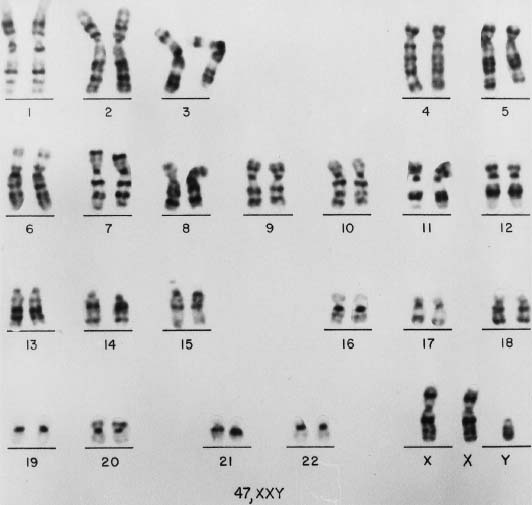
47,XXY GENITAL AND GONADAL PATHOLOGY. External and Internal Genitalia. In Klinefelter syndrome, the external genitalia are usually well differentiated (Fig. 2), but development of the penis and scrotum may be delayed. Problems of
penile development in the first 12 to 14 weeks’ gestation can
give rise to incomplete morphogenesis associated with anomalies such as
hypospadias, chordee, and micropenis. Problems after 14 weeks’ gestation
usually result in a well-formed but unusually small penis.30 The penis is normal in size in 80% to 90% of 47,XXY patients31,32; nevertheless, 47,XXY Klinefelter syndrome is the most commonly known
testicular disorder associated with micropenis. The cause is probably
a moderate deficiency in testosterone production,33 and the penis is only occasionally incomplete in morphogenesis. The prostate
is smaller than usual, also presumably owing to decreased testosterone
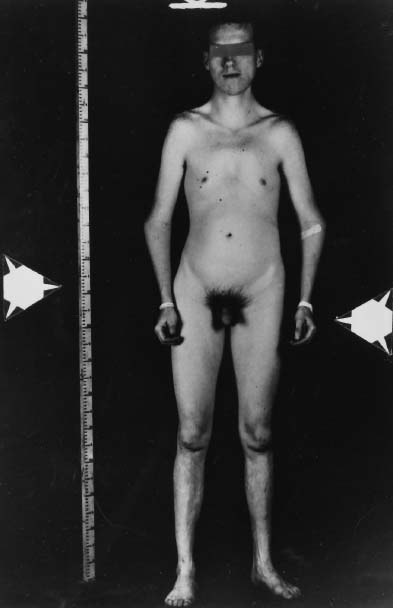
levels.34 Cryptorchidism is infrequent. 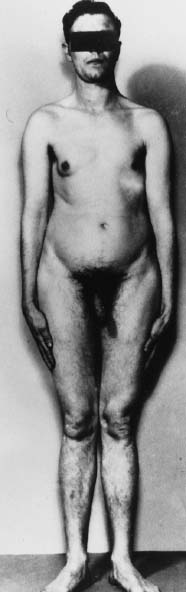 Fig. 2. Photograph of a 47,XXY Klinefelter syndrome patient showing gynecomastia.(Ferguson-Smith: Testis and intersexuality. In Hubble D [ed]: Paediatric
Endocrinology, p 359. Oxford, Blackwell Scientific Publication, 1969) Fig. 2. Photograph of a 47,XXY Klinefelter syndrome patient showing gynecomastia.(Ferguson-Smith: Testis and intersexuality. In Hubble D [ed]: Paediatric
Endocrinology, p 359. Oxford, Blackwell Scientific Publication, 1969)
|
Testes. Although the prepubertal testes of 47,XXY patients are usually described
as normal in size, Laron and Hochman35 reported that even before puberty the testes are smaller than those of
normal boys. Prepubertal 47,XXY patients display neither seminiferous
tubule atrophy nor apparent Leydig cell hyperplasia, but primary spermatogonia
are reduced in number.36,37 The testes of 47,XXY adults are rarely more than 2 cm in greatest diameter, compared
with 3.5 cm in normal males. The testes are usually firmer
than normal but are sometimes softer.32 The differences in consistency probably reflect differences in the extent
of seminiferous tubule hyalinization. The typical histologic pattern
of the adult testes of 47,XXY patients consists of hyalinization and
fibrosis of the seminiferous tubules and a relative increase in the
number of Leydig cells, with variable degrees of clumping (Fig. 3). Interstitial areas also contain increased numbers of fibroblasts and
fat cells.38 Leydig cells are abnormal both histologically and functionally: histologically, they
lack crystalloids of Reinke; functionally, their steroid
production is decreased.39 Abnormal Leydig cell function is evidenced by decreased plasma testosterone, subnormal
response to administration of human chorionic gonadotropin, and
increased gonadotropin levels.40 Azoospermia has been reported in at least 90% of these patients, and
except for a few instances, paternity has not been established.41 It is quite likely that most if not all cases of paternity in these patients
represent instances of undetected mosaicism. 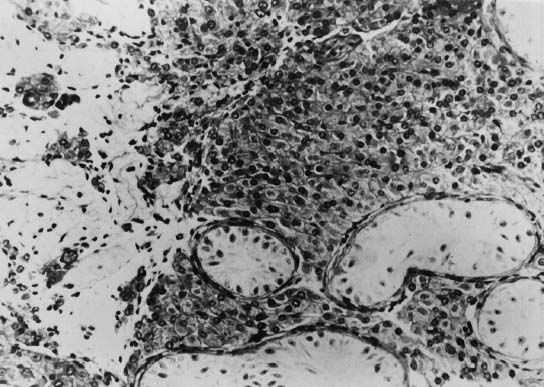 Fig. 3. Photomicrograph of a testicular biopsy of a 47,XXY patient. Seminiferous
tubules contain Sertoli cells but no spermatogonia. Leydig cells are
hyperplastic (×200).(Ferguson-Smith MA: Testis and intersexuality. In Hubble D [ed]: Paediatric
Endocrinology, p 359. Oxford, Blackwell Scientific Publication, 1969) Fig. 3. Photomicrograph of a testicular biopsy of a 47,XXY patient. Seminiferous
tubules contain Sertoli cells but no spermatogonia. Leydig cells are
hyperplastic (×200).(Ferguson-Smith MA: Testis and intersexuality. In Hubble D [ed]: Paediatric
Endocrinology, p 359. Oxford, Blackwell Scientific Publication, 1969)
|
SECONDARY SEXUAL DEVELOPMENT. Klinefelter syndrome becomes more conspicuous in adolescence. There is
a delay in the onset of puberty in about half of 47,XXY patients, and
reduced penile size in about 20%.42 Pubic hair is usually feminine in distribution; facial and body hair are
scanty, and few patients shave daily. Adolescent acne is rare, and
temporal hair recession usually does not occur. Obesity, gynecoid fat
distribution, and poor muscular development are also frequent. Sexual
activity is often reduced,43 although libido and potency may be improved with testosterone therapy.44 Increased parenchymal breast tissue, as determined by palpation, is found
in 50% to 75% of 47,XXY patients,39,45 but only approximately 20% have overt gynecomastia.32 Breast enlargement in 47,XXY patients is characterized by increased collagenous
material in interglandular spaces; the ductal epithelium is
only slightly hyperplastic.6 ENDOCRINE STUDIES. Prepubertal 47,XXY boys show no significant abnormalities in gonadotropin
or testosterone serum levels.44 In adults with 47,XXY complements, levels of both follicle-stimulating
hormone (FSH) and luteinizing hormone (LH) are usually elevated,40,45 and testosterone concentrations are usually below normal or in the lower
part of the normal range (5 to 86 mg/100 mL).46 The gonads are the principal source of testosterone in patients with Klinefelter
syndrome, as in normal males. The total plasma testosterone
level, however, is affected by peripheral metabolism, alterations in
binding proteins, and diurnal changes and may not be a sensitive indicator
of testicular androgen synthesis. These factors contribute to variable
reports regarding the plasma testosterone levels in Klinefelter
syndrome patients. As noted earlier, both plasma LH and FSH values are elevated in Klinefelter
syndrome patients. These elevations result from the deficient testicular
function and the consequent absence of feedback inhibition by
sex steroids and other testicular products (e.g., inhibin), probably acting at both the pituitary and hypothalamic levels.47,48 Smals and colleagues49 found elevated gonadotropins that responded to a bolus of LH-releasing
hormone (LHRH). In addition, an 8-hour infusion of LHRH elicited an increase
in testosterone as well, indicating that despite the resting hypogonadism, the
Leydig cells in Klinefelter syndrome have a functional
reserve. Abnormal glucose tolerance test (GTT) results and diabetes mellitus are
more frequent among Klinefelter syndrome patients and their parents than
in the general population.50,51 Of 157 cases of Klinefelter syndrome reviewed by Nielsen,51 28 (29%) had a diabetic GTT result and 13 (18%) had overt
diabetes mellitus, compared to a 6.2% incidence of an abnormal
GTT result in a random population less than 50 years of age and 16.1% in
those older than 50. Diabetes mellitus is usually mild in
Klinefelter syndrome patients.51 Engelberth and colleagues52 reported high levels of autoantibodies against insulin and against pancreatic
tissue in some patients with Klinefelter syndrome. Nielsen,51 however, found no such antibodies in his Klinefelter syndrome patients
with diabetes mellitus. Although clinical hypothyroidism is rare, decreased 131I uptake and poor responsiveness to thyroid-stimulating hormone have been
reported.53,54 Thyroid-binding globulin levels are usually normal,55 and antithyroid antibodies are usually absent.55 SOMATIC ANOMALIES. The height of 47,XXY boys under age 3 falls within normal distributions. After
age 3, however, the height distribution is skewed, with significantly
fewer boys than expected having height percentiles below 25.56 Adult 47,XXY patients are frequently taller than average and often have
abnormal skeletal proportions, with relatively long legs and a decreased
upper:lower segment ratio.56,57 Klinefelter syndrome patients differ from other eunuchoid patients in
that their arm span is usually only 2 to 3 cm greater than their height, if
at all greater; in most other types of eunuchoidism, the arm span
is usually at least 4 cm greater than the height. The cellular cause
of the abnormal proportions is unknown. It is possible that the epiphyses
respond abnormally to androgens,39 or an early acceleration of growth rather than a later epiphyseal closure
may cause excessive length in the lower limbs. The epiphyses, in fact, close
earlier in Klinefelter syndrome boys than in girls.58 The bone age is usually normal. Robinson and colleagues59 summarized the findings of 63 neonates with 47,XXY complement prospectively
ascertained through various chromosome surveys of unselected newborns. In 18% of
these boys, one or more major congenital abnormalities
were found: Cleft palate: four cases
Inguinal hernia: four cases
Undescended testes: four cases
Unilateral kidney agenesis and ureter deformities: one case
Microcephaly: one case
Corneal opacity: one case
Aortic stenosis: one case
Nerve deafness: one case
Hypospadias: one case
Funnel chest: one case
Small forehead: one case.
Minor anomalies (e.g., clinodactyly, squint, external rotation of legs, third fontanel, small
penis, antimongoloid slants, recurvated knees) were found in 26% of
the boys. Of 224 siblings, only 2 (1%) had major anomalies; minor
abnormalities were found in 15 (7%). Simpson and colleagues60 have tabulated from the literature a comparison of some clinical features
of 47,XXY, 48,XXXY, and 49,XXXXY patients (Table 1). These tabulations suggest that somatic anomalies are more frequent in 49,XXXXY
than in 48,XXXY and even less frequent in 47,XXY. The spectrum
of anomalies in 48,XXXY appears to be the same as the spectrum in 49,XXXXY, although
anomalies occur less often. The most frequent skeletal
anomalies in 47,XXY (e.g., scoliosis, kyphosis, pectus excavatum, clinodactyly V) are the same that
are frequently present in 48,XXXY and 49,XXXXY patients. The frequency
of congenital heart defects may be higher.61,62 No single anomaly, however, is consistently present. Autoimmune diseases, such
as systemic lupus erythematosus,63,64,65 ankylosing spondylitis,66 and Sjögren’s syndrome, characterized by keratoconjunctivitis, dry
mouth, and arthritis,65 occur with increased frequency in Klinefelter syndrome patients. It has
been hypothesized that testosterone protects males from autoimmune phenomena; therefore, hypogonadal males are prone to defects in T-cell
activity that lead to autoimmune disorders.65 Chronic leg ulcerations have also been associated with Klinefelter syndrome, with
a reported prevalence as high as 13%.67,68,69 Platelet hyperaggregability, rather than venous disease, appears to be
the causative factor.69 In addition, Klinefelter syndrome patients are said to be prone to varicose
veins and chronic pulmonary diseases (e.g., emphysema, bronchitis, asthma).32,70,71 TABLE 1. Comparison of Some Clinical Features of Patients With 47,XXY, 48,XXXY, and 49,XXXXY
Klinefelter’s Syndrome
| | 47,XXY† | 48,XXXY† | 49,XXXXY§ |
Anomaly* |
Cases |
N |
Cases |
N |
Cases |
N |
Mental retardation |
6 |
141∥ |
29 |
29 |
28 |
28 |
Small testes |
143 |
143 |
26 |
26 |
28 |
28 |
|
Hypoplastic penis |
11 |
44¶ |
11 |
24 |
24 |
28 |
Gynecomastia |
26 |
44¶ |
9 |
24 |
5 |
10# |
Wide-set eyes |
0 |
143 |
2 |
25 |
20 |
23 |
Epicanthal folds |
1 |
143 |
6 |
25 |
16 |
19 |
Upturned nose |
0 |
143 |
0 |
25 |
5 |
23 |
Low-set ears |
0 |
143 |
1 |
25 |
2 |
21 |
Malformed ears |
0 |
143 |
1 |
25 |
11 |
21 |
Strabismus |
0 |
143 |
2 |
25 |
11 |
21 |
Prognathism |
0 |
143 |
2 |
25 |
8 |
16 |
Webbed neck |
0 |
143 |
2 |
25 |
4 |
19 |
Short neck |
0 |
143 |
0 |
25 |
14 |
14 |
Kyphosis |
1 |
143 |
3 |
25 |
9 |
13 |
Scoliosis |
0 |
143 |
2 |
25 |
4 |
9** |
Radioulnar synostosis |
0 |
143 |
3 |
25 |
8 |
19 |
Abnormal ulnar or radius |
0 |
143 |
0 |
25 |
4 |
19 |
Clinodactyly V |
2 |
143 |
7 |
25 |
23 |
26 |
Coxa valga |
0 |
143 |
1 |
25 |
12 |
14 |
Genu valgum |
0 |
143 |
0 |
25 |
6 |
24 |
Pes planus |
0 |
143 |
1 |
25 |
10 |
24 |
Malformed toes |
0 |
143 |
0 |
25 |
5 |
24 |
Pes cavus |
2 |
143 |
0 |
25 |
0 |
24 |
Wide gap, first and second toes |
0 |
143 |
0 |
25 |
2 |
26 |
Talipes equinovarus |
0 |
143 |
0 |
25 |
2 |
26 |
*An anomaly is listed if, in the data surveyed, it was found in any two
persons with the same chromosomal complement. A number of minor radiographic
anomalies detected in 49,XXXY patients are not listed.
†Tabulated from patients of Court-Brown et al,93 except as noted.
†Tabulated from the study by Simpson et al.60
§Tabulated from Zaleski et al,109 except as noted.
∥Ascertained in surveys of the mentally retarded.
¶Estimate of Froland.34
#Estimate of Ferguson-Smith92
**Estimate based on radiographic studies tabulated by Zaleski et al.109
N = total sample.
(Simpson JL, Morillo-Cucci G, Horwith M et al: Abnormalities of human sex
chromosomes: VI. Monozygotic twins with the complement 48,XXXY. Humangenetik 21:301, 1974) Certain types of malignancies, particularly germ cell tumors, both gonadal72,73 and extragonadal,74,75 appear to occur with increased frequency in Klinefelter syndrome patients. Lee
and Stephens74 suggested that dysgenic germ cells might arrest in their migration along
the urogenital ridge and result in formation of mediastinal and retroperitoneal
germ cell tumors; there have also been rare reports of cerebral
germ cell tumors.75,76 In addition, breast carcinoma is 20 times more frequent among Klinefelter
syndrome patients than among normal males; 3% to 4% of
males with breast cancer are 47,XXY patients44,77,78 however, only 27 cases of breast cancer in these patients have been reported.79,80 Because of the risk of malignancy, as well as for cosmetic purposes, some
investigators have advocated mastectomy if gynecomastia is pronounced.32 Lymphoma81 and leukemia82,83 have been reported in 47,XXY patients as well, although no clear association
between these malignancies and Klinefelter syndrome has been established. INTELLIGENCE AND PSYCHOSOCIAL DEVELOPMENT. Although 47,XXY patients are more likely to be retarded or socially maladjusted
than normal (46,XY) males, the exact risk is uncertain. That 47,XXY
patients have an increased probability of being retarded can be
deduced from observations that 1% of mentally retarded males have 47,XXY.22,84 The prevalence of 47,XXY is higher among patients with an IQ between 50 and 85 than
among those with a lower IQ.32,85 Robinson and colleagues,59 reviewing the data of 63 infants with 47,XXY complements prospectively
ascertained in several chromosome surveys of neonates, found the distribution
of full-scale IQ levels was significantly skewed to the left, with 29% of
the boys having IQ levels below 90. The difference
between siblings and controls was significant (p < 0.001); however, performance tests correlated well with siblings
and controls. Graham and colleagues86 suggested that low IQ scores in patients with Klinefelter syndrome are
the result of specific linguistic deficits, rather than a global mental
deficiency. 47,XXY boys demonstrated significant impairment in expressive
language skills, causing difficulties in word finding, syntactic
production, and narrative formulation. Netley87 found a 10- to 20-point reduction in verbal skills persisting through
adolescence82 and into adult life.42 Performance IQ scores, however, did not significantly differ from those
of controls.87 Early referral for speech therapy may help prevent reading and writing
disabilities.88 Persons with Klinefelter syndrome frequently exhibit certain behavioral
peculiarities, such as poor self-motivation, passivity, absence of anxiety, and
bland facies.89,90,91 They adapt poorly to new situations and may display inappropriately aggressive
behavior when confronted with stressful situations. Few, however, are
overtly sociopathic. Among 47,XXY patients studied after being
prospectively ascertained in newborn chromosome surveys, Robinson and
colleagues59 determined that 32% had delayed emotional development, compared
to 9% of the siblings (p < 0.002). Maladjustment to structured school situations was found
in 20 of 45 cases (44%) among 47,XXY boys, compared to 32 of 136 cases
among siblings and controls (24%) (p < 0.025). Psychiatric problems were no more frequent in 47,XXY
boys than in siblings and controls. |