
of consanguinity was high, consistent with 81% of patients being homozygous
for the mutation. The first three exons are most consistently
involved.78 The AMH receptor gene is located on 12q13 and consists of 11 exons. Imbeaud
and colleagues79 were the first to report a mutation in the AMH receptor in PMD, finding
at that time one case among 21 AMH-positive cases. Later, this group
reported 16 AMH receptor mutations. In contrast to AMH-negative cases, AMH-positive
cases were not Arab or Mediterranean, but French or European. A
total of 45% were homozygous for a given mutation, but consanguinity
was still infrequent. In 10 of the 16 in which a mutation was
found, it involved a deletion of 27 bp in exon 10. The relative molecular
homogeneity is attractive diagnostically. Miniature Schnauzer dogs
provide an informative animal model for AMH receptor mutations.80 Wolffian Aplasia and Congenital Absence of the Vas Deferens Wolffian ducts differentiate in vas 9 deferentia, epididymides and seminal
vesicles. Absence of wolffian derivatives (i.e. wolffian aplasia) may be an isolated defect, or it or may be associated
with absence of the upper urinary tract. The latter circumstance—absence
of wolffian duct derivatives and the upper urinary tract—implies
total failure of mesonephric development. Absence of wolffian
derivatives without upper urinary tract anomalies implies resorption
of wolffian elements after the wolffian duct reaches the cloaca. Regardless
of whether absence of wolffian derivatives is accompanied by
abnormalities of the upper urinary tract, the gonads are only rarely involved. More
frequently, upper urinary tract is normal in individuals
who lack an epididymis, vas deferens, or seminal vesicle. If wolffian
aplasia is bilateral, affected individuals are infertile because of azoospermia. If
the defect is unilateral, patients are usually asymptomatic. Cystic fibrosis is the result of a mutation in the cystic fibrosis transmembrane
regulation (CFTR) gene on chromosome 7, which functions as a chloride channel. It has long
been known that almost all cystic fibrosis (CF) homozygotes are infertile, usually
because of congenital absence of the vas deferens (CAVD). Cystic
fibrosis correlates with the absence of the vas deferens. Up
to 70% of males with CAVD have cystic fibrosis,84 usually in the form of compound heterozygosity. Two mutant CFTR alleles should be assumed to be present, even if only one can be detected
molecularly. The most common mutations are ΔF508 and R117H. If
only one (or no) CF for CF mutations are evident, a polymorphism may
exist in which five thymidines are present in a particular sequence
of intron 8.85 The presence of seven or nine thymidines has no effect. This 5-thymidine
polymorphism at the site results in low (10%) transcription of CFTR
protein on that (cis) chromosome. This is the result of improper exon-intron
splicing loss of exon 9 and CFTR mutant protein incapable of functioning
as a chloride channel. If neither CFTR mutants nor a 5-thymidine polymorphism is evident in CAVD, a rare mutation (“private”) may still exist.85 Several older reports have described affected sibs having congenital absence
of the vas deferens.86,87,88 These familial aggregates could reflect polygenic/multifactorial cause, but
it seems more likely that mutations in the CFTR locus explain familial aggregates of CAVD. If upper tract anomalies coexist, this
statement would not apply. Failure of Fusion of Epididymis and Testis Another relatively common urologic defect is failure of the testicular
rete cords of the testis to fuse with the mesonephric tubules destined
to form the ductule efferentia. Spermatozoa cannot exit. If the defect
is bilateral, infertility results. One or both testes may also fail
to descend. Fusion defects of this type occur in about 1% of cryptorchid and in about 1% of
azoospermic men. Familial aggregates have not been reported. Fertility
is achievable by aspirating sperm from the testes or epididymis
and using assisted reproductive technologies like intracytoplasmic
sperm injection. |