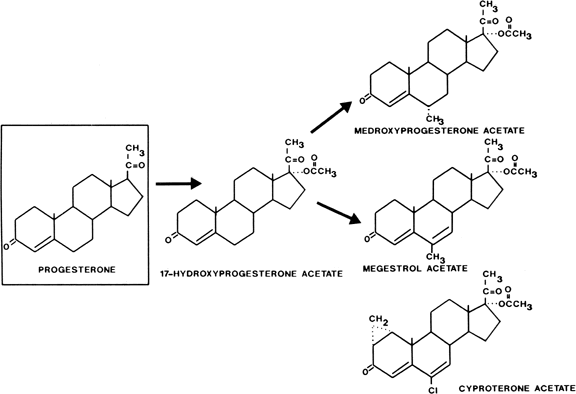
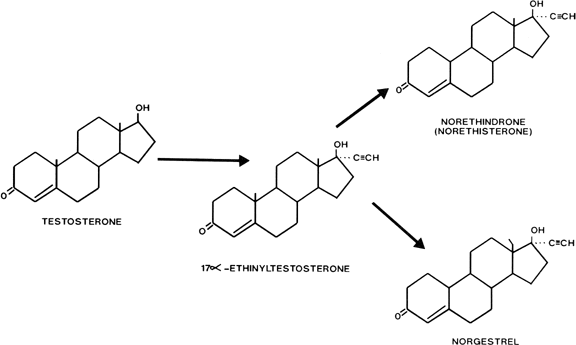
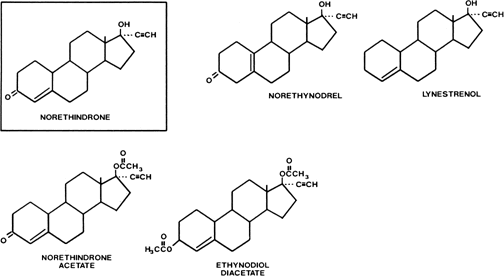
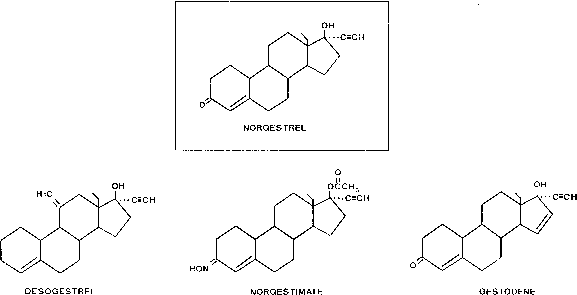
Hypothalamus-Pituitary Although contraceptive steroids interfere with the reproductive process
at several levels, the primary site of action is at the level of the
hypothalamus and pituitary to suppress the midcycle luteinizing hormone (LH) surge, thereby
preventing ovulation. Basal secretion of LH is not
suppressed, even with preparations of synthetic estrogens in a 100-μg/day
dose.81 With combinations containing 50 μg or less, irregular LH peaks can
be observed throughout the treatment cycle.82 There is some evidence that pituitary reserve of gonadotropins is depleted
by OC steroids. This was shown by a sequential pituitary stimulation
test (SST) consisting of an infusion of thyrotropin-releasing hormone (TRH) followed
by gonadotropin-releasing hormone (GnRH). The pituitary
response to stimulation was significantly depressed in OC users compared
with control subjects, even after GnRH priming, after ingestion
of OC formulations containing both high- and low-dose synthetic estrogens.83 This effect appears to be dose related in that patients receiving less
than 50 μg of estrogen had a lesser LH suppression after a bolus of
GnRH.84 Responses of both growth hormone and thyroid-stimulating hormone (TSH) to
the SST were unaffected by ingestion of contraceptive steroids, whereas
the prolactin response was significantly elevated after treatment
with either high- or low-dose estrogen therapy. Estrogen and the Pituitary Lactotroph Coincident with the advent of widespread OC use, laboratory techniques
for the measurement of prolactin and radiographic technologies for the
detection of pituitary lesions improved dramatically, and an association
between the use of contraceptive steroids and the development of prolactin-secreting
pituitary adenomas arose. Several observations fueled speculation that the estrogen contained in
OCs caused pituitary adenomas in women. There is in vitro evidence of a specific receptor of estrogen on the lactotroph that regulates
the transcription of the prolactin gene.85 Endogenous estrogen stimulates prolactin secretion, causing hyperprolactinemia
and lactotroph hyperplasia throughout human gestation.86 In addition, pharmacologic doses of exogenous estrogen were shown in animals
studies to induce pituitary adenomas in rodents.87 However, the Pituitary Adenoma Study Group88 reporting the data from a large multicenter case-control study found no
casual relationship between OC use and the risk of subjects having a
prolactin-secreting adenoma develop, consistent with the findings of
two large prospective studies conducted by the Royal College of General
Practitioners and the Oxford Family Planning Association reported earlier.89 As the authors discuss, the data collected were not designed to address
the issue of whether estrogen may exacerbate a pre-existing adenoma. A
Swedish group studied 70 women with prolactinomas and compared those
who had previously used OCs (61%) with those who had not.90 Patients who had used OCs had a shorter duration of symptoms, lower serum
prolactin levels, and less-pronounced enlargement of the sella turcica, suggesting
that the use of OCs either did not promote the growth
of the pituitary adenoma or may have led to earlier detection of the
lesion. It has been reported in a number of studies that basal levels of prolactin
are slightly elevated in healthy women taking OCs.91 Mishell and colleagues83 performed TRH stimulation tests on a group of women taking various doses
of contraceptive estrogens and found the initial prolactin response
to TRH, as well as the maximal prolactin response, to be significantly
greater in subjects using high or low doses of estrogen formulations. Episodic hyperprolactinemia, probably stress-related, is not uncommon in
healthy women,92,93 and the diagnostic precision of finding elevated prolactin levels associated
with an organic lesion is improved by frequent sampling.94 Luciano and colleagues95 reported that the incidence of hyperprolactinemia as determined by multiple
blood sampling was significantly greater in a group of OC users
than in a group of women who used barrier methods of contraception (12% versus 5%), but
that the hyperprolactinemia was often transient and
resolved spontaneously in approximately 50% of both OC users and control
subjects. They also identified a subset of patients who were more sensitive
to the lactogenic effects of exogenous estrogens and who therefore
might be at greater risk of having hyperprolactinemia develop. Consistent
with other reports, neither the duration of pill use nor the
dose of estrogen in the pill had any effect on the development of hyperprolactinemia. Menstrual Dysfunction Although the Pituitary Adenoma Study Group88 reported that the relative risk of subjects having a prolactinoma develop
was not increased in OC users with a history of menstrual disorders, Luciano
and colleagues95 noted that the patients in their study in whom hyperprolactinemia developed
on OCs had a significantly higher prevalence of menstrual dysfunction
in their histories. Regardless of whether they take OCs, women who
have irregular menstrual periods are more likely to have hyperprolactinemia
and are more likely to have secondary amenorrhea develop. Therefore, each
individual case should be investigated before beginning OC
therapy. Return of Fertility In a review by MacLeod,96 it was found that the incidence of postpill amenorrhea and that of secondary
amenorrhea unrelated to OC use were similar (0.2% to 2.2% and 0.1% to 0.8%, respectively). The rate of return of fertility after the
discontinuation of OCs is lower than for women who have used barrier methods
for 2 to 3 years, but the percentage of women who eventually conceive
after ceasing either form of contraception becomes the same.97 Neither the rate of spontaneous abortion98 nor the incidence of chromosomal abnormalities99 in abortuses is increased in women who conceive in the first or subsequent
months after discontinuing OCs. More important, in contrast to earlier
published reports, a large cohort study reported that ingestion
of OC steroids in the first few months of pregnancy does not significantly
increase the risk of congenital malformations among the offspring
of users overall or among those of nonsmoking users.100 Thyroid Thyroid hormone circulates as both thyroxine (T4) and triiodothyronine (T3). Most
of the circulating thyroid hormone is bound to thyroid-binding
globulin, whereas less than 0.1% circulates in the unbound or free
form. It is the unbound hormone that is thought to have a biologic effect. As
we have noted in the discussion of other metabolic parameters, ingestion
of synthetic estrogens stimulates hepatic production of globular
proteins, including thyroid-binding protein. Therefore, total thyroxine
levels are elevated with OC use, reflecting an increased bound
fraction. However, levels of free thyroxine are unaffected by estrogen
ingestion and, together with measurement of TSH, provide an accurate
assessment of a patient's thyroid state. Adrenals Similar to thyroid-binding globulin, estrogen increases hepatic production
of corticosteroid-binding globulin (CBG). As early as 1963, women
taking OCs were noted to have significantly elevated levels of total plasma
cortisol.101 It was later reported that levels of both free and bound cortisol are
elevated by combination OCs.102 Both components of combination pills are involved in this increase in
free cortisol.102 Estrogen reduces the ability of the liver to metabolize cortisol and progesterone, and
certain progestogens can displace cortisol from CBG.103 The secretion of adrenocorticotropic hormone (ACTH) is diminished during
OC therapy, possibly by negative feedback of unbound cortisol.104 Dehydroepiandrosterone sulfate (DHEAS) levels are also decreased,105 depending on the type of progestogen used. Administration of estrogen
alone results in an increase in DHEAS,106 whereas OCs containing norethindrone and levonorgestrel show a suppressive
effect on adrenal androgen secretion.107 Despite this putative suppression of adrenal function, the adrenal glands
respond normally to ACTH in women taking OCs, and the pituitary-adrenal
response to stress appears unaffected. Skin The triphasic EE2/norgestimate combination OC pill is the first and only OC pill indicated
for the treatment of moderate acne vulgaris in menarchial women unresponsive
to topical anti-acne medication. This is the first time the
FDA has stated that an OC can be indicated for noncontraceptive use.108 However, more recent studies show that low-dose OCs improve acne,107 and it appears that one OC is not any better than another in improving
this clinical manifestation. The mechanism by which OCs improve acne and hirsutism can be explained
by considering androgen effects at the level of the pilosebaceous unit
in skin. Within these cells, androstenedione and testosterone are converted
to DHT via the pivotal enzyme, 5∝-reductase.109 The latter hormone is markedly more potent than testosterone. OCs suppress
bioavailable testosterone, which is the non-SHBG-bound fraction of
total testosterone, thereby reducing the availability of precursor for 5α-reductase.107 Studies also show that in skin of hirsute women, there is an increased
conversion of testosterone to DHT compared with non-harsute women, suggesting
that increased activity of 5α-reductase underlies hirsutism
more so than elevated levels of testosterone.109 Lobo and colleagues have shown in vitro that levonorgestrel and northeldrone inhibit 5α-reductase activity
by 50% to 60%, thereby suggesting a mechanism by which progestogens
may achieve their observed improvements in acne and hair growth.9 Furthermore, it throws into question the concept that certain progestogens
are inherently androgenic. |