
Anemia Associated with Pregnancy
Authors
INTRODUCTION
Anemia among women of reproductive age is so common that healthcare providers may consider it the norm, while epidemiologists note it as a marker for healthcare quality, and sociologists index it to disadvantaged populations. Anemia during pregnancy, as noted in this chapter, is even more likely to be labelled as normal since hemodilution takes place during the first and second trimester as plasma volume increases disproportionately to red cell mass. Frequently it is described as “physiologic anemia” which again serves to disabuse healthcare providers as well as patients and their families from the notion that anemia during gestation is abnormal. On the other hand, pregnant women with even mild anemia have increased perinatal mortality and early neonatal mortality largely associated with preterm birth and growth restriction.1 Even when anemia is noted early in pregnancy and treated promptly, there is an increased risk of preterm birth.2 More severe cases of anemia (Hb <8 g/dL) are associated with greater risks of preterm birth and low birth weight.3 Delivery itself is a factor as up to 20% of women with normal hemoglobin values in the last trimester of pregnancy, were discovered to have anemia at their first postpartum visit.4 Among low income women, the Centers for Disease Control and Prevention (CDC) found the prevalence of anemia to increase with gestation, noting 8%, 12%, and 29% in the first, second, and third trimester, respectively.5 Additionally, there is a real risk of transfusion, particularly among patients who have cesarean delivery. More than a third (36%) of those women with a preoperative hematocrit of 25% or less required a postoperative blood transfusion.6
There are also quality of life issues associated with maternal anemia that are comparable to those seen in individuals with serious chronic diseases, such as difficulty in concentration, cognition, disturbed mother–infant interactions, and depression.7 For example, the physical functioning and vitality scores in women with postpartum anemia compare to scores seen with congestive heart failure, chronic renal disease and cancer patients.8, 9, 10 After correction of the postpartum anemia with carboxymatose iron infusion, physical function and vitality scores equalled or exceeded those in the general population without anemia.7 Postpartum fatigue and reduced immune function associated with increased risks for infection were also shown to have a relationship with anemia.11, 12, 13 Anemia has also been linked to postpartum depression14 and, if severe, can be related to cardiovascular symptoms, dizziness, and need for prolonged hospitalization.15 Cognitive functions and emotional distress have also been noted in women with anemia,16 while more symptoms of depression such as “postpartum blues” and a reduced sense of well being are demonstrated compared to nonanemic women.17 Unfortunately, available data suggest that most pregnant women are judged to be at low risk for anemia during pregnancy or during the postpartum period and therefore, the CDC, the American Congress of Obstetricians and Gynecologists (ACOG), and the Institute of Medicine recommend anemia screening at 4–6 weeks of postpartum only for those considered to be at “high risk” of developing anemia.18 In summary, as providers we seem to have a problem with pregnancy associated anemia as we do not consider it to be an abnormality until it becomes quite severe. Although as noted above, there are significant maternal–fetal implications even with mild reductions in hemoglobin. The goal of this chapter is to raise the index of suspicion in pregnant women who are or may become anemic and suffer from these problems.
Anemia: Differential Diagnoses
Decreased erythrocyte production (hypoproliferative)
Disturbance in maturation
Hemoglobin synthesis
Heme (iron deficiency)
Globin (thalassemia)
DNA synthesis (megaloblastic)
Folic acid deficiency
Vitamin B12 deficiency
Bone marrow failure
Other (neoplasia, inflammation, drug)
Blood loss
Early gestation (miscarriage)
Late gestation (placental abnormalities)
Puerperium (uterine atony)
Surgery (cesarean section)
Intestinal (hemorrhoids)
Parasites (hookworms)
Increased erythrocyte destruction (proliferative)
Extrinsic mechanism (acquired)
Hypersplenism
Mechanical (microangiopathic)
Coombs-positive anemia
Intrinsic mechanism (inherited)
Membrane abnormalities (spherocytosis)
Enzyme abnormalities (G6PD)
Globin chain abnormalities (sickle hemoglobin)
Other (paroxysmal nocturnal hemoglobinuria)
For the diagnosis of anemia, it is important to remember that there may be more than one factor involved. Anemia due to folic acid deficiency (FAD), chronic infection, and iron deficiency anemia (IDA) all yield different red blood cell (RBC) indices in their pure form. If combined, however, a mixed pattern results, which may render these diagnostic tests less useful. Several deficiencies may cause the same type of anemia. Nutritional deprivation, reduced inter-pregnancy interval, and adolescence are common etiologic factors in IDA in pregnancy. Treatment of one cause may not result in total compensation. Finally, pregnancy itself may alter several common laboratory tests, complicating anemia assessment techniques. During antepartum evaluation of anemia, each laboratory result should be interpreted carefully, since many factors affect the diagnostic and therapeutic modalities used, and thus maternal–fetal outcome.
PLASMA VOLUME
The plasma volume (PV) begins to rise slowly above nonpregnant levels around the 6th week of pregnancy (Fig. 1). At 16 weeks, it is approximately 10% above normal and rises rapidly until approximately 26 weeks to levels greater than 50% above baseline at which time a constant plateau is maintained until near term.
|
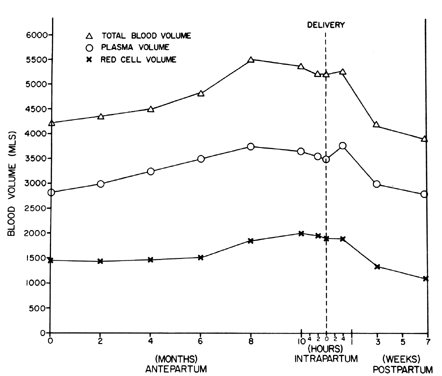
In the early postpartum period, PV decreases, only to increase again 2–5 days after delivery (see Fig. 1). The increase may be related to the rise in aldosterone observed at approximately 72 h postpartum. This elevation usually abates by 6 days postpartum, and the PV begins its final decrease to normal levels. By 3 weeks postpartum, the PV is still elevated 10–15% above non-pregnant levels, but when measured at 6 weeks, has usually returned to normal.
There are several advantages to the physiologic increase in PV during pregnancy. The apparent decrease in plasma cellular volume (PCV) reduces viscosity, decreasing the risk of thrombosis. Lowering of peripheral resistance and expansion of the blood volume appear to promote more oxygen exchange at the tissue level for a given cardiac output, reducing cardiovascular work. Also, there is moderate impairment of venous return late in pregnancy and since arteriolar sympathetic sensitivity is reduced in the normal pregnancy; an increase in PV may prevent syncope during positional change. It also may afford protection against acute blood loss, allow a graduated response to the hemoconcentration characteristics of such maternal disorders as preeclampsia, and enable the parturient a margin of tolerance for various anesthetic procedures. Finally, the failure of the dilution of hemoglobin to occur in early pregnancy has been related to stillbirth and intrauterine growth restriction (IUGR).19 The only disadvantage appears to be related to the timing of maximal PV increase (i.e., during the second trimester and at delivery) in patients with cardiac or renal disorders who may experience decompensation if their clinical disease is severe.
RED BLOOD CELL MASS
The increase in RBC mass does not begin until approximately 20 weeks, but then increases more rapidly than the PV until 28 weeks (see Fig. 1). From 28 weeks to term, the RBC mass rises only slightly, but the slope of erythrocyte increase begins to exceed that of the PV (a situation opposite that found earlier in pregnancy). The RBC mass is approximately 30% higher than its maximum in the non-pregnant state. In the early postpartum period, the RBC mass remains approximately 10% above non-pregnant levels for 1–2 weeks but returns to normal by 6 weeks. The decrease is principally related to blood loss at delivery and a decline in erythrocyte production. There is no evidence of increased RBC destruction during the puerperium. Bone marrow erythropoiesis assumes a normal level of RBC production by the end of the postpartum period.
The increase of RBC mass during pregnancy is accomplished by a complex interaction of several hormonal and physiologic factors, but in general it follows the erythropoietin production. In normal pregnancy, the erythropoietin level begins to rise slowly at 15 weeks, but the effects of this stimulation on RBC mass are delayed until 18–20 weeks. The maximal activity for erythropoietin occurs between 20 and 29 weeks, corresponding with the maximal increase in uterine blood flow and basal oxygen consumption. The level of erythropoietin begins to decrease slowly after birth in spite of blood loss at delivery. Studies in mice, but not humans, have shown that the RBC mass is increased in lactating animals compared with non-lactating controls. Increased erythropoietin levels are also prominent during hypoxia, phlebotomy, polycythemia, specific anemias (iron deficiency, vitamin B12 deficiency, or folic acid deficiency) but not those due to starvation, infection, most malignancies, and chronic renal disease. It is decreased by hyperoxygenation, ordinary transfusion, uremia, and malnutrition.
The significance of all the aforementioned changes are related to the diagnostic and therapeutic considerations of the specific anemias later in the chapter. It must be remembered, however, that failure to take these physiologic changes into account may confuse the clinician in cases of pathologic anemias.
ANEMIA DETECTION
As in most diseases, the diagnosis of anemia rests on a thorough history, physical examination, and laboratory assessment. Most of the symptomatology and physical signs of anemia can be attributed to a reduction in the oxygen-carrying capacity of the blood. Tissue hypoxia usually does not occur in the patient with anemia except in the most severe cases, although relative hypoxia can be observed as a result of increased oxygen consumption during pregnancy. In most cases, however, a compensatory expansion of RBC mass occurs to offset this process. The decrease in peripheral resistance, modest elevation of cardiac output, increased tissue perfusion, and hypocapnia seen during pregnancy may ameliorate anemia during pregnancy. Obviously, precursors such as iron, vitamins, and folic acid must be present or the anemia may proceed uncompensated. It should be remembered that severe, primary hematologic disease usually occurs infrequently during the childbearing years; however, hematologic manifestations secondary to other diseases occur as frequently in the pregnant patient as in the non-gravid patient.
History
The history should include general symptoms (e.g., evaluation of the performance status of the patient), which may be helpful in establishing the magnitude of the anemia as well as in delineating the effect of therapy. Symptoms of mild anemia, such as easy fatigability and malaise, are common in normal pregnancy. Patients with anemia during pregnancy of mild-to-moderate degree may have no additional manifestations. Patients presenting with the classic symptoms of tachycardia, exertional dyspnea, pallor, and palpitations should be carefully evaluated. Not only are these manifestations of moderate-to-severe anemia but these symptoms may also herald a rare underlying hematologic disorder such as leukemia or cardiorespiratory disease. Adolescent pregnancy, short interconceptual spacing, nutritional deficiencies, and concurrent medical diseases may contribute to an anemia process. A history of reaction or exposure to various drugs and chemicals may be an important factor in the diagnosis of hemolytic anemias. Finally, a family history may be helpful, particularly as it relates to hemoglobinopathies and other inherited hematologic disorders.
Physical examination
On the physical examination, a central feature of anemia appears to be the pallor caused by the reduced hemoglobin level. This is most helpful in Caucasians and Eurasians, but examination of the mucous membranes may be used for similar purposes of African descent. During pregnancy, however, examination of the skin and mucous membranes in any race may be misleading owing to the hyperemia of these areas. Palmar creases, which usually are white in the fully open hand if the hemoglobin level is less than 10 g/dL, may appear pink in anemic pregnant patients because of hyperemic effects of human placental lactogen (HPL) and progesterone. Pallor of the nail beds, however, is a reliable indicator of anemia during pregnancy in any racial group. Nails that are ridged longitudinally and flattened (koilonychia) rather than convex are present in chronic IDA. Other observations that may be helpful include cyanosis (congenital methemoglobinemia) and jaundice (hemoglobinopathies and hemolytic processes). An enlarged, smooth tongue is associated with pernicious anemia, but is quite rare in the United States. Glossitis related to IDA is more common; the tongue is coated, enlarged, and painful. With severe IDA, the lips may reveal cracks, particularly at the edges (chelosis). Neurologic examination may be needed if vitamin B12 deficiency or IDA is suspected, since both may give rise to peripheral neuropathies. With the exception of pallor, these aforementioned signs, although not altered by pregnancy, are usually not present unless the anemia is severe (hemoglobin <6–8 g/dL). It is also important that the skin, liver, spleen, and lymph nodes be evaluated for enlargement, excoriation, or other abnormalities that may indicate primary hematologic disease or a secondary response to other disease states.
Laboratory examination
Since anemias are so common in women of reproductive age and since most women with mild-to-moderate anemias during pregnancy are asymptomatic, it is recommended that all patients be assessed for anemia during their initial prenatal visit, late in pregnancy (28–32 weeks), and at the postpartum visit. Laboratory assessment includes a complete blood count (red blood cell indices, hematocrit, hemoglobin, white blood cell count, and platelet count). A peripheral smear and a reticulocyte count should be considered if the initial complete blood count is abnormal. The laboratory should examine carefully the RBC morphology in all anemic prenatal patients, because it may reveal several hematologic abnormalities. Because there is a demonstrated association between maternal anemia and unfavorable pregnancy outcome, these screening tests for anemia appear to be cost effective and beneficial for the mother and the baby. Normal screening and specific values for the pregnant and the non-pregnant state are shown in Table 1.
Table 1. Laboratory norms for the non-pregnant and pregnant patient
| Non-pregnant | Pregnant | |
| General assessment |
| |
| Hemoglobin | 13–15 g/dL | 11.5–12.5 g/dL |
| Hematocrit | 37–47% | 33–38% |
| RBC count | 4.2–5.4 million/mm3 | 3.8–4.4 million/mm3 |
| Mean corpuscular volume (MCV) | 80–100 µm3/cell | 70–90 µm3/cell |
| Mean corpuscular hemoglobin | 27–34 pg/cell | 23–31 pg/cell |
| Mean corpuscular hemoglobin concentration | 31–36 g/dL | Unchanged |
| Reticulocyte count | 0.5–1.0% | 1–2% |
| Specific diagnostic tests |
| |
| Serum iron | 50–110 µg/dL | 40–100 µg/dL |
| Unsaturated iron binding capacity | 250–300 µg/dL | 280–400 µg/dL |
| Transferrin saturation | 25–35% | 15–30% |
| Serum ferritin | 75–100 µg/L | 55–70 µg/L |
| Free erythrocyte protoporphyrin | 25 µg/L | 35 µg/L |
| Estimated sedimentation rate | 0–15 mm/h | 40–50 mm/h |
| Serum folate (fasting) | 6.5–19.6 ng/ml | 5–10 ng/ml |
| Serum B12 | 150–450 pg/ml | Unchanged |
The laboratory assessment of anemia is more difficult during pregnancy. In general, iron-sufficient, disease-free women with relative anemia during pregnancy have a hemoglobin level above 11 g/dL and a hematocrit above 35%. The average hemoglobin levels during pregnancy are between 11.5 and 12.5 g/dL instead of the normal 13–15 g/dL found in the non-pregnant state (see Table 1). The hemoglobin content during pregnancy tends to be further reduced, since the derivative actually measured by most laboratory techniques (cyanmethemoglobin) is slightly lower during gestation. The hematocrit likewise is lower in pregnancy, averaging 33–38% compared with the 37–47% range associated with the normal non-pregnant female. The MCV appears to be a good discriminator of the various types of hypoproliferative anemias (Fig. 2). All indices reflect average cell values and do not detect abnormalities in mixed cell populations. A patient with an IDA and concomitant megaloblastic process may reveal normal indices rather than the classic presentation of either disease.
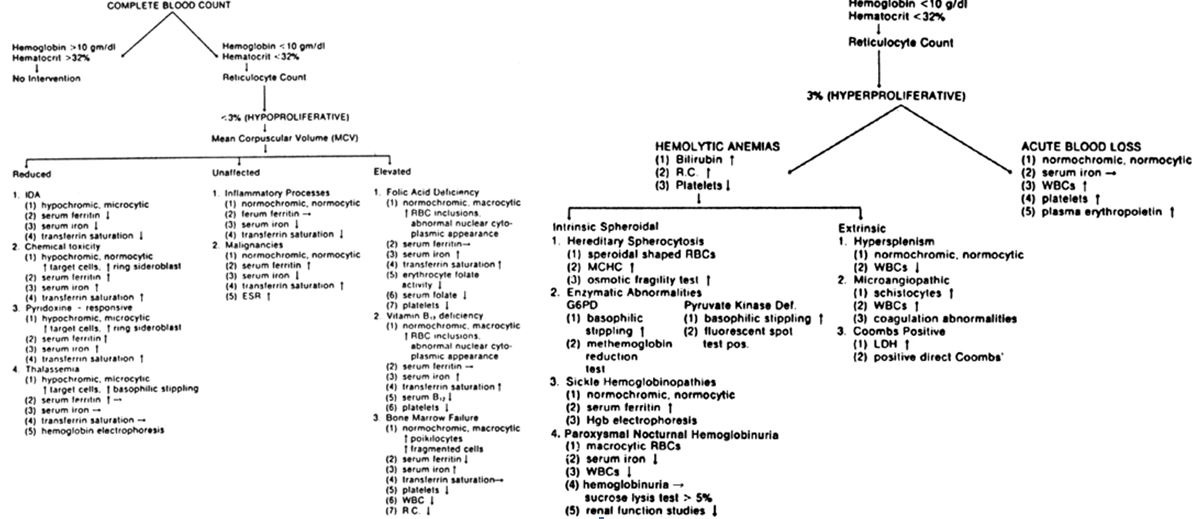 Fig. 2. Anemias and related laboratory findings
Fig. 2. Anemias and related laboratory findings
If the initial assessment of the patient's hematologic state indicates an anemia, the classification by mechanism as outlined in Fig. 2 can allow the physician to arrive at a diagnosis by an orderly method of laboratory analysis. The reticulocyte count (RC) divides anemias into categories in which the marrow is proliferative or hypoproliferative. Methylene blue is used to stain the reticulocytes (young, nonnucleated RBCs) on the peripheral smear. Usually if the RC in the presence of anemia is below 3%, the mechanism is one of diminished erythropoiesis. If it is above 3%, excessive hemolysis or acute blood loss is usually the cause. Therefore, a normal RC of 1–2% during pregnancy in a patient with anemia indicates a hypoproliferative process in which the patient cannot respond with new RBC production. Examination of the bone marrow is usually not performed during pregnancy owing to hypervascularity and subsequent maternal risk. However, if serious hematologic disease is suspected, then this diagnostic technique is warranted. Specific laboratory methods used during the diagnosis of anemia are discussed under the particular anemia processes. A flow sheet as shown in Fig. 2 allows the orderly procurement of essential laboratory tests in the diagnosis of anemia.
DECREASED ERYTHROCYTE PRODUCTION
Disturbance in maturation: hemoglobin synthesis
IRON DEFICIENCY ANEMIA
Iron deficiency anemia (IDA) afflicts 6% of the 60 million women of reproductive age in the United States and disproportionately represented are those who are multiparous, socioeconomically disadvantaged as well as those who are of African or Hispanic descent.20 IDA accounts for up to 75% of all the anemias diagnosed during pregnancy and, along with acute blood loss, comprises most of the anemias during the puerperal period.21 IDA complicates over 10% of all postpartum women up to 6 months postpartum.22 It is known that one in eight American women have iron deficiency up to 12 months after delivery.23 As previously noted, particularly IDA, is associated with maternal adverse affects such as dizziness, fatigue, infections, lactation problems, and a need for prolonged hospitalization.14 In addition, cognitive function as well as postpartum depression and a reduced sense of well-being are co-morbidities associated with IDA.15, 16 The infants of women with IDA have also been found to have smaller placental weights, preterm birth, fetal growth restriction, lower hemoglobin values, lower birth weights, and lower Apgar scores.24, 25 Understandably, given these adverse maternal–fetal effects of IDA, it is incumbent upon the provider to understand iron absorption, metabolism and replacement.26
Iron compartments
Not only is iron necessary for RBC formation it is also intimately involved with proteins integral to intermediary metabolism as nearly half of the enzymes and co-factors in the Kreb's cycle either contain iron or require its presence. The basic compartments of iron distribution include hemoglobin iron, storage iron, myoglobin iron, labile pool iron, other tissue iron, and transport iron. Table 2 illustrates the significant differences in the total body iron distribution between the adult male and the female during the reproductive years. Pregnancy normally increases the amount of iron but not the percentage distribution to each compartment. However, socioeconomic status, nutritional status, and concurrent disease processes can modify the dispersion of iron in each compartment.
Table 2. Iron compartments*
| Iron content (mg) | |||
| Compartment | Male | Female† | Total body iron (%) |
| Hemoglobin iron | 2500 | 1700 | 67 |
| Storage iron | 1000 | 700 | 27 |
| Myoglobin iron | 130 | 130 | 3.5 |
| Labile pool | 80 | 80 | 2.2 |
| Other tissue iron | 8 | 8 | 0.2 |
| Transport iron | 3 | 3 | 0.08 |
* These values represent estimates for an iron-sufficient person
† Nonpregnant
Hemoglobin iron
Assessments with radioactive tracers have shown that hemoglobin iron makes up 65–70% of total body iron and averages 1700 mg in a normal adult woman. Iron deficiency is precipitated during pregnancy because of expansion of the RBC mass during the second trimester. IDA is compounded by the preferential fetal utilization of iron from maternal storage compartments. Iron loss during late pregnancy and after delivery further contributes to postpartum IDA. Approximately 1000 mg of elemental iron are needed during pregnancy with 300 mg being used for the fetus and placenta, while 700 mg is added in the expansion of maternal hemoglobin.26 In addition, approximately 200 mg is lost in bleeding during and after delivery, but some of the 500 mg of iron from metabolized maternal RBC is returned to the iron storage postpartum, resulting in at least 500–600 mg of iron debt. Thus, this amount of iron reserve is considered the minimum for pregnant women. However, a recent report indicates that only 20% of women of reproductive age have such a reserve of iron.27 As IDA in the postpartum period may impair the woman’s ability to participate in child care, household tasks and social activities, it also diminishes their productivity in regard to physical and intellectual work.28 These changes are worrisome because they have been shown to lead to disturbed maternal–infant interactions as shown by one study comparing infants of IDA women to non-anemic parturient controls.29
Storage iron
Storage iron exists in two forms: ferritin and hemosiderin. In healthy, iron-sufficient women, storage iron totals approximately 600–800 mg and makes up approximately 27–30% of total body iron. Depletion of this storage compartment occurs when iron loss or use exceeds iron absorption; it is usually decreased during pregnancy even in iron-sufficient women.
Ferritin contains approximately half the storage iron and is found in plasma as well as in most of the cells in the body. The role of ferritin in iron absorption is direct; a low concentration of this compound in the intestinal mucosal cell enhances the biosynthesis of additional apo-ferritin, which results in more iron absorption. The life span of apo-ferritin is only a few days; the degeneration and re-synthesis provides an available intracellular iron pool. The measurement of serum ferritin has become one method of delineating iron stores without having to resort to bone marrow sampling.
Hemosiderin is found only in cells of the reticuloendothelial system, such as those of bone marrow, liver, and spleen (about one-third in each organ). This storage iron is attached to a substrate called apo-hemosiderin, which is amorphous, thus lacking the firm, crystal-like molecular arrangement of ferritin. The mechanism of mobilization, use, and contribution of storage iron in an anemia process is discussed in the sections on iron absorption and homeostasis. While the measurement of ferritin is involved with an assessment of iron absorption capabilities, the assessment of hemosiderin is a measure of iron balance; until hemosiderin becomes depleted, no signs of iron deficiency develop (Fig. 3). Therefore, as long as storage iron is present and released normally, no change in the peripheral blood smear is seen.
|
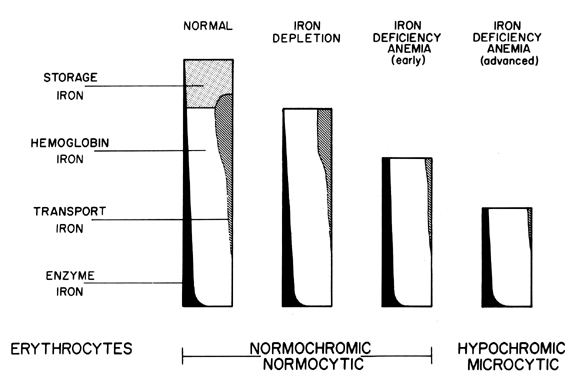
During pregnancy, the increase in RBC mass gives an indirect measurement of iron stores. The principal cause for a failure of the expansion of the hematocrit is the absence of bone marrow hemosiderin. This finding indicates exhaustion of storage iron, its absence being the earliest sign of iron deficiency. If IDA is clinically evident by a decreased hemoglobin concentration, by a characteristic microcytic, hypochromic blood smear, and by altered RBC indices, the iron stores will be nonexistent. On the other hand, bone marrow stores decrease to minimal levels during pregnancy, even in iron-sufficient women, in the absence of iron supplementation. Since a bone marrow examination is not practical during pregnancy unless severe hematologic disease is suspected, reductions in serum ferritin, hemoglobin concentration, and RBC indices are the first laboratory indications of inadequate iron stores. Iron stores are reduced in IDA, blood loss, and nutritional anemias; however, hemolytic processes, hereditary anemias, and ineffective iron utilization during infection or inflammation may be associated with normal to increased storage iron levels and concomitant reductions in hemoglobin levels, and RBC indices.
Myoglobin iron
Myoglobin iron accounts for approximately 3–4% of total body iron (130 mg) and is relatively constant between men and women, even in pregnancy. Each myoglobin molecule consists of a heme moiety surrounded by a helical chain of 150 amino acids. The myoglobin is less than 1% iron and has a molecular weight of approximately 17,000 Da. It is found in most muscle cells and appears to serve as an oxygen reservoir when cellular damage from hypoxia occurs. Function of this iron component in pregnancy has not been reported.
Labile iron pool
The labile iron pool represents approximately 80 mg of iron constantly in exchange between the plasma, interstitial, and intracellular compartments. The labile iron pool offers a method of studying the clearance rate, incorporation data, and daily iron turnover; ferrokinetic studies indicate that in IDA the incorporation of iron into hemoglobin and subsequently into normal RBCs is almost 100%. In contrast, in hemolytic or megaloblastic anemia, the incorporation of iron is rapid and complete, but there is ineffective erythropoiesis and early destruction of the erythroblasts. Patients with aplastic anemia or thalassemia also demonstrate an incorporation rate that is markedly reduced in the presence of normal erythrocytes.
Enzymatic iron
Tissue iron is constant during pregnancy and represents approximately 6–8 mg of iron, or between 0.2% and 0.5% of total body iron. Although extremely small, this compartment is very important because it includes enzymes such as the cytochrome peroxidase catalase dehydrogenase, as well as acyl-CoA and various other oxidases. Changes in enzymatic iron are reflected in decreased efficiency of the mitochondrial systems; thus, severe anemias may affect fetal growth and development by such a mechanism. In addition, maternal–fetal steroidogenesis and host response to various disease states may be modified by functional changes in these critical enzyme systems. As shown in Fig. 3, the amount of parenchymal iron is altered only in severe IDA, and it is one of the last compartments to become depleted.
Transport iron
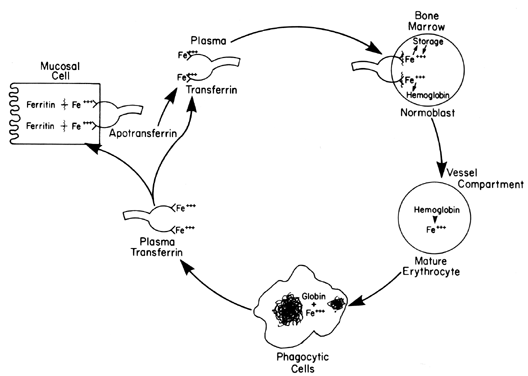
Transport iron represents less than 0.1% of the total iron (3 mg) in both men and women, and yet, kinetically, it is the most active compartment, being replaced approximately every 2.5 h. The metal in this category represents the interchange between all compartments previously mentioned and is not altered during pregnancy. Transport iron is loosely bound to a specific protein (i.e., transferrin), as shown in Fig. 4.
|
Iron absorption
Iron absorption depends on many factors (Table 3), reflecting the diet, the status of the bowel lumen, and mucosal abnormalities, as well as systemic factors. Iron is principally absorbed in the duodenum and proximal small intestine. The iron presented to the gastrointestinal tract is usually in one of three forms: the ferrous form (from elemental iron), hemoglobin (from animal protein sources), and the trivalent or ferric form (from vegetable complexes) (Fig. 5). The ferrous salts are best, since they need no conversion to be absorbed; the ferric iron in vegetable protein complexes must be reduced to the divalent state before it can pass into the mucosal cell. Hemoglobin iron is readily absorbed after being hydrolyzed in the gut lumen into heme and globin. The globin, although degraded by intestinal enzymes into small peptides, remains an integral factor in absorption, since it continues to stabilize the heme iron in the ferrous state.
Table 3. Conditions affecting iron absorption
| Increased absorption | Decreased absorption | |
| Iron content |
|
|
| Form of iron | Heme iron | Enteric-coated capsules |
| Intraluminal factors |
|
|
| Intestinal secretions | Hydrochloric acid | Achlorhydria |
| Stomach contents | Ascorbic and other acids, cysteine | Oxalates, phytates, phosphorus, carbonate |
| Intestinal motility | Atropine | Cathartics |
| Chelators |
| EDTA, desferrioxamine |
| Metallic cations |
| Cobalt, nickel |
| Mucosal factors |
|
|
| Disease states | Intermittent outlet obstruction | Gastrectomy, lymphoma |
| Cellular | Decreased mucosal iron | Increased mucosal iron |
| Systemic factors |
|
|
| Erythropoiesis | Acute blood loss | Aplastic anemia |
| Iron requirements | Pregnancy, growth | Weight loss, thalassemia |
|
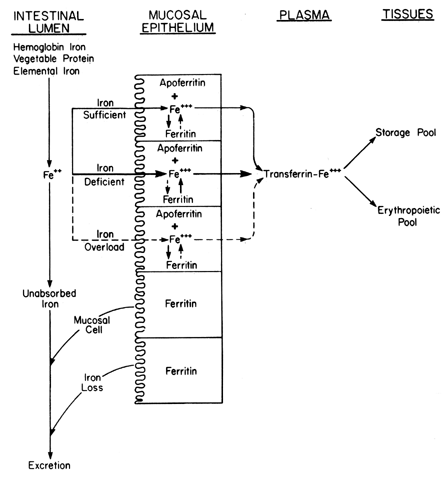
Gastric acid degrades the organic iron complexes (vegetable protein) and reduces the trivalent mineral to ferrous iron. Moreover, hydrochloric acid serves to prevent alkaline dietary constituents from forming insoluble complexes with ferrous iron, to prevent various agents from oxidizing iron to the ferric state, and to prevent iron precipitating as insoluble ferric hydroxide. While gastric secretions may facilitate elemental iron absorption, it does not appear to have any effect on hemoglobin iron absorption. The alkaline milieu of the duodenum and upper small bowel favors absorption, since the ferrous iron has greater solubility in these surroundings; this is true for all three sources of iron, but it is particularly necessary for hemoglobin iron. Therefore, agents that form alkaline-soluble complexes promote iron absorption; these include the reducing agents ascorbic acid, fructose succinic acid, lactate, pyruvate, and sorbitol and certain amino acids such as cysteine. Certain drugs such as hydroquinone and alcohol may also increase iron absorption by increasing gastric acid production, while medications that decrease intestinal motility, such as atropine or reserpine, also enhance iron absorption.
In contrast, oxidants and certain medications reduce iron absorption by increasing the concentration of the trivalent form. Other substances form insoluble complexes with the ferrous iron; chelators have a similar mode of action. Metallic cation competitors decrease absorption by competing for binding sites, while other agents reduce absorption by reducing gastric acidity. Cathartics and disease processes reduce iron absorption by increasing intestinal motility, reducing the time of exposure of mucosal cells to iron. Surgical disruption of the gastric (i.e. gastric bypass surgery) or duodenal mucosa limits the surface area for absorption.
Finally, another factor, pica, has long been associated with IDA. Pica is the ingestion of various substances that have no dietary value; pagophagia (ice), geophagia (clay), and amylophagia (starch) are examples. In some ethnic groups, it is a common presenting symptom in patients with proven IDA. The “cravings” these patients experience are not always for non-foods, but may be for vegetables (carrots or celery) or commercial items such as “fast foods” (invariably not a good source of iron). Formerly all forms of pica were thought to delay iron absorption. Now it is clear that pica is a sign of IDA rather than a cause. A history of pica in an adult suggests that the patient has IDA, since the abnormal craving usually disappears in a few days after the institution of iron supplementation. Although not diagnostic, certain food cravings during pregnancy may represent pica due to the increased iron demands of pregnancy. This hypothesis is supported by the observation that most pica occurs after the 20th week, a time during which the iron demands increase most rapidly. The relationship of pica to IDA is not completely understood; however, it seems to be related to a decrease of iron-containing enzymes in the oral mucosal cells of these patients.
Iron homeostasis
The conservation of iron in humans is tenacious, with only 0.1% of the total amount of body iron lost each day. This amount is easily replaced in the non-pregnant adult if the dietary source is adequate. The average amount of iron excreted by the adult averages 0.9 mg/day, with most being lost in the intestinal tract as desquamated gastrointestinal cells, blood, and bile. Additionally, epidermal cell loss and sweat produce a daily iron loss of 0.2 mg. In areas of high temperature and humidity, an additional 0.5 mg/day may be lost, but this loss rarely produces IDA. Finally, a small amount (0.1 mg) of iron is excreted daily in the urine. In women during their reproductive years, menses compounds iron loss. Although the blood loss is relatively constant in successive periods, the individual variation between women is large. Most normal, iron-sufficient women lose an average of 25–45 ml of blood through menses each month, which approximates to 0.7–1.4 mg/day in terms of iron loss. The blood loss is lower in the younger age groups, in women taking oral contraceptives, in women with good nutrition, and in higher socioeconomic groups. Blood loss is exaggerated in women over 40 years old, those with intrauterine contraceptive devices, patients after tubal sterilization, females of high parity, women of lower socioeconomic groups, and those with poor nutrition. A menstrual blood loss of 50–60 ml seems to be the upper limit of normal, since women whose losses have exceeded this amount eventually develop IDA.
Pregnancy has a marked effect on iron homeostasis (Table 4). During the first half of pregnancy, iron requirements are not increased. After the 20th week, however, the RBC mass begins to expand and the fetus requires more iron. Even with increased absorption, the amount of dietary iron is not adequate to prevent a reduction in iron stores. Obviously, if dietary iron does not meet the requirements, then the storage iron must supply the needs. Since the average North American diet provides about 6 mg of iron per 1000 calories and the pregnant woman of ideal weight should consume 1800–2200 calories each day, the average pregnant woman should receive between 10 and 12 mg of elemental iron daily. Even though the absorption rate increases from 10% in the first trimester to 30% during the latter half of pregnancy, the iron acquired from the diet alone during this time ranges from 450 to 500 mg; thus, approximately 400–500 mg must be supplied by storage iron during pregnancy. In women who are deficient in storage iron prior to pregnancy, this further requirement may lead to overt IDA. It should also be noted that if the storage iron is insufficient at the beginning of pregnancy, the maternal hemoglobin mass will not be expanded until the fetal demands are met. The lack of expansion of the RBC mass thus may be an indication of inadequate iron stores. Postpartum, the amount of iron in the expanded hemoglobin mass not lost at delivery can be returned to the iron stores and the anemia can be partially balanced. However, in most women this is not sufficient replacement, and almost all women will remain deficient in storage iron unless they receive supplementation during gestation.
Table 4. Iron homeostasis in pregnancy, expressed in milligrams of elemental iron
| Maternal loss | Maternal gain | Borrowed |
| 500 (RBC mass expansion) | 490 (diet)† |
|
| 300 (fetal/placental) | 270 (returned to storage after delivery) |
|
| 190 (basal loss) |
|
|
| 230 (loss at delivery) |
|
|
| TOTAL: 1220 | TOTAL: 760 | 460 |
† Average of 3–4 mg/day actual absorption
Laboratory assessment
The laboratory diagnosis of IDA depends on the severity of iron depletion. In the mildest stage, iron deficiency is manifested by a decrease in serum ferritin, but serum iron, hematocrit, and hemoglobin values are usually normal (see Fig. 3). Iron deficiency without anemia is the next stage; absence of storage iron, manifested by reduced serum ferritin, low serum iron, and decreased transferrin saturation without anemia are characteristic. IDA is first reflected by a reduced RBC mass, then reductions in hematocrit and hemoglobin levels following hypochromasia and microcytosis (see Fig. 3).
Iron assessment tests must be altered during pregnancy. A relative decrease in serum iron occurs from approximately 12 weeks through 32–34 weeks owing to the increase in plasma volume. However, as the RBC mass approaches the increase in plasma volume, the serum iron rises to normal non-pregnant levels. During the early postpartum period, serum iron again rapidly decreases over the first 4–5 days before returning to normal at the end of the first week. This is probably related to ineffective release of storage iron owing to the change in hormonal milieu. Transferrin begins to increase from 12 weeks through 34–36 weeks, but a slight decrease occurs toward term. During the first 7 days after delivery, the transferrin concentration increases before it returns to normal levels approximately 10 days postpartum. These changes are also thought to be hormonally mediated, since similar changes have been observed in women taking oral contraceptives. These observations are present only in iron sufficient pregnant women and are not significant enough to affect the diagnosis of IDA.
The most consistent laboratory findings in a patient with IDA are a decrease in the hematocrit and hemoglobin concentration, with a concomitant decrease in the mean corpuscular volume (MCV); typically the MCV will be <80 fL. Other laboratory evidence of IDA includes hypochromia and microcytosis observed on the peripheral smear. Serum iron, serum ferritin, total iron binding capacity (TIBC), and transferrin saturation may be used to confirm IDA, although they are not routinely obtained during antepartum screening.30 IDA is usually suspected if the serum iron is below 60 µg/dL, the serum ferritin is below 30 µg/L, the transferrin saturation is below 20%, and the TIBC is above 350 µg/dL; the normal values are shown in Table 1. Values of less than 30 µg/dL, less than 10 µg/L, less than 10%, and greater than 400 µg/dL in the serum iron, ferritin, transferrin saturation, and TIBC, respectively, are diagnostic of IDA.
Differential diagnosis
Although iron depletion can be easily treated in most cases, it is important to rule out more severe hematologic or systemic diseases. Hypochromic anemia, caused by the following conditions, may be confused with IDA: (1) chemical toxicity related to the intake of chloramphenicol, lead, alcohol, or isoniazid; (2) inflammatory processes; (3) malignancy; (4) pyridoxine-responsive anemia; and (5) hemoglobinopathies.
Drug effects may be toxic (lead, alcohol) or idiosyncratic (isoniazid). Maternal encephalopathy and basophilic stippling of the normal red cells may be observed with lead poisoning. On the other hand, toxic dosages of alcohol or reactions to drugs (chloramphenicol) usually cause a deficiency in porphyrin synthesis, which leads to sideroblastic anemia. On the peripheral smear, the red cells are hypochromic, but the presence of target cells and ring sideroblasts helps differentiate this process from IDA. Characteristically, in toxic anemias the serum iron is elevated and the transferrin saturation is increased; the opposite is found in IDA. Examination of the bone marrow, if performed, reveals the presence of ring sideroblasts, along with increased iron stores common in porphyrin synthesis deficiencies.
Chronic diseases, such as malignancies, and inflammatory processes, including arthritis and the collagen-vascular diseases, may cause anemia, but usually the peripheral smear reveals normochromic and normocytic cells. Unfortunately, the symptoms from these processes are protean, and they may be difficult to differentiate from those of IDA if the peripheral blood picture is similar. As with IDA, patients with chronic disorders will characteristically have low serum iron levels. Elevated-to-normal serum ferritins are seen in these disorders, while patients with IDA exhibit decreased ferritin values. In those affected with inflammatory processes, the transferrin saturation is decreased and TIBC is increased, as seen in IDA. On the other hand, patients with malignancies exhibit an increased transferrin saturation and decreased TIBC (see Fig. 2). Another screening device to differentiate IDA from acute and chronic inflammation is the erythrocyte sedimentation rate. It almost always is increased in neoplasia or acute and chronic inflammation over that found during normal pregnancy, whereas in severe IDA it is normal. Erythrocyte survival times are slightly decreased in IDA and may also be shortened in chronic diseases. Infestation (e.g., hookworms), although rare in the United States, must be included in the differential diagnosis in developing countries, in world travelers, or in areas of poor sanitation. Other causes of excess intestinal iron loss, such as diverticulitis, intestinal cancer, and peptic ulcer disease, are rare during pregnancy.
Pyridoxine (vitamin B6)-responsive anemias are characterized by hypochromic, microcytic erythrocytes with increased ring sideroblasts and prominent target cells. Unlike IDA, serum iron levels, transferrin saturations, and serum ferritins are increased in patients with pyridoxine-responsive anemia. The TIBC is characteristically decreased.
In hemoglobin synthesis abnormalities, such as alpha and beta thalassemias, a microcytic, hypochromic pattern may also be demonstrated, and these abnormalities may be confused with IDA. RBC morphology usually shows more basophilic stippling and target cells in thalassemia patients; these are not common in patients with IDA. In a person of African or Mediterranean origin, thalassemia should be ruled out by hemoglobin electrophoresis. Other aids in diagnosis include the erythrocyte count, the MCV, and the reticulocyte count (RC). Almost 85% of patients with heterozygous thalassemia have an RBC count greater than 5 million/mm3 despite a reduced hemoglobin concentration. In contrast, only 3% of adults with IDA have RBC counts over 5 million/mm3. The MCV is reduced in thalassemia; values of 55–70 µm3/cell are the rule, whereas values below 70 µm3/cell are very uncommon in IDA. Reticulocytosis of greater than 5% is much more commonly noted in thalassemia patients than in those with IDA. In addition, serum iron concentration and transferrin are not usually altered in the hereditary anemias; however, serum ferritin levels are characteristically normal to increased.
In the final analysis, the response to iron therapy may prove to be diagnostic. This method is commonly used in the low-risk patient and can be harmful only in patients who have allergic reactions or in those who have iron overload due to other hematologic diseases. In any case, in “therapeutic” diagnosis of IDA, the patient should be followed carefully to detect iron-unresponsive anemia. If the patient with IDA receives sufficient medication, an intense reticulocytosis should occur between the 7th and 10th day after the initiation of therapy. A significant increase in hemoglobin values should be evident in 3–4 weeks, and the hemoglobin concentration should approach normal values within 2 months. This may not occur during pregnancy, however, since the increase in RBC mass and transfer of iron to the fetus may continue the iron-depletion process. In this case, if other factors such as inflammatory diseases, chronic infections, or hemoglobinopathies are not suspected, the iron therapy should be continued until the end of the pregnancy, since eventually there will be a response.
Iron therapy
There are basically two methods by which iron can be administered to correct IDA. Traditionally, the most common approach to the treatment of IDA has been oral iron supplementation as the elemental salt, parenteral iron therapy or in severe cases particularly those associated with blood loss, direct infusion of blood components. The most common and least expensive method of supplementation is oral iron supplementation in the form of ferrous sulfate, ferrous gluconate or carbonyl iron. The latter is more expensive but better tolerated from a gastrointestinal standpoint. Some iron preparations have been combined with vitamins, including prenatal vitamins, but are no more effective than simple elemental iron and are more expensive. In addition, they are usually given once per day, a dosage not likely to eradicate IDA. Sustained-released capsules of iron given in an effort to avoid upper gastrointestinal side-effects such as nausea and vomiting have a reduced absorption rate (80%) compared to elemental iron. These preparations do not appear to be associated with any less symptomatology than carbonyl or fumarate elemental iron. Most oral agents contain 200–300 mg of the iron complex yielding approximately 40–60 mg of elemental iron.31, 32 This ideal dosage (40–60 mg) of elemental iron per day should be given in three divided doses to take advantage of the absorption rate which is maximal about 4–6 h after each dose. It should be administered with meals during pregnancy as this may reduce the incidence of gastric irritation, nausea, vomiting, and diarrhea. The frequency and timing of the dosage does not appear to affect the side-effect of constipation is observed in 10–12% of pregnant women. One study observed common adverse gastrointestinal side-effects including abdominal pain, dyspepsia, and constipation when a ferrous iron agent was administered daily for up to 12 weeks.33 In this study, 33% of women given oral iron, gastrointestinal side-effects resulted in their inability to take the iron as prescribed. Similarly, Wulff and Ekstrom34 noted that the recommendations for iron supplementation amongst their pregnant women were adhered to less than 40% of the time. In another investigation, using several types of elemental iron preparations, up to 10% of patients were not compliant with therapy after just 2 weeks, 25% after 1 month and 32% after 2 months of therapy.35 Obviously in some cases, the treatment is worse than the disease as it interferes with the woman’s quality of life. Finally, one of the most common causes of fatality from poisoning in young children includes elemental iron preparations.36 Indeed, this has led many to suggest that food fortification, particularly in developing countries may enhance the ingestion of enough iron to prevent IDA.37 In any case, if oral iron therapy is well tolerated, it should be continued throughout the pregnancy and for up to 3 months postpartum.38 During the therapy in the patient with IDA progress should be monitored and if the hemoglobin level remains unchanged after 6 weeks of therapy other hematologic problems should be considered.
The CDC recommends that all pregnant women take a 30 mg/day iron supplement by the first prenatal visit.39 Intermittent iron supplementation (one to three times per week) appears to be as effective as daily supplementation for preventing anemia at term and is better tolerated.40
A 2015 systematic review for the United States Preventive Services Task Force observed that routine iron supplementation had inconsistent effects on a variety of pregnancy outcomes, but noted a consistent reduction in the frequency of iron deficiency anemia at term (relative risk [RR] 0.29, 95% CI 0.17–0.49; four trials).41 However, there is no convincing evidence that iron supplementation in non-anemic pregnant women improves maternal or child clinical outcomes.
Women with iron deficiency anemia (first- or third-trimester hemoglobin [Hb] <11 g/dL or second-trimester Hb ≤10.4 g/dL and low serum ferritin) should receive an additional iron supplement of 30–120 mg per day until the anemia is corrected.42 Iron absorption decreases with increasing dose, thus larger supplementation amounts are best split into several doses during the day.
A second option, is intravenous iron therapy which has the potential to both quickly correct the anemia by providing sufficient iron to replenish storage and also holds promise of averting or minimizing blood transfusion in the postpartum anemic patient.43, 44 Dextran irons are not typically used due to significant adverse side effects that are associated with this form of IV iron. Non-dextran-containing IV iron agents such as sucrose or gluconate parenteral products are limited by the FDA approved dosing guidelines. Iron gluconate is only approved by the FDA for use in hemodialysis patients (maximum dose 125 mg IV over 10 min). Sucrose iron has been used and when compared in a randomized open-label study, it restored iron stores faster and more effectively than oral iron.45 However, iron sucrose products have a maximum dose limited to 100 mg intravenously. These agents may require 5–20 IV doses be administered to allow for adequate replacement of iron stores.
Another IV form of iron is ferric carboxymatose (FCM). FCM has been compared to oral iron in a group of open-label randomized trials that included over 2000 women who received FCM vs. a control group of women using oral iron. In these studies women assigned to receive IV FCM achieved 12 g/dL hemoglobin levels (91 vs. 67%), a rise of ≥3 g/dL hemoglobin (91 vs. 65%) and had less postpartum fatigue, depression and child care issues.6 Therefore, those women who do not respond to oral iron or who are not compliant with oral iron are great candidates for IV iron infusions.
In summary, patients with iron depletion or deficiency will rarely be detected prior to changes in the hemoglobin or hematocrit level. Supplemental iron therapy is recommended in most pregnant women because (1) iron depletion is so common, (2) therapy is safe and inexpensive, (3) lack of response to supplements is an indication for further testing, (4) other testing is avoided, in most cases, and (5) the effects of an undetected common disorder like IDA on the mother and fetus are not fully known.
THALASSEMIA
Another disorder in hemoglobin production is thalassemia, which is caused by an alteration in the rate of synthesis of the α- or β-chain. The hemoglobinopathy is classified based on which chain is affected. Clinical symptoms may also be used to classify thalassemia as thalassemia major, intermedia, and minor. In general, only the homozygous forms are in the major category, while the heterozygous forms demonstrate variable degrees of symptomatology.46
Homozygous forms
α-Thalassemia major (hemoglobin Bart's disease) appears to be the most severe, with fetal death occurring in most cases. The poor outcome is due to the increased production of α-chains, which combine to form Bart's hemoglobin, a high-oxygen-affinity hemoglobin that functions poorly for oxygen transport. Electrophoresis on fetal blood shows 85–100% Bart's hemoglobin, with only small amounts of hemoglobin A and hemoglobin F. The infant develops severe anemia and hydrops similar to that noted in those with Rh alloimmunization. Homozygous α-thalassemia is the most common form of hydrops fetalis in Southeast Asia, but is rare in the United States. Homozygous β-thalassemia (Cooley's anemia) is characterized by severe anemia, usually leading to death in early childhood. Patients with homozygous beta (0) thalassemia are unable to make any Hb A. In untransfused patients only Hb F and Hb A2 are present on hemoglobin electrophoresis.47 Treatment involves repeated transfusions, and pregnancy is infrequent.
Heterozygous forms
Patients heterozygous for thalassemia rarely manifest signs or symptoms of the disease. Heterozygous β-thalassemia is the most common form; patients usually exhibit only mild hypochromic, microcytic anemia, with elevation of the hemoglobin A2 (above 3.5%) on the haemoglobin electrophoresis as well as a mildly increased hemoglobin F percentage (2–5%).
Heterozygous α-thalassemia is difficult to detect by laboratory means because the affected globin chain is common to all types of hemoglobin. Therefore, there is a reduction in the amount of hemoglobin A, F, and A2, but the percentage of these compounds remains the same as in normal persons. Often times the existence of alpha thalassemia trait is recognized in family members because of the birth or prior family history of an individual with more severe alpha thalassemia (e.g., HbH disease or hydrops fetalis with Hb Barts). Therefore, family history is important in raising suspicion of this condition in family members. In the absence of a positive family history, the condition should be considered in individuals from ethnic groups with a very high incidence of alpha thalassemia (e.g., Asians and people of Mediterranean origin; African Americans almost never have severe alpha thalassemia despite a high incidence of a single gene deletion). Anemia, hypochromia and microcytosis may be minimal or non-existent, especially if only one locus is deleted (i.e., alpha thalassemia minima, silent carrier state).
The presence of microcytosis with minimal or mild anemia is a major clue, but, in many individuals, hypochromic and/or microcytic cells on peripheral blood films, in the absence of overt microcytic indices or anemia is the only clue. In newborns, small amounts of Hb Barts may be present; in children and adults, low levels of HbH may be found, although the analysis of hemoglobin in circulating red cells can be normal. Unlike the beta thalassemic disorders, hemoglobin A2 levels are not increased in the alpha thalassemias. The only certain way to determine the alpha thalassemia genotype of an individual with suspected alpha thalassemia is direct sequencing DNA analysis of the globin genes.
Heterozygous thalassemia in pregnancy has few adverse maternal or fetal effects, except for a slight increase in spontaneous abortions. In general, the maternal outcome is related to the severity of the anemia; those more severely affected have an increased maternal morbidity, although fertility does not appear to be affected. The double heterozygote (α-β-thalassemia) is also difficult to diagnose and may be more common than previously suspected.
Prenatal diagnosis
The prenatal diagnosis of thalassemia syndromes should be offered to any couple with a prior affected child or those at risk because of ethnic origin or pedigree. Prospective parents not having an affected child previously need verification of carrier status based on MCV, hemoglobin electrophoresis, and pedigree analysis before fetal assessment is undertaken. A sample of fetal DNA can be obtained by chorionic villus sampling, amniocentesis, or percutaneous umbilical (cord) blood sampling. DNA analysis of that portion of the α-globin or β-globin gene containing the deletion or mutation can be identified using restriction fragment length polymorphisms and Southern blotting techniques. Direct DNA sequence analysis is available by performing polymerase chain reaction amplification of fetal DNA. Using this method of detection, prenatal diagnosis can usually be confirmed within 7 days of fetal sampling.48
Management
Management of the thalassemia traits during pregnancy is much the same as for the patient with normal hemoglobin. Proper diagnosis is difficult, since the mild microcytic, hypochromic anemia can be easily confused with IDA, a condition that frequently coexists. Folic acid supplementation is recommended because of the increased utilization and high erythrocyte turnover. Blood transfusion or other invasive therapy should be avoided, if possible, unless severe anemia or other problems intercede.49
Disturbance in maturation: DNA synthesis (megaloblastic anemia)
Although not specific, the term megaloblastic anemia is used by most hematologists to describe a group of hypoproliferative disorders that have a characteristic morphologic appearance, ineffective erythropoiesis, and a moderate hemolysis of the circulating RBCs. Pernicious anemia (lack of vitamin B12) and folate deficiencies are the prototypes of this disorder. The underlying biochemical defect is impaired thymidylate formation, an essential rate-limiting initial step in the DNA synthesis of the body's cells, which require tetrahydrofolic acid as a coenzyme.
The megaloblastic changes occur as a result of a maturation defect in the marrow affecting erythrocytic, leukocytic, and thrombocytic cell lines. The lack of vitamin B12 or folate slows DNA synthesis and delays the nuclear maturation of chromatin of the immature cell into the dense cyanotic figure associated with the normoblast during normal erythropoiesis. Although the nucleus remains large and immature, the cytoplasmic mass decreases normally as it does during maturation. Therefore, the large macrocytes found in the peripheral blood of those with megaloblastic anemia represent a delay in nuclear maturation in the erythrocytic series and are diagnostic by their abnormal nuclear cytoplasmic appearance. Likewise, in the leukocytic series, the abnormal granulocyte is referred to as a giant metamyelocyte because of the condensation failure in the large nucleus. Due to granulocyte marrow turnover, leukopenia develops and the cells found in the peripheral blood are hypersegmented, with the nucleus having six or more lobes rather than the normal three to five. Finally, inadequate thrombopoiesis occurs, which results in an increase in bone marrow megakaryocyte mass with fewer platelets in the peripheral blood. The platelets that are present may function poorly, and a bleeding diathesis may be present if the vitamin B12 or folate deficiency is severe.
ETIOLOGY
Folic acid deficiency (FAD) appears to be the most common cause of megaloblastic anemia. However, vitamin B12 appears to catalyze the conversion of 5-methyltetrahydrofolate to its active form, as well as to affect the storage and transport of folic acid within the body.50 Therefore, a deficiency in vitamin B12 may lead to megaloblastic anemia singularly or by its effect on folate metabolism.51 Causes of folate or vitamin B12 deficiency include decreased intake and conditions in which requirements are increased (e.g., pregnancy, hyperthyroidism, hyperparathyroidism, and malignancies). Causes that are not related to vitamin B12 or folate therapy include the purine and pyrimidine synthesis inhibitors (cancer chemotherapy), pyridoxine-responsive megaloblastic anemia, and Di Guglielmo's syndrome (erythremic myelosis), in which platelet production is more severely affected than erythrocyte or leukocyte maturation. Finally, in various inborn errors of metabolism, such as Lesch–Nyhan syndrome and hereditary orotic acid deficiency, either a lack of enzymatic catalases or metabolic blockage to folate incorporation can cause megaloblastic anemia. All the infrequent causes are difficult to diagnose, and megaloblastic anemia due to these causes is unresponsive to folate therapy. Fortunately, vitamin B12 deficiency and FAD are the most common etiologies of megaloblastic anemia, accounting for 98% of reported cases. FAD is a much more common cause during pregnancy (99%), and it causes 92% of all cases in any age group.52 Vitamin B12 deficiency is extremely uncommon during pregnancy (1:8000 pregnancies); the lack of intrinsic factor almost always occurs in persons beyond the childbearing age unless significant gastric surgery has been performed.53
DIAGNOSIS
The major clinical signs and symptoms of megaloblastic anemia are variable and are usually not detectable until the anemia is severe. As in other anemia processes, the symptoms increase in severity with the progression of anemia, from pallor, through weakness, malaise, dizziness, and shortness of breath, and finally to congestive heart failure. Patients with megaloblastic anemias may appear jaundiced more frequently than those with IDA because of the rapid cell turnover and their increased propensity for bleeding diathesis. If anemia is present, the appearance of the peripheral smear is usually diagnostic, demonstrating several hyper-segmented granulocytes as well as RBC inclusions (i.e., stippling, Howell–Jolly bodies, Cabot's rings, and non-hemoglobin iron). The most sensitive indicator of early folate deficiency appears to be the hyper-segmentation of neutrophilic nuclear material (average low value >3.27 or more than 4% with more than five lobes in 100 consecutive polymorphonuclear neutrophils). Serum folate values in pregnant women are usually lower than in the non-pregnant patient, and these decrease progressively toward term. In the absence of IDA, however, a low fasting serum folate (<3 ng/ml) is virtually diagnostic of folate deficiency. In addition, a low erythrocyte folate activity (<20 ng/ml) is probably the best biochemical index of FAD. Although the anemia is macrocytic, it is usually normochromic with normal MCH and MCHC. The MCV, however, is strikingly elevated in contrast to IDA and may exceed 150 µm3/cell (see Table 1). The high MCV is helpful because it is in contrast to that found during pregnancy, IDA, and other microcytic processes. In general, the more severe the anemia, the more bizarre the erythrocytic changes, with nucleated RBCs appearing in the peripheral blood smear when the hematocrit is less than 20%. Unfortunately, if folate is deficient, there is usually a concomitant IDA, which may confuse the diagnosis.54 However, the reticulocyte count (RC) is lower in a megaloblastic anemia compared with the level in IDA.
INCREASED ERYTHROCYTE LOSS
Anemias due to increased loss of erythrocytes can be divided into acute and chronic types. The chronic types exist in patients with intestinal disease, such as parasite infestation, significant hemorrhoids, or peptic ulcer disease. Since the loss of blood is chronic, the actual number of RBCs lost is probably not as important as the amount of iron lost in excess of what is absorbed. This, coupled with the increasing demand of the fetus and expansion of the maternal blood volume, can convert mild iron depletion to IDA during pregnancy. Although these causes are relatively rare in the reproductive age group in the United States, careful assessment of iron unresponsive anemias in women from developing countries or with histories suggestive of intestinal disorders should be undertaken. Diagnosis and treatment are directed toward the specific disorder.
Anemias resulting from acute blood loss during pregnancy usually have evident etiologies, since external blood loss usually occurs and symptoms are sudden. These disorders can include multiple trauma and spontaneous splenic rupture, as well as disorders of the gastrointestinal, pulmonary, or urinary tract, which may or may not be related to obstetric conditions. Obstetric disorders may occur during early pregnancy (e.g., abortion, ectopic pregnancy, molar pregnancy) or in late pregnancy (abruptio placentae and placenta previa).
Blood loss at delivery may also account for significant losses of iron. Estimation of these losses has been measured with dye and isotope techniques, but large errors are common. Factors that lead to an underestimation of external blood loss at delivery include failure to take placental blood volume into account, incomplete measurement of hemoglobin in solution, incomplete extraction of hemoglobin from clots, and hidden bleeding. The average loss, determined by Cr techniques, is between 150 and 250 mL, depending on the patient's parity and the use of episiotomy, while an additional 30 mL is usually sequestered in the placenta. Therefore, approximately 200 mL of packed RBCs, equivalent to 500 mL of whole blood, is lost at the time of delivery. Approximately 230 mg of the 500 mg of iron used to expand blood volume is lost, while the remaining 270 mg is returned to the iron stores.
The placental “loss” of iron is variable, averaging 25 mg per pregnancy. The transfer of iron appears to take place by the same iron–transferrin complex used in the mother. The placenta, however, appears to function independently, since it stores iron as ferritin and hemosiderin even in the absence of a fetus and after fetal death. The transport of iron across the placenta is unidirectional and preferential, with the fetus accepting iron even when maternal levels are low. This maternal–fetal transfer involves an active transport mechanism, since the saturation of transferrin and the plasma iron level are considerably higher in the fetus. Most of the iron transferred across the placenta (70–80%) goes to the fetal liver, which serves in a storage capacity as well as a hematopoietic organ. The placenta also functions as a barrier to iron overload; thus, iron toxicity in the fetus by maternal overdose does not occur. Study of the incorporation of placental iron into fetal hemoglobin using radioactive material cannot be undertaken because in early investigations, use of labeled iron was associated with infant malignancies. However, indirect evidence shows heme incorporation into fetal RBCs to be similar to maternal utilization. Diseases in the mother (e.g., infection or inflammation) may decrease the amount of iron transferred, owing to poor storage mobilization and transferrin release. On the other hand, anemia due to iron loss during lactation, FAD, or chronic blood loss does not affect transfer of iron across the placenta.
The iron loss in lactation is small and usually averages 1 mg/day. Breast milk contains an iron binding protein similar to transferrin, called lactoferrin. This protein is synthesized in the mammary gland and stored in the ductal lining cells. The secretion of lactoferrin into the breast milk accounts for the iron loss noted in iron sufficient women during lactation. This compound is also bacteriostatic, and thus assists the infant in combating early infection and possibly prevents maternal breast infections. Moreover, since many lactating women do not have menses, the excretion of 1 mg of iron per day is offset by the avoidance of menstrual loss. Therefore, dietary iron supplementation as prescribed during and after pregnancy is usually sufficient to replenish iron stores during lactation.
During the postpartum period, acute bleeding problems may also occur owing to uterine atony, genital lacerations, or retained placenta. The acute causes, both obstetric and non-obstetric, are usually best treated with blood transfusion and removal of the offending agent. Obviously, the diagnosis depends on the specific disorder, as does the proper therapy. After the correct diagnosis is made and acute therapy is started, treatment with oral iron is usually indicated. Patients with acute blood loss usually show a normochromic, normocytic cell pattern with normal blood indices, until after equilibration occurs, a process that may take from 12 to 24 h. Iron studies in the immediate postpartum period are typically within normal ranges.
INCREASED ERYTHROCYTE DESTRUCTION
Diseases caused by increased destruction of erythrocytes present a hematologic profile characterized by persistent reticulocytosis, a finding that would indicate that there is increased erythrocyte destruction in excess of compensatory erythropoiesis by the bone marrow.55 The etiology of these disorders can be (1) extrinsic hemolytic anemia in which a mechanical, drug, infectious, or immune factor is responsible for the decreased life of the red cell; (2) intrinsic hemolytic anemia in which the red cell loss is due to an inherited defect in the blood cell or membrane; or (3) unknown.56, 57
Extrinsic hemolytic anemia
Extrinsic hemolytic anemia is a proliferative disorder that involves erythrocytes that are structurally and metabolically normal but owing to environmental factors, suffer early death. Most frequent causes are hypersplenism, Coombs-positive hemolytic anemia, and microangiopathic anemia.
HYPERSPLENISM
Enlargement of the spleen usually leads to excessive RBC sequestration and destruction, and is most common in its severe and fatal form during childhood. In adulthood, the degree of anemia is usually mild, with a reticulocyte count rarely greater than 6%. There are several mechanisms of hemolysis that stems from spenomegaly. Stasis and trapping in the spleen is associated with macrophage attack which leads to red cell remodelling and a subsequent reduction in the surface area to volume area of the RBC which leads to spherocytosis. Prolonged movement of RBCs through the spleen can also lead to distortion of their shape and thus lead to further trapping and hemolysis. The anemia is usually normochromic and normocytic, and the overall prognosis is dependent on the underlying cause. Many conditions can cause splenomegaly in adults including: chronic myeloid leukemia, primary myelofibrosis, lymphoma, Gaucher disease, kala-azar, and hyperactive malarial splenomegaly.58 In some cases, corticosteroids are helpful, and removal of the spleen, particularly during pregnancy, is used only as a last resort.
MICROANGIOPATHIC HEMOLYTIC ANEMIA
In microangiopathic hemolytic anemia (MHA), the products of destroyed red cells such as schistocytes are seen in the peripheral smear. Autoimmune diseases such as lupus erythematosus and polyarthritis have been associated with this disorder, as has preeclampsia, HELLP (hemolysis, elevated liver enzymes and low platelets) syndrome, acute glomerular nephritis, and idiopathic thrombocytopenia purpura. In addition, other disorders associated with disseminated intravascular coagulation (DIC), such as fetal death, amniotic fluid embolus, and severe infection, may demonstrate similar signs of MHA. In most of these diseases, small-vessel damage or abnormal coagulation leads to a network of fibrin strands that partially occlude the microcirculation. It is this basic pathophysiologic change that causes the fragmentation and lysis of the RBC. Therapy for those patients with MHA involves diagnosis and treatment of the underlying disorder and principally the reestablishment of the integrity of the microcirculation. The prognosis for both mother and fetus depends upon the promptness with which the underlying disorder can be overcome.
COOMBS-POSITIVE ANEMIA
In some patients, a positive direct Coombs test is found in conjunction with hemolytic anemia. In these patients, usually a complement of the IgG antibody variety is irreversibly fixed to the erythrocyte membrane. This leads to premature destruction of the RBCs. These types of disorders usually are categorized as having “warm” or “cold” antibody hemolytic anemia. Those with warm antibody hemolytic anemias are usually associated with an IgG antibody and noted in many collagen-vascular diseases, patients with lymphomas, or drug reactions (particularly α-methyldopa). Those with cold autoantibodies are usually of the IgM class and are most commonly noted after a viral or mycoplasmal infection. The IgM antibody does not affix to the RBC membrane but rather stimulates complement, which is responsible for the positive antibody test. If the underlying disorder is treated or removed, the hemolytic process is usually resolved in 2–3 weeks, although the Coombs test may remain positive for up to a year. Transfusion is not used unless absolutely necessary because the transfused cells likewise undergo fragmentation. Frequently, corticosteroids are used in those with warm antibody or IgG-mediated disorder. The prognosis for mother and fetus is dependent on the prognosis of the underlying disease process.
Intrinsic hemolytic anemia
Intrinsic hemolytic anemias are those in which there are inherited disorders leading to premature destruction of the RBCs. Included in these types of anemias are hereditary spherocytosis, various RBC enzyme deficiencies, and the hemoglobinopathies.
HEREDITARY SPHEROCYTOSIS
This anemia is inherited as an autosomal dominant trait, and the RBC membrane is abnormally permeable to sodium. This leads to cellular disruption and also to premature cell death in the spleen. The diagnosis of hereditary spherocytosis is confirmed with the osmotic fragility test, and the treatment is usually transfusions or splenectomy. The prognosis for mother and fetus is usually excellent unless splenectomy must be performed during pregnancy. In most cases, treatment of this disorder with splenectomy has already been carried out prior to conception.
ENZYMATIC ABNORMALITIES
Various enzymatic defects inherited in RBCs can also lead to premature destruction of the erythrocyte. The two most common disorders involve pyruvate kinase deficiency and glucose-6-phosphate dehydrogenase (G6PD) deficiency. The pyruvate kinase deficiency is inherited as an autosomal recessive trait and is usually diagnosed during childhood because of persistent low-grade anemia and jaundice. The diagnosis for pyruvate kinase deficiency is made when there is evidence of hemolytic anemia and at least one of the following: low levels of erythrocytic PK enzymatic activity and/or the presence of a mutation of PK LR gene that impairs enzymatic activity. The advent of tests that detect mutations at the cDNA level have allowed for prenatal testing to be performed.59 This is usually a mild disorder and does not disturb maternal or fetal homeostasis.
G6PD deficiency, on the other hand, is inherited as an X-linked disorder and is more commonly found in patients of African descent, with 13% of African-American males being affected. The use of oxidizing drugs, such as nitrofurantoin and primaquine, stimulates acute hemolysis of red cells with accompanying hemoglobinuria. In addition, viral or certain bacterial infections may also stimulate this hemolytic event. The fluorescent spot test is the simplest, most reliable and most sensitive of the G6PD screening tests and confirmatory testing can be performed by adding a measured amount of red cell hemolysate to assay mixture that contains glucose-6 phosphate and a cofactor (NADP); the rate of NADPH production is then measured.60, 61 It is rare that this disorder would adversely affect the mother or developing fetus. Nevertheless, G6PD deficiency should be kept in mind prior to prescribing drugs to African parturients, since 25% of African-American women are carriers of this trait.
HEMOGLOBIN ABNORMALITIES
Most of the disorders regarding hemoglobin usually involve the abnormal globin structure of the RBC. Fortunately, most of these abnormal hemoglobins, called hemoglobinopathies, are very rare; of the more than 300 types reported, only a few are clinically very common. Most of these disorders involve a single amino acid substitution in one of the globin chains, although some involve deletions, additions, or fusions in the α- or β-chains. All of these hemoglobinopathies, when clinically significant, lead to the production of erythrocytes that either are structurally unable to perform duties, such as oxygen transport, or are prematurely destroyed. All are associated with an increased reticulocytosis, indicating that the body is attempting compensation. To understand these, familiarity with hemoglobin synthesis and structure is warranted.
Erythrocyte composition
The erythrocyte is extremely complex; its membrane is composed of lipids and proteins, while the interior of the cell contains primarily hemoglobin, which is intimately associated with oxygen transport. The erythrocyte develops from the multipotential stem cell or myeloblast, which differentiates along the erythroid line in the bone marrow. During maturation, it loses most of the metabolic and biosynthetic capabilities inherent in most other cells. The capacity to synthesize nucleoproteins, such as DNA, is lost at the basophilic normoblast stage, and stainable DNA itself is removed before reticulocyte release. Ribonucleic acid (RNA) synthesis is lost as the peripheral reticulocyte matures, and loss of electron transport mechanisms prevents the use of phosphate bonds as an energy vehicle, requiring the mature erythrocyte to employ anaerobic glycolysis by the hexose and penrose phosphate shunt pathways. Thus, the RBCs only metabolic function is the process of gaseous and ionic transfer, using the remaining pathways for purine and pyrimidine metabolism. Although these critical functions are performed well in the mature RBC, the lack of RNA and DNA is extremely important, since it renders the cell incapable of repair or repetition. Therefore, in certain disease processes that alter this maturation sequence, the production rate and functioning of the cell lines are impaired.
Hemoglobin structure
The structure of hemoglobin is shown in Fig. 6 and consists of heme, which is composed of a divalent iron atom connected to four pyrrole rings through their substituent nitrogen atoms.62 This polyvalent compound is surrounded by two pairs of dissimilar polypeptide chains, each linked to one of the pyrrole rings. Each pair of polypeptide chains has different sequences of amino acids and is stereochemically arranged in four helical configurations. The physical properties of heme, as well as its association with the iron atom, classify it as a metalloporphyrin, a structure similar to chlorophyll or vitamin B12. The conjugated double bonds of heme readily absorb visible light and are responsible for the red color of hemoglobin. Functionally, it is the iron atom that carries the oxygen; in its ferrous state (Fe2+ ), six binding sites are present. Four are attached to the heme, while another is attached to a histidine residue of a globin chain. The other coordination position of iron is unoccupied in deoxyhemoglobin under proper physiologic conditions. This final binding point is protected from oxidation by the globin chains surrounding the heme moiety. Separation of heme from the globin during destruction results in the oxidation of the iron atom and the formation of hematin. If hemoglobin iron is oxidized to its ferric state (Fe3+), methemoglobin is formed; this cannot serve as the oxygen carrier since the final coordination site is bound.
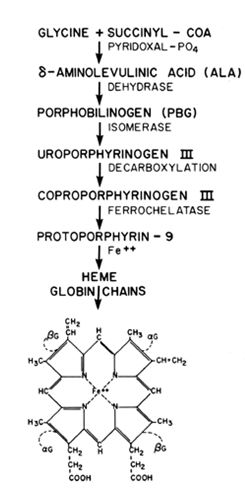 Fig. 6. Biosynthesis of hemoglobin
Fig. 6. Biosynthesis of hemoglobin
Heme synthesis
Heme synthesis takes place in the particulate and soluble fraction of the erythrocyte (see Fig. 6). Initially, δ-aminolevulinic acid (ALA) is synthesized from succinyl-CoA and glycine in the particulate fraction of the cell.63 Next, two moles of ALA combine to form porphobilinogen, and subsequently, four of these moieties converge to form one mole of uroporphyrinogen III. Through several intermediate steps, this compound is converted into coproporphyrinogen III. By successive decarboxylations, it is converted into the heme precursor protoporphyrin IX, a reaction catalyzed by a mitochondrial enzyme called ferrochelatase or heme synthetase. A ferrous iron moiety is inserted into the protoporphyrin as a final step in the pathway of formation of heme. Current data suggest that heme biosynthesis is unidirectional and the control mechanisms for the pathway are necessarily located in the first enzymatic step (ALA synthesis). Positive feedback inhibition of the ALA pathway by certain metabolic byproducts such as hematin or hemin occurs, while other degradatory products such as billverdin and bilirubin do not appear to affect heme synthesis. Finally, the lack of precursors or cofactors, such as Fe2+, folic acid, vitamin B6, and vitamin B12, may impair heme production at various reaction sites during hemoglobin synthesis.
Globin chain synthesis
The sequence of amino acids in the globin chains is determined by genetic input. Once initiated, the sequence of the nucleotide bases in DNA is transcribed to messenger RNA (mRNA). The mRNA is transported from the nucleus to the ribosomal RNA (rRNA), which is the actual site of protein synthesis. The mRNA, by collation with transfer RNA (tRNA) on the rRNA template, translates the message from a language of nucleotide bases into that of amino acids in the proper sequence. The initiation of the translation always begins with the same code word (AUG) from mRNA regardless of the globin chain synthesized; therefore, the first amino acid is always methionine. The initial phase, called transcription, proceeds through approximately 40 amino acid subunits. The elongation phase extends nearly to the termination point and is catalyzed by a protein called elongation factor I, which is glucose triphosphate (GTP) and tRNA dependent. Finally, the parent mRNA gives the signal to terminate transcription and the newly formed globin chain is released, while the ribosome searches for the initial phase of another mRNA. The three nucleotide codons that signal for the chain termination of all proteins are UAA, UAG, and UGA. These are decoded by tRNA and by protein releasing factors in the particulate cell fraction. Once complete, each polypeptide chain contains 141–146 amino acids. After synthesis, two pairs of globin chains interact with the four pyrrole portions of the heme and the assembly of the hemoglobin moiety is complete (see Fig. 6).
Hemoglobin nomenclature
The polypeptide chains in normal adult hemoglobin are termed alpha (α), beta (β), gamma (γ), and delta (δ). The nomenclature is based on various alterations in the globin chain and is summarized in Table 5. The α-chains are found in all normal adult hemoglobin and contain 141 amino acids. They combine with the β-, γ-, or δ-chains, each containing 146 residues, to form the two pairs of globin chains found in normal hemoglobin. For instance, when two β-chains accompany two α-chains (α2Aβ2A) hemoglobin A (HbA) is formed; this accounts for 95% of the total hemoglobin in the normal adult. Hemoglobin A2 (HbA2) is formed by two δ-chains accompanying the α-chains (a2Aδ2A) and accounts for 2–3.5% of normal hemoglobin. Fetal hemoglobin (HbF) is formed by two γ-chains coupled with two α-chains (α2A γ2F) and accounts for the remainder of hemoglobin found in the adult.
Table 5. Nomenclature of normal and abnormal hemoglobins
| Hemoglobin classification | Globin | Chain | Common | Official |
| Adult hemoglobin* |
| 2α | HbA | α2Aβ2A |
| Adult hemoglobin* |
| 2α | HbA2 | α2Aδ2A |
|
|
| 2δ |
|
|
| Adult hemoglobin* |
| 2α | HbF | α2Aγ2F |
| Embryonic fetal hemoglobin* |
| 2α | HbGower II | α2Aε2G |
| Sickle hemoglobion | β-chain substitution | 2α | HbS | α2Aβ2S(6 glu ® val) |
| C-hemoglobin† | β-chain substitution | 2α | HbC | α2Aβ2C(6 glu ® lys) |
| Methemoglobin† | α-chain substitution | 2α | HbMBoston | α2M(58 his ® tyr) β2A |
| Increased oxygen affinity† | α-chain substitution† | 2α | HbChesapeake | α2Ches(92 arg ® leu) β2A |
| Unstable hemoglobin† | β-chain substitution‡ | 2α | HbSaki | α2Aβ2Saki(14 leu ® pro) |
| β-Thalassemia | None (decreased synthesis) | 2α | HbBThai | α2Aβ2Thal |
* Structurally normal variants
† Structural abnormalities include substitution, inversion, or deletions of amino acids in either chain
‡ Other abnormalities may affect either chain
An amino acid deletion, inversion, or substitution structurally alters the globin chains to cause most of the abnormal hemoglobins. The location of an amino acid abnormality (if known) is designated by a superscript notation (in parentheses) next to the involved globin chain: α2Aβ2S(6 glu ® val).25 Abnormal hemoglobins may also involve the α-chains and are called α-hemoglobinopathies. If several abnormal hemoglobins are similar in their biochemical properties, a subscript is used to indicate the geographic location where the hemoglobinopathy was first described (e.g., HbM-Boston, HbM-Milwaukee). The changes in the amino acid sequence of the globin chains may evoke a wide spectrum of clinical symptoms. Abnormal hemoglobins may also be classified according to the changes they elicit in oxygen carrying capacity, stability, erythrocyte longevity, or synthesis rate (see Table 5).
Hemoglobin genetics
The structure and function of the globin chains are determined by polygenetic inheritance, whereas there appears to be genetic control for heme synthesis common to all hemopoietic tissue. The four globin chains contain hundreds of amino acids as possible mutagenic sites, and since there may be at least four mutations per position by nucleotide sequencing, the number of changes in hemoglobin structure by genetic malfunction is enormous. Four separate genetic loci have been identified for the α, β, γ and δ polypeptide chains. The use of marker chromosomes in cytogenetic studies indicates that the genetic loci for the α-chain are on chromosome 16, while the β-, γ-, δ-chain loci are closely associated on chromosome 11. The structural changes in the amino acid sequencing characterizing most abnormal hemoglobins appear to be related to genetic mutation. In contrast, thalassemias are a result of defective mRNA.
The cause of the various mutations responsible for the globin chain sequence for most hemoglobinopathies has not been determined; however, the mutation in hemoglobin S (HbS) may be a genetic response to malaria.
Sickle hemoglobinopathies
Sickle hemoglobin is rather common among African-Americans. HbS-S (sickle cell anemia) and other variants, HbS-C and HbS-Thal, comprise a group of hemoglobinopathies called sickle cell disease (SCD). These patients are often severely affected clinically during pregnancy or in the non-pregnant state. Those affected with sickle cell trait typically experience no pregnancy-associated complications.
Hemoglobin S represents a structural defect caused by a genetic mutation, possibly as an adaptive response to falciparum malaria. Heterozygosity (HbA-S) gives a protective effect, while those persons who are HbA-A or HbS-S are more likely to die during childhood in areas endemic for malaria. The substitution of the amino acid valine for glutamic acid at the sixth position from the N-terminal of both β-chains gives S hemoglobin its unique sickling characteristic compared with normal adult HbA. Likewise, the substitution of lysine at the identical position results in hemoglobin C (HbC). Otherwise, these hemoglobins are exactly the same as normal adult hemoglobin, and this one change is responsible for all of the symptomatology. The amino acid substitution also causes the differences in electrophoretic migration of HbS and HbC, HbA, HbF, and HbA2. Changes caused by amino acid substitutions, deletions, or inversions in either the α- or β-chain give rise to other structurally abnormal variants, and although some of these hemoglobins have an adverse effect on pregnancy outcome, they are fortunately quite rare. Therefore, hemoglobinopathies listed in the SCD category are the only ones that are clinically important. Mild forms of the disease are important only when the patient is under severe stress or for genetic counselling purposes, since the clinical presentation is usually benign.
CLASSIFICATION
The terminology of sickle hemoglobinopathies has undergone many revisions since 1960. The currently accepted classifications include: (1) sickling disorder, any red cell that undergoes in vitro/in vivo sickling in the deoxygenated state; (2) sickle cell trait (HbA-S), the heterozygous form of HbS infrequently associated with clinical symptoms; (3) sickle cell anemia (HbS-S), the homozygous form of HbS frequently associated with severe clinical symptoms; (4) sickle cell disease (HbS-S, HbS-C, and HbS-Thal), disorders in which HbS is all or part of the abnormal hemoglobin composition and that are usually associated with severe symptoms; and (5) mild sickle cell states (HbS-D, HbS-E, HbS-Memphis), those in which HbS is present but clinical symptomatology is usually mild. Numerically, HbS, both homozygous and in combination with other hemoglobins, is the most common abnormal structural defect; HbC is the second most common hemoglobinopathy in the USA, and HbE is next in frequency. Hemoglobin D is similar in its other properties to HbS but does not sickle. Both HbE and HbD can combine with HbS to give mild clinical symptomatology.
PATHOPHYSIOLOGY
 Fig. 7. The sickle cell crisis cycle. (Martin JN Jr, Morrison JC. Managing the parturient with sickle cell crisis. In Huddleston JF (ed). Clinical Obstetrics and Gynecology: Sickle Cell in the Gravida Woman. Philadelphia, JB Lippincott, 1984)
Fig. 7. The sickle cell crisis cycle. (Martin JN Jr, Morrison JC. Managing the parturient with sickle cell crisis. In Huddleston JF (ed). Clinical Obstetrics and Gynecology: Sickle Cell in the Gravida Woman. Philadelphia, JB Lippincott, 1984)
LABORATORY ASSESSMENT
Anemia is the most constant feature of subjects with severe hemoglobinopathy. Generally, patients with a hematocrit less than 20% or a hemoglobin level below 6 g/dL have homozygous β-thalassemia or HbS-S. Patients with a hematocrit of 20–25% may have a HbS-S, HbS-C, or HbS-Thal hemoglobinopathy, while subjects with a hematocrit of 25–30% may have any hemoglobinopathy except homozygous β-thalassemia and HbS-S. In most of the mild forms (HbS-D, HbS-E, HbA-S), the hematocrit is 30–35%. When the hematocrit is 25–35%, diagnosis is particularly difficult because many disorders are included in the differential diagnosis. The RBCs are usually normochromic, normocytic, but concomitant IDA may confuse the issue. A reticulocytosis of up to 20% and thrombocytosis are not uncommon in HbS-S patients during crisis. The serum bilirubin is usually mildly elevated in the asymptomatic patient with HbS-S but may reach 15–20% during crisis. The urinalysis usually shows hyposthenuria (with specific gravity <1.005), hematuria, and bile and direct bilirubin. Asymptomatic bacteriuria is more common in these patients, and pyuria usually leads to active symptomatic infection due to poor opsonization of bacteria. The liver enzymes are usually elevated and reach high levels during crisis; abnormalities of coagulation, such as a decrease in the platelet count and fibrinogen with presence of fibrin split products, are more common during a crisis but may be present at any time. The prothrombin, partial thromboplastin, and clotting times are normal in patients with sickle hemoglobinopathies.
Hemoglobin electrophoresis is the only reliable method that aids in the diagnosis of the specific hemoglobinopathy. By electrophoresis, HbA-S usually has 25–35% HbS, 60% HbA, normal HbA2, and increased HbF; HbS-S demonstrates 90–95% HbS, with small concentrations of HbA2 and normal to increased amounts of HbF; HbS-C usually demonstrates normal percentages of HbA2 and HbF, with an equal division of the remainder between HbS and HbC (45–48%). Hemoglobin S-Thal is very difficult to diagnose even by electrophoresis but appears to be much like HbA-S with slightly more sickle hemoglobin.
CLINICAL PRESENTATION
Hemoglobin S-S, HbS-C, and HbS-Thal can be differentiated from the benign hemoglobinopathies, such as HbA-S, by a history of recurrent painful vaso-occlusive crises in the former. These crises usually last 2–6 days, may be associated with fever as well as leukocytosis, and may lead to unnecessary surgical procedures unless the hematologic diagnosis is suspected. These attacks are more common in HbS-S patients, usually average one to four a year, and are commonly associated with fever, pregnancy, trauma, or physical stress. The crisis is rarely a true hemolytic crisis and is more commonly termed vaso-occlusive, although other types of crises are possible (see Fig. 7). Finally, cerebral manifestations are not extremely common but are ominous, since they may be associated with severe morbidity and death. Sudden deaths with SCD have been reported to involve both coronary and cerebral vessel occlusions. Coma, convulsions, and death due to cerebral infarction are most common in these patients.
SICKLE CELL DISEASE IN PREGNANCY
The hemoglobinopathies comprising SCD are more common than they appeared to be in the past. Hemoglobin S-S was said to occur in 1 in 600 African-American descent; however, the incidence is closer to 0.5–0.7% in certain areas in the United States. Hemoglobin S-C is present in about 1 in 800 persons of African descent.64 The incidence of HbS-Thal is said to be 1 in 1250, but this is probably underreported, since the anemia is mild. When it comes to fertility issues, evidence demonstrates that the incidence of pregnancy in these subjects is the same as in the population at large.65
MATERNAL EFFECTS
The prevailing attitude of most perinatologists is that the patient with SCD appears to be at significantly greater risk for crisis and other dire complications when pregnant.66 Only infection, trauma, hypoxia, and acidosis approach pregnancy as a frequent cause of crisis and severe morbidity/mortality in patients with SCD. Factors associated with pregnancy that complicate the clinical picture include the hypercoagulable state of pregnancy, the increased susceptibility to infections, the increased vascular stasis in the pelvis and lower extremities, the increased metabolic and hematologic demands of pregnancy, and the stress of parturition. The culmination of all of these factors leads to significant risks for morbidity during pregnancy.
Several large review series prior to 1969 show a morbidity rate of 50–80% and maternal mortality figures ranging from 2 to 25% for SCD patients during pregnancy.64, 66 Painful crises were among the most common symptoms, being reported by all investigators. In addition, most patients had a reduction in their hematologic values during gestation. Other major complications such as pneumonia (3–15%), pyelonephritis (5–12%), and endometritis (72–10%) are frequent causes of crisis. Pulmonary embolus (1–9%) and congestive heart failure (1–5%) are also common causes of severe morbidity, as is preeclampsia. Since 1970, the maternal death rate has dropped considerably (i.e., 0–1% in most large series), as has the morbidity rate in this country. These changes probably reflect better antepartum scrutiny as well as new management techniques.
Although some authors feel that the clinical course of SCD in pregnancy tends to be similar to its course before pregnancy, many previously asymptomatic patients may suffer a serious, sometimes fatal, crisis during pregnancy. Several investigators have associated an increased level of hemoglobin F with a reduction in the number of complications. Those rare persons with persistent hereditary fetal hemoglobin who have no crises support this hypothesis. However, with persistent hereditary fetal hemoglobin, the hemoglobin F is homogeneously distributed in each cell, whereas in patients with SCD, the hemoglobin F, although present in larger quantities, is unequally distributed in the cell. Several studies have shown that the level of hemoglobin F varies greatly in both asymptomatic patients and those in crisis. Therefore, it is unwise to rely on a negative history or absolute hemoglobin F percentage to guide therapeutic judgment in pregnant patients with SCD.
Although hemoglobin C is more stable than hemoglobin S, the combination of hemoglobin C with hemoglobin S appears to facilitate sickling more than does a combination containing hemoglobin A. This is particularly true in late pregnancy, when venous stasis and maternal hypercarbia are usually present. It has been shown that the number of sickled cells remains constant for a given PO2 if hemoglobin S is present alone. However, if both hemoglobin C and hemoglobin S are present, the number of distorted cells increases to a greater degree under similar conditions. The red cells appear to have a shorter survival time in HbS-C patients due to the crystallization of hemoglobin C within the red cell. Symptomatically, patients with HbS-C appear to be healthier and have fewer crises than do those with HbS-S, but complications resulting from stress, such as pregnancy, infection, or trauma, can be more severe and sudden (e.g., serious eye lesions, renal papillary necrosis, aseptic bone necrosis, acute pulmonary thrombosis, marrow emboli, and hemolytic episodes). In general, pregnancy results in a similar risk for patients with HbS-C and HbS-S with regard to morbidity and mortality, while patients with HbS-Thal have a clinical course intermediate between those with HbS-S and HbA-S.
FETAL EFFECTS
The incidence of spontaneous abortion is high (20–35%) and has been related to the “chronic disease state” or malnourished condition of the patient, sickling in the infundibulopelvic vessels (decreased progesterone), and distorted cells in the arcuate arteries of the uterus (decreased placental blood flow). These changes may also lead to intrauterine growth restriction and premature labor, which is also increased (10–55%) in gravid patients with SCD. The decrease in mean birth weight of these infants (250–500 g) has also been related to the high incidence of maternal infection and premature labor. Stillbirths are also increased (5–13%) and appear to be related to severe maternal crises with evidence of placental sickling. Although fetal deaths occur in asymptomatic subjects, there have been no reported stillbirths due to sickling in the HbS-S fetus. The overall neonatal mortality rates are not increased for progeny of women with SCD, provided asphyxia and growth related complications do not occur.
The perinatal salvage rates are decreased in patients with HbS-C, ranging from 40 to 85%. There is an increase in miscarriages and fetal deaths. Both prematurity and small-for-gestational-age infants are more common, but not to the extent found in HbS-S patients. Hemoglobin S-Thal patients show even greater reduction in miscarriages and stillbirths compared with HbS-S and HbS-C patients.
The percentage of small-for-gestational-age and premature infants is increased in HbS-Thal and near average for HbS-C patients.
ANTEPARTUM MANAGEMENT
Intensive antepartum management of SCD patients, with visits every 2 weeks for the first 20 weeks and then weekly until delivery, appears to be the best method of preventing decompensation and crisis. During each visit a thorough evaluation for infection and historical and physical evidence of sickling. Vigilance for crisis is important, and the patient should be hospitalized if pain, fever, infection, or other signs of sickling are noted. Hematocrit and hemoglobin determinations should be performed at each visit, since rapid reductions in these indices may occur even in the asymptomatic patient. A reticulocyte count, peripheral smear, haptoglobin, lactate dehydrogenase or serum bilirubin determination may also be important in assessing the patient's hematologic risk for crisis. Methods for assessment of fetal well being including serial growth ultrasounds, non-stress tests, and biophysical profiles, should be undertaken due to increased risks for placental insufficiency.67
Growing evidence suggests that women with SCD have vitamin and micronutrient deficiencies.68, 69 Folic acid, 4 mg daily, is recommended in pregnant SCD patients due to ongoing folate consumption from ongoing hemolytic anemia. Iron therapy generally is avoided in women with SCD due to chronic RBC destruction and subsequent free iron being released into the maternal circulation which can lead to a state of hemochromatosis if administered continually. However, iron deficiency anemia should not be overlooked as one investigation demonstrated a deficiency in as many as 65% of pregnant HbS-S patients.70
Therapy of patients with SCD has been largely targeted toward symptoms. Analgesics, hydration, vasodilatation, oxygen, bed rest, and anticoagulation have all been used with variable success. Tolazine, cobaltous chloride, carbon monoxide, and carbonic anhydrase inhibitors have not been found useful. Hyperbaric oxygen, although effective, is cumbersome and most often unavailable. In general, only oxygen and mild analgesics are still used clinically in the treatment of crisis during pregnancy.
Antitumor drugs such as hydroxyurea increase production of hemoglobin F in primates and in patients with SCD.71, 72, 73 The drug, decitabine has shown efficacy in short- and long-term trials in increasing HbF levels and total hemoglobin levels in patients failing to respond to hydroxyurea.74, 75, 76, 77 However, animal studies have demonstrated teratogenicity, embryotoxicity, stillbirths, and intrauterine growth restriction, and therefore it is advised to avoid becoming pregnant during treatment with any of these agents. On the other hand, the use of erythropoietin during pregnancy is considered safe. In non-pregnant sickle cell patients, initial reports suggested that recombinant human erythropoietin (rhEpo) stimulated HbF production; however, subsequent clinical studies with both rhEpo alone and in combination with standard doses of hydroxyurea have produced conflicting results.78, 79, 80, 81 Because of its documented safety, and owing to good, personal clinical experience with this medication during pregnancy, these authors continue to use erythropoietin in pregnant patients with significant anemia that may have relative contraindications to blood transfusions (e.g., multiple red blood cell alloantibodies).
Red blood cell dehydration plays a key role in the SCD process due to the excess activity of potassium chloride and calcium activated potassium transport channels.82, 83 Research has focused on medications that inhibit the activity of these channels including oral supplementation with magnesium which has demonstrated some promise in regard to effectively treating sickle cell patients.84 Nicosan is an herbal plant extract that has been successfully used in Nigeria to prevent painful crises associated with SCD, but the safety of this medication during pregnancy is not known.85, 86, 87 Other promising areas of investigation include hematopoietic cell transplantation and gene therapy, neither of which would likely be utilized in the pregnant patient population.88
Controversy exists regarding the role of prophylactic blood transfusion in the management of SCD in pregnancy.89, 90, 91, 92 In the only randomized controlled trial published to date, prophylactic transfusion was associated with a decreased risk for painful crisis and severe anemia, but no difference was observed for pregnancy outcome.89 However, there are some experts that believe selective use of prophylactic transfusion may be beneficial in pregnant women with sickle cell disease at highest risk of complications, such as those with previous perinatal mortality or severe anemia.90, 91, 92
By limiting transfusion to situations in which it is clinically indicated, patients are not subjected to the increased risk for blood-borne infections, iron overload, and alloimmunization. The rate of alloimmunization in sickle cell patients is estimated to be between 18% and 36%.93, 94 It is estimated that six to eight deaths per year in the United States occur as a result of delayed transfusion reactions.95 It appears from the available evidence that the reduction in morbidity and mortality of SCD in pregnancy may be attributable to improvements in general management of pregnancy rather than prophylactic transfusion per se.96 In contrast, therapeutic transfusions are indicated in patients who have particularly severe disease manifestations or for symptomatic patients who are unresponsive to conservative management.97 Pregnancies impacted by acute symptomatic or severe anemia (Hgb <5.0–6.0 g/dL), intrapartum complications including hemorrhage and septicemia, refractory painful crisis (relapse <1 week, no improvement >10 days), and those women with reticulocytopenia (e.g., those affected by parvovirus B19 infection) often benefit from intervention with a blood transfusion. There is no consensus regarding the exact hematocrit value below which transfusion should be considered. However, when a transfusion is clinically indicated in the patient with SCD, the objective is to lower the percentage of HbS to approximately 40% while simultaneously raising the total hemoglobin concentration to about 10 g/dL.97
Exchange transfusion, also known as erythrocytapheresis, allows the HbS-containing cells and irreversibly sickled cells to be removed by extracorporeal differential centrifugation. It affords the simultaneous return by venous access in the other arm of the patient’s own plasma, leukocytes, platelets, and clotting factors along with donor, leukocyte-poor washed HbA-containing red cells. This type of transfusion occurs via a machine that allows for automated continuous erythrocytapheresis. In addition, this method maintains euvolemia at all times and allows the provider to accurately monitor the patient’s hematologic indices such as hemoglobin levels and hematocrit. It also has the advantage of administration on an outpatient basis. If such a device is not available, the manual “push–pull” mechanism can be considered.98 Generally, six units of donor packed red cells are exchanged by this process and there is rapid resolution of crisis symptomatology and ongoing sickling. In addition to being more euvolemic, automated continuous erythrocytapheresis requires less transfusion time than the manual method.99 Most of the complications associated with this approach are related to the risk of the blood products. Blood products given to these patients should be from cytomegalovirus-seronegative donors or leukoreduced. Careful cross-matching to minimize minor blood incompatibilities and alloimmunization is critical in avoiding problems later for these patients who may need blood products at various points during their life. The use of blood from family members or friends matched for recipient and donor red cell antigens are clinically helpful in reducing the number of post-transfusion crises. Such episodes are known as delayed hemolytic transfusion reactions. The exact mechanism of hemolytic transfusion reaction is not well understood, and the pathophysiology seems to be far more complex because it involves the destruction of both patient and donor RBCs. A subcategory of this problem is known as a delayed transfusion reaction, which is classically characterized by a clinical triad of fever, hyperbilirubinemia, and anemia occurring 3–10 days after transfusion. Laboratory tests that aid diagnosis include increased reticulocyte count, free hemoglobin in the urine, fragmented RBCs on peripheral smear, decreased post-transfusion HbA on electrophoresis, or discovery of previously undetected alloantibodies.100
INTRAPARTUM MANAGEMENT
During labor and delivery, an epidural block appears to be the best method of analgesia if performed by the obstetric anesthesiologist or skilled perinatologist; hypotension and resultant hypoxia greatly increase the risk of sickling. Alternatively, small amounts of opioids or other analgesic agents may be used with pudendal or spinal anesthesia for delivery. Oxygen is administered at 4 L/min with the patient laboring in the semi-Fowler, lateral recumbent position. The patient’s status may be followed by blood smears to detect sickling, and the fetus is assessed by continuous electronic monitoring. Delivery is usually spontaneous at term, and cesarean deliveries are performed only for obstetric indications. Oxytocin has not been found harmful in the patient with SCD and is used when indicated.
POSTPARTUM MANAGEMENT
Postpartum SCD patients should be monitored closely for excess blood loss, infection, or thrombophlebitis. Antithrombotic stockings, if properly applied, and early ambulation are encouraged; prophylactic antibiotics are usually not necessary. Frequent visits (bi-weekly) to the clinic assure normality through the 6-week postpartum adjustment phase. The infant should be tested using cord blood for electrophoresis to detect heterozygotes and to identify those with SCD so that counseling and education of the parents may lead to a more favorable outlook for the child.
In the recent past, there has been much controversy in regard to safe contraceptive methods in patients with SCD. In general, patients with this disease should be considered to have a condition that exposes them to increased risk in the event of an unintended pregnancy and therefore the benefits of most available contraceptive options is considered to outweigh potential risks. In 2010, the Centers for Disease Control modified the World Health Organization recommendations for medical eligibility criteria for contraceptive use.101 The use of combined oral, hormonal or progestin only pills, depot medroxyprogesterone acetate or intrauterine devices utilizing copper or levonorgestrel should all be considered methods that generally outweigh the theoretical or proven risks. The barrier contraceptives such as condoms, diaphragms, and foams are extremely safe but are not very effective; therefore, if they are to be used, intensive patient education is mandatory to ensure compliance.
Much has been written about patients with severe hemoglobinopathies concerning surgical sterilization or abortion. Based on earlier data, some have recommended sterilization prior to pregnancy, while others have urged abortion and sterilization or sterilization after one pregnancy. More recent statistics, however, indicate that the danger to the patient from pregnancy is less than in earlier years. For this reason, the patient should be able to make a decision based on her own desires as to family size, taking into account the genetic disability of the infant, the risk of the therapy during pregnancy, and her potentially decreased longevity. Although all the maternal and fetal statistics are improved, it is still much more dangerous for SCD patients to endure pregnancy than it is for those subjects without a hemoglobinopathy.
Sickle cell trait (HbA-S)
Sickle cell trait is a common hematologic abnormality and occurs in 5–14% of African Americans in the US. The patient is usually not anemic unless a concomitant IDA is present. The benign clinical course and inadequate screening lead to under-diagnosis of the HbA-S. The fertility of these patients is thought to be normal. Sickle trait has been associated with an increased susceptibility to renal problems, such as hematuria, frequent infections, and hyposthenuria. Splenic infarction has been reported, but actual crisis is rare unless hypoxia or infection is present. Studies in the literature concerning pregnant patients with HbA-S are less well documented and less numerous than those involving SCD.102, 103, 104 These patients appear to have an increased incidence of pyelonephritis, asymptomatic bacteriuria, and chronic renal disease when studied during pregnancy. The renal problems may be related to sickled cells lodging in the hypertonic portions of the renal medulla, leading to stasis, ischemia, and structural damage, followed by frequent infections. The persistent hyposthenuria may involve a similar mechanism, but these theories are unproven. Pneumonia and preeclampsia may be more common in pregnant HbA-S patients, but most data do not support this contention. Therefore, the maternal morbidity is much less than in patients with SCD and near that for similarly matched patients with HbA-A. The effect of sickle trait on the neonate is minimal, with most studies demonstrating that the mean birth weight, Apgar scores, prematurity rate, and abortions are identical to a matched control population of the hemoglobin A subjects.103, 104
Antepartum management of these patients should be directed toward screening programs for detection, close scrutiny for positive urine cultures, and avoidance of intrapartum complications such as hypotension or blood loss, which may lead to hypoxia. Routinely, iron and vitamin supplementation are given to these women as to the normal parturient, since nutritional IDA may exist. If asymptomatic bacteriuria is present and persists after treatment or if a single episode of cystitis or pyelonephritis occurs, prophylactic antibiotic administration is recommended during the entire pregnancy.
Other sickling hemoglobinopathies
Hereditary persistence of fetal hemoglobin (HPFH) occurs in 0.1% of African Americans in the US. The inheritance pattern for HPFH is considered autosomal codominant, with allelic genes for hemoglobin A, S, and C. Whether pregnant or non-pregnant, persons with hemoglobin-S and HPFH will have are more likely to have a benign clinical course as the higher concentration of HgbF in the red cell means that the concentration of HgbS will be lower. Lower levels of HgbS in the red cell leads to lower degrees of polymerization within the cell, which in turn leads to a lower likelihood of the red cell becoming sickled.105 Persons with HPFH should be distinguished from other subjects with SCD who may have higher concentrations of hemoglobin F. In HPFH-HbS the electrophoretic pattern is hemoglobin F, 25–30%; hemoglobin S, 70–75%; and hemoglobin A2, 2–3%. Persons with HbS-S rarely have over 20% hemoglobin F. More importantly, the hemoglobin F is heterogeneously distributed in routine hemoglobin S patients, with some erythrocytes having no hemoglobin F and others having 30–40%. HPFH patients, in contrast, have the hemoglobin F evenly distributed (approximately 30%) in each RBC, accounting for the lack of clinical symptoms.106 Hemoglobin S patients with HPFH should be reassured concerning their longevity and the benign clinical course.
Hemoglobin Lepore-S and hemoglobin O Arab-S are caused by a structural defect in the β-chain. They resemble HbS-S or HbS-C in symptomatology but their laboratory assessment mimics homozygous β-thalassemia. The therapy during pregnancy is similar to that for SCD because of the clinical severity.107
Structural hemoglobinopathies without hemoglobin S
Other clinically significant hemoglobin variants include unstable hemoglobins, the M or cyanotic hemoglobins, as well as those with abnormal oxygen affinity.108 Diagnosis of the unstable hemoglobins can be assisted by analysis of a peripheral blood smear. Normocytic, hypochromic cells with some poikilocytosis and basophilic stippling are seen. Inclusion bodies may be difficult to distinguish prior to splenectomy but appear as finely distributed dots in their early stages. Characteristically, the reticulocyte count and serum iron values are high. Electrophoretic separation can confirm the diagnosis. When the degree of hemolysis is severe and an acute hemolytic crisis occurs, transfusions and splenectomy may be performed. Clinical management is dependent on the clinical manifestations as well as the severity of the disease.109
Congenital methemoglobinemia, or hemoglobin M, can be linked to two etiologies. A single amino acid substitution in one of the globin chains of hemoglobin is the more common cause. Five varieties of this abnormal hemoglobin have been identified. Substitutions in the α-chain have been identified in two varieties leading to cyanosis in infants at birth. The remaining three variants involve substitutions in the β-chains; these induce cyanosis by the third or fourth month of life. However, chemically induced or congenital deficiency of the enzyme methemoglobin reductase has also been linked to this disorder.110 Normally, oxygen dissociates from the hemoglobin molecule, leaving the iron with the hemoglobin in the ferrous state. When oxidation to the ferric form results, methemoglobin is produced and an ineffective molecule for oxygen binding results. When the level of methemoglobin exceeds 2 g/dL, a state of cyanosis occurs.
Methemoglobinemia, resulting from amino acid substitution in a globin chain, has autosomal dominant transmission, whereas methemoglobin reductase deficiency is the result of autosomal recessive error. Although those affected are cyanotic, they remain asymptomatic. Diagnosis of hemoglobin M can be made by spectroscopy; the abnormal hemoglobins should first be identified by electrophoresis. More specific differentiation of the normal and abnormal hemoglobins can then be made using electron paramagnetic resonance to identify the amino acid substitution. Those affected with methemoglobin reductase deficiency reveal normal spectral analysis. When hemolysis occurs, hemoglobin M can induce slight jaundice.110 As with the other hemolytic anemias, sulfonamides can exacerbate the hemolysis. Methylene blue and ascorbic acid have been useful in treating those with the enzyme deficiency; however, all modes of therapy are ineffective in treating those with the structural defect. Because the disease is benign and lacks symptomatology, these abnormal hemoglobins do not affect pregnancy.
Abnormalities of oxygen affinity have been reported, with almost 50 variants already identified. High-affinity disorders account for three-quarters of the variations, with 32 having been identified. Because of the increased affinity of the hemoglobin molecule to oxygen, smaller amounts of oxygen are released to the tissues. A compensatory polycythemia results in response to increased levels of erythropoietin. These variants are transmitted by autosomal recessive means.111
Hemoglobin functions quite well in the oxyhemoglobin form with respect to oxygen affinity, heme interaction, Bohr effect, and reactivity with 2,3-diphosphoglycerate (2,3-DPG). In the deoxygenated state, the abnormal valine forms hydrophobic bonds with adjacent amino acids. This aggregation results in the formation of tetramers, which are relatively insoluble and coalesce to form microcables. These structures “gel” with similar aggregates to form a cable that distorts the cell into the classic sickled erythrocyte. As this process continues, the cell membrane is affected by loss of cations and phosphorylation capability; thus, the cell becomes irreversibly sickled. The age of the cell, concentration of S and F hemoglobin, extent of oxygenation, status of the cell membrane, temperature, pH, and osmotic pressure of the milieu in the microcirculation all play a role in determining whether a given cell will sickle. The initiation of the crisis may be the formation of sickled cells, with further deoxygenation in the microvasculature, creating a vicious cycle (Fig. 7). Continued sequestration of the cells may lead to the acute and chronic morphologic changes found in the organs of patients with sickle cell anemia. Clinically, this sequence of events causes the pain, leg ulcers, hepatomegaly, autosplenectomy, and chronic anemia found in these subjects. As a rule, hemoglobinopathies not associated with HbS, unless homozygous, usually present fewer management problems to the clinician than do those in the SCD category. Many of these (HbA-C, HbC-C, HbA-D, heterozygous thalassemia) are usually diagnosed only during the workup of a patient with mild, iron-unresponsive anemia.112
REFERENCES
Little MP, Brocard P, Elliott P, Steer PJ. Hemoglobin concentration in pregnancy and perinatal mortality: A London-based cohort study. Am J Obstet Gynecol 2005;193:220-6. |
|
Xiong X, Buekens P, Alexander S, Demianczuk N, Wollast E. Anemia during pregnancy and birth outcome: A meta-analysis. Am J Perinat Vol 17, No 3, 2000. |
|
Steer P, Alam MA, Wadsworth J, Welch A. Relation between maternal hemoglobin concentration and birth weight in different ethnic groups. BMJ 1995;310:489-491. |
|
Bodnar LM, Scanlon KS, Freedman DS, Siega-Riz AM, Cogswell ME. High prevalence of postpartum anemia among low-income women in the United States. Am J Obstet Gynecol 2001;185:438-43. |
|
Centers for Disease Control and Prevention. Pregnancy nutrition surveillance, 1996 full report. Atlanta: US Department of Health and Human Services, Centers for Disease Control and Prevention, 1998. |
|
Rouse DJ, MacPherson C, Landon M, Varner MW et al. Blood transfusion and cesarean delivery. Obstet Gynecol 2006;108:891-7. |
|
Van Vyck DB, Martens MG, Seid MH et al. Intravenous ferric carboxymaltose compared with oral iron in the treatment of postpartum anemia. A randomized controlled trial. Obstet Gynecol 2007;110:267-78. |
|
Beusterien KM, Nissenson AR, Port FK, Kelly M, Steinwald B, Ware JE et al. The effects of recombinant human erythropoietin on functional health and well-being in chronic dialysis patients. J Am Soc Nephrol 1996;7(5):763-73. |
|
Perlman RL, Finkelstein FO, Liu L, Roys E, Kiser M, Eisele G et al. Quality of life in chronic kidney disease (CKD): a cross-sectional analysis in the Renal Research Institute-CKD study. Am J Kidney Dis 2005 Apr;45(4):658-66. |
|
Jones M, Schenkel B, Just J, Fallowfield L. Epoetin alfa improves quality of life in patients with cancer: results of meta-analysis. Cancer 2004 Oct 15;101(8):1720-32. |
|
Corwin EJ, Arbour M. Postpartum fatigue and evidence-based interventions. Abstract MCN Vol 32 No 4. |
|
Weiss G. Modification of iron regulation by the inflammatory response. Best Practice In Research in Clinical Hematology 2005;18:183-201. |
|
Weyermann M, Rothenbacher D, Gayer L, Bode G et al. Role of helicobacter pylori infection in iron deficiency during pregnancy. Am J Obstet Gynecol 2005;192:548-53. |
|
Troy NW. Is the significance of postpartum fatigue being over looked in the lives of women? MCN The Am J Maternal/Child Nursing 2003;28:252-257. |
|
Breymann C, Zimmermann R, Huch R, Huch A. Use of recombinant human erythropoietin in combination with parenteral iron in the treatment of postpartum anemia. European J Clin Invest 1996;26:123-130. |
|
Beard J, Hendricks M, Perez E, Murray-Kolb L, Berg A, Vernon-Feagans L, Irlam J, Isaacs W, Sive A, Tomlinson M. Maternal iron deficiency anemia affects postpartum emotions and cognition. Am Society for Nutritional Sciences 2005;267-271. |
|
Meyer J, Eichorn Karl-Heinz, Vetter K, Christen S, Schleusner E, Klos A, Huch R. J Perinat Med 1995;23:99-109. |
|
Iron deficiency anemia: recommended guidelines for the prevention, detection, and management among U.S. children and women of childbearing age. Institute of Med (IOM) and The National Academy of Sciences, 2000. |
|
Stephansson O, Dickman PW, Johansson A, Cnattingius S. Maternal hemoglobin concentration during pregnancy and risk of stillbirth. JAMA 2000;284:2611-7. |
|
Iron deficiency—United States, 1999-2000. MMWR Morb Mortal Wkly Rep 2002 Oct 11;51(40):897-9. |
|
Müngen E. Iron supplementation in pregnancy. J Perinat Med 31 (2003) 420-426. |
|
Bodnar LM et al. Have we forgotten the significance of postpartum iron deficiency? Am J Obstet Gynecol 2005;193:36-44. |
|
Bodnar LM, Siega-Riz AM, Miller WC et al. Who should be screened for postpartum anemia? An evaluation of current recommendations. Am J Epidemiol 2002;156:903-912. |
|
Hindmarsh PC, Geary MPP, Rodeck CH, Jackson MR, Kingdom JCP. Effect of early maternal iron stores on placental weight and structure. Obstet Gynecol Surv Vol 56(2), Feb 2001, 66-67. |
|
Lee HS, Kim MS, Kim MH, Kim YJ, Kim WY. Iron status and its association with pregnancy outcome in Korean pregnant women. European J Clin Nutrition 2006;60:1130-1135. |
|
Milman N, Byg Keld-Erik, Agger AO. Hemoglobin and erythrocyte indices during normal pregnancy and postpartum in 206 women with and without iron supplementation. Acta Obstet Gynecol Scand 2000;79:89-98. |
|
Milman N, Bergholt T, Keld-Erik Byg, Eriksen L Graudal N. Iron status and iron balance during pregnancy. A critical reappraisal of iron supplementation. Acta Obstet Gynecol Sand 1999;78:749-757. |
|
Baker WF. Iron deficiency in pregnancy, obstetrics, and gynecology. Hematol Oncol Clin North Am 2000;51:s2-s9. |
|
Perez EM, Hendricks MK, Beard JL, Murray-Kolb LE, Berg A, Tomlinson M et al. Mother-infant interactions and infant development are altered by maternal iron deficiency anemia. J Nutr 2005 Apr;135(4):850-5. |
|
Casanova BF, Sammel MD, Macones GA. Development of a clinical prediction rule for iron deficiency anemia in pregnancy. Am J Obstet Gynecol 2005;193:460-6. |
|
Haram K, Nilsen ST, Ulvik RJ. Iron supplementation in pregnancy – evidence and controversies. Acta Obstet Gynecol Scand 2001;80:683-688. |
|
Milman N, Bergholt T, Eriksen L et al. Iron prophylaxis during pregnancy – how much iron is needed? A randomized dose – response study of 20-80mg ferrous iron daily in pregnant women. Acta Obstet Gynecol Scand 2005;84:238-247. |
|
Patterson AJ, Brown WJ, Roberts DC. Dietary and supplement treatment of iron deficiency results in improvements in general health and fatigue in Australian women of childbearing age. J Am Coll Nutr 2001 Aug;20(4):337-42. |
|
Wulff M, Ekström EC. Iron supplementation during pregnancy in Sweden: to what extent is the national recommendation followed? Acta Obstet Gynecol Scand 2003;82:628-635. |
|
Bonar J, Goldberg A, Smith JA. Do pregnant women take their iron? Lancet 1969 Mar1;1:457-8. |
|
Berkovitch M, Matsui D, Lamm SH, Rosa F, Koren G. Recent increases in numbers and risk of fatalities in young children ingesting iron preparations. Vet Hum Toxicol 1994 Feb;36(1):53-5. |
|
Sloan NL, Jordan E, Winikoff B. Effects of iron supplementation on maternal hematologic status in pregnancy. Am J Public Health 2002;92:288-293. |
|
Mára M, Zivný, Eretová V, Kvasnićka J et al. Changes in markers of anemia and iron metabolism and how they are influenced by antianemics in postpartum period. Acta Obstet Gynecol Scand 2001;80:142-148. |
|
Tolkien Z, Stecher L, Mander AP, et al. Ferrous sulfate supplementation causes significant gastrointestinal side-effects in adults: a systematic review and meta-analysis. PLoS One 2015; 10:e0117383. |
|
Bregman DB, Morris D, Koch TA, et al. Hepcidin levels predict nonresponsiveness to oral iron therapy in patients with iron deficiency anemia. Am J Hematol 2013; 88:97. |
|
Werner E, Kaltwasser JP, Ihm P. [Oral iron treatment: intestinal absorption and the influence of a meal (author's transl)]. Dtsch Med Wochenschr 1977; 102:1061. |
|
Auerbach M, Ballard H, Glaspy J. Clinical update: intravenous iron for anaemia. Lancet 2007; 369:1502. |
|
Martin SR, McKee R, Melseth R et al. Intravenous iron improves anemia in pregnancy. Am Col Obstet Gynecol 98S |
|
Bayoumeu F, Subiran-Buisset C, Baka NE et al. Iron therapy in iron deficiency anemia in pregnancy: Intravenous route versus oral route. Am J Obstet Gynecol 2002;186:518-22. |
|
Al RA, Unlubilgin E, Kandemir O et al. Intravenous versis oral iron for treatment of anemia in pregnancy. Obstet Gynecol 2005;106:1335-40. |
|
Kumar RM, Rizk DE, Khuranna A. Beta-thalassemia major and successful pregnancy. J Reprod Med 1997;42:294-8. |
|
Couto FD, De Albuquerque AB, Adorno EV et al. alpha-Thalassemia 2, 3.7 kb deletion and hemoglobin AC heterozygosity in pregnancy: a molecular and hematological analysis. Clin Lab Haematol 2003;25:29-34. |
|
Kazazian HH Jr. Prenatal diagnosis of â-thalassemia. Semin Perinatol 1991;15:15 |
|
Sheiner E, Levy A, Yerushalmi R, Katz M. Beta-Thalassemia minor during pregnancy. Obstet Gynecol 2004;103:1273-7. |
|
Garcia-Casal MN, Osorio C, Landaeta M et al. High prevalence of folic acid and vitamin B12 deficiencies in infants, children, adolescents and pregnant women in Venezuela. Eur J Clin Nut 2005;59(9):1064-70. |
|
Lindbald B, Zaman S, Malik A et al. Folate, vitamin B12, and homocysteine levels in South Asian women with growth-retarded fetuses. Acta Obstet Gynecol Scand 2005;84(11):1055-61. |
|
Stark KD, Pawlosky RJ, Beblo S et al. Status of plasma folate after folic acid fortification of the food supply in pregnant African American women and the influences of diet, smoking, and alcohol consumption. Am J Clin Nutrition 2005;81(3):669-77. |
|
Koc A, Kocyigit A, Soran M et al. High frequency of maternal vitamin B12 deficiency as an important cause of infantile vitamin B12 deficiency in Sanliurfa province of Turkey. Eur J Nutr 2006;45(5):291-7. |
|
Milman N, Byg KE, Hvas AM et al. Erythrocyte folate, plasma folate and plasma homocysteine during normal pregnancy and postpartum: a longitudinal study comprising 404 Danish women. Eur J Haematol 2006;76(3):200-5. |
|
Natelson EA. Pregnancy-induced pancytopenia with cellular bone marrow: distinctive hematologic features. Am J Med Sci 2006;332(4):205-7. |
|
Tichelli A, Socié G, Marsh J et al. Outcome of pregnancy and disease course among women with aplastic anemia treated with immunosuppression. Ann Intern Med 2002;137:164-72. |
|
Hopenhayn C, Bush HM, Bingcang A, Hertz-Picciotto I. Association between arsenic exposure from drinking water and anemia during pregnancy. J Occup Environ Med 2006;48:635-43. |
|
Ackerman L. Hyperreactive malarial syndrome. J Am Board Fam Pract. 1996; 9(5): 356. |
|
Baronciani L, Beutler E. Analysis of pyruvate kinase-deficiency mutations that produce nonspherocytic haemolytic anemia. Proc Natl Acad Sci USA. 1993; 90(9): 4324. |
|
Beutler E., Mitchell M. Special modifications of the fluorescent screening method for glucose-6-phosphate dehydrogenase deficiency. Blood. 1968;32(5): 816. |
|
Beutler E. Red cell metabolism. In: A Manual of Biochemical Methods, 3rd ed, Grune and Stratton, New York 1984. |
|
Kaplan BH, Bank A: Synthesis of heme: the synthesis of globin. In Williams WJ, Beutler E, Erslen AJ et al., eds. Hematology, 3rd edn. New York: McGraw Hill, 1993:287-300 |
|
NHLBI. Evidence-Based Management of Sickle Cell Disease: Expert Panel Report, 2014. p.24. http://www.nhlbi.nih.gov/guidelines (Accessed on April 30, 2016). |
|
Dauphin-McKenzie N, Gilles JM, Jacques E, Harrington T.Sickle Cell Anemia in the Female Patient Obstet Gynecol Survey 2006;61(5):343-52. |
|
Serjeant GR, Hambleton I, Thame M. Fecundity and pregnancy outcome in a chohort with sickle cell disease followed from birth. BJOG. 2005 Sep; 112:1308-14 |
|
Powars DR, Sandhu M, Niland-Weiss J et al: Pregnancy in sickle cell disease. Obstet Gynecol 1986;67:271. |
|
American College of Obstetrics and Gynecology. Hemoglobinopathies in pregnancy. ACOG Practice Bulletin No. 64. ACOG 2007;802-809. |
|
Claster S, Wood JC, Noetzil L, Carson SM, ofstra TC, Khanna R, Coates TD. Nutritional deficiencies in iron overloaded patients with hemoglobinopathies. Am J Hematol. 2009; 84:344-8. |
|
Arruda MM, Mecabo G, Rodrigues CA, Matsuda SS, Rabelo IB, Figueiredo MS. Antioxidant vitamins C and E supplementation increases markers of haemolysis in sickle cell anaemia patients: a randomized, doble-blind, placebo-controlled trial. Br J Haematol. 2013; 160:688-700. |
|
Rao JN, Sur AM. Iron deficiency in sickle cell disease. Acta Paediatr Scand 1980;69:337. |
|
Platt OS, Orkin SH, Dover G, et al. Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia. J Clin Invest 1984; 74:652. |
|
Dover GJ, Humphries RK, Moore JG, et al. Hydroxyurea induction of hemoglobin F production in sickle cell disease: relationship between cytotoxicity and F cell production. Blood 1986; 67:735. |
|
Charache S, Dover GJ, Moyer MA, Moore JW. Hydroxyurea-induced augmentation of fetal hemoglobin production in patients with sickle cell anemia. Blood 1987; 69:109. |
|
Koshy M, Dorn L, Bressler L, et al. 2-deoxy 5-azacytidine and fetal hemoglobin induction in sickle cell anemia. Blood 2000; 96:2379 |
|
DeSimone J, Koshy M, Dorn L, et al. Maintenance of elevated fetal hemoglobin levels by decitabine during dose interval treatment of sickle cell anemia. Blood 2002; 99:3905 |
|
Saunthararajah Y, Hillery CA, Lavelle D, et al. Effects of 5-aza-2'-deoxycytidine on fetal hemoglobin levels, red cell adhesion, and hematopoietic differentiation in patients with sickle cell disease. Blood 2003; 102:3865. |
|
Saunthararajah Y, Molokie R, Saraf S, et al. Clinical effectiveness of decitabine in severe sickle cell disease. Br J Haematol 2008; 141:126. |
|
al-Khatti A, Umemura T, Clow J, et al. Erythropoietin stimulates F-reticulocyte formation in sickle cell anemia. Trans Assoc Am Physicians 1988; 101:54 |
|
Nagel RL, Vichinsky E, Shah M, et al. F reticulocyte response in sickle cell anemia treated with recombinant human erythropoietin: a double-blind study. Blood 1993; 81:9. |
|
Rodgers GP, Dover GJ, Uyesaka N, et al. Augmentation by erythropoietin of the fetal-hemoglobin response to hydroxyurea in sickle cell disease. N Engl J Med 1993; 328:73. |
|
el-Hazmi MA, al-Momen A, Kandaswamy S, et al. On the use of hydroxyurea/erythropoietin combination therapy for sickle cell disease. Acta Haematol 1995; 94:128 |
|
Brugnara C, Bunn HF, Tosteson DC. Regulation of erythrocyte cation and water content in sickle cell anemia. Science 1986; 232:388. |
|
Schwartz RS, Musto S, Fabry ME, Nagel RL. Two distinct pathways mediate the formation of intermediate density cells and hyperdense cells from normal density sickle red blood cells. Blood 1998; 92:4844. |
|
De Franceschi L, Bachir D, Galacteros F, et al. Dietary magnesium supplementation reduces pain crises in patients with sickle cell disease (abstract). Blood 1997; 90:264a. |
|
Pawliuk R, Westerman KA, Fabry ME, et al. Correction of sickle cell disease in transgenic mouse models by gene therapy. Science 2001; 294:2368 |
|
Wambebe C, Khamofu H, Momoh JA, et al. Double-blind, placebo-controlled, randomised cross-over clinical trial of NIPRISAN in patients with Sickle Cell Disorder. Phytomedicine 2001; 8:252 |
|
Cordeiro NJ, Oniyangi O. Phytomedicines (medicines derived from plants) for sickle cell disease. Cochrane Database Syst Rev 2004; :CD004448. |
|
Fawibe AE. Managing acute chest syndrome of sickle cell disease in an African setting. Trans R Soc Trop Med Hyg 2008; 102:526. |
|
Koshy M, Burd L, Wallace D et al. Prophylactic red cell transfusion in pregnant patients with sickle cell disease: a randomized cooperative study. N Engl J Med 1988;319:1447. |
|
Okusanya BO, Oladapo OT. Prophylactic versus selective blood transfusion for sickle cell disease in pregnancy. The Cochrane database of systematic reviews 2013;12:CD010378. |
|
Malinowski AK, Shehata N, D'Souza R, Kuo KH, Ward R, Shah PS, et al. Prophylactic transfusion for pregnant women with sickle cell disease: a systematic review and meta-analysis. Blood 2015 Nov 19;126(21):2424-35. |
|
Asma S, Kozanoglu I, Tarım E, et al. Prophylactic red blood cell exchange may be beneficial in the management of sickle cell disease in pregnancy. Transfusion 2015; 55:36. |
|
Aygun B, Padmanabhan S, Paley C, Chandrasekaran V. Clinical significance of RBC alloantibodies and autoantibodies in patients who received transfusions. Transfusion 2002; 42: 37–43 |
|
Proudfit CL, Atta E, Doyle NM. Hemolytic transfusion reaction after preoperative prophylactic blood transfusion for sickle cell disease in pregnancy. Obstet Gynecol 2007; 110: 471–4. |
|
Brumfield CG, Huddleston JF, DuBois LB, Harris BA Jr. A delayed hemolytic transfusion reaction after partial exchange transfusion for sickle cell disease in pregnancy: a case report and review of literature. Obstet Gynecol 1984; 63: 13s–15s |
|
ACOG Committee on Obstetrics. ACOG Practice Bulletin No. 78: Hemoglobinopathies in pregnancy. Obstet Gynecol 2007; 109: 229–37. |
|
Koshy M, Burd L. Management of pregnancy in sickle cell syndromes. Hematol Oncol Clin North Am 1991; 5: 585–96. |
|
Morrison JC, Whybrew WD, Bucovaz ET. Use of partial exchange transfusion preoperatively in patients with sickle hemoglobinopathies. Am J Obstet Gynecol 1978; 132: 59–63. |
|
Cabibbo S, Fidone C, Garozzo G, et al. Chronic red blood cell exchange to prevent clinical complications in sickle cell disease. Transfus Apher Sci 2005; 32: 315–21. |
|
National Institutes of Health, National Heart, Lung, and Blood Institute, and Division of Blood Diseases and resources. The Management of Sickle Cell Disease, 4th edn. Bethesda: NIH Publication No 02-2117, 2002. |
|
U.S. Medical Eligibility Criteria for Contraceptive Use, 2010 — Adapted from the World Health Organization Medical Eligibility Criteria for Contraceptive Use, 4th edition. MMWR 2010; 59. Available at www.cdc.gov/mmwr/preview/mmwrhtml/rr59e0528a1.htm. (Accessed September 3, 2011). |
|
Taylor MY, Wyatt-Ashmead J, Gray J et al. Pregnancy loss after first-trimester viability in women with sickle cell trait: time for a reappraisal? Am J Obstet Gynecol 2006;194(6):1604-8. |
|
Bryant AS, Cheng YW, Lyell DJ et al. Sickle cell trait and preterm delivery. Obstet Gynecol 2007;109:870-4. |
|
Tita ATN, Biggio JR, Chapman V, Neely C, Rouse DJ. Perinatal and maternal outcomes in women with sickle or hemoglobin C trait. Obstet Gynecol 2007;110:1113-19. |
|
Poillon WN, Kim BC, Rodgers GP, Noguchi CT, Schechter AN. Sparing effect of hemoglobin F and hemoglobin A2 on the polymerization of hemoglobin S at physiologic ligand saturations. Proc Natl Acad Sci 1993; 90:5039. |
|
Forget B. Molecular Basis of Hereditary Persistence of Fetal Hemoglobin. Annals of the New York Academy of Sciences. 1998; 850: 38-44. |
|
Ghosh K, Colah R, Ross C, et al. Guidelines for diagnosis and management of hemoglobinopathies. Indian Journal of Human Genetics. 2014; 20: 101-119. |
|
Nagel, RL. Unstable hemoglobins; hemoglobins with latered O2 affinity; Hb M. In: Disorders of haemoglobin: Genetics, Pathophysiology, Clinical Management, Steinberg MH, Forget BG, Higgs DR, et al. (eds), Cambridge University Press, Cambridge 1999 |
|
Werner EJ, Stockman JA. Red cell disturbances in the feto-maternal unit. Semin Perinatol 1983;7:139. |
|
DeBraun MR, Frei-Jones M, Vichinsky E. Hereditary methemoglobinemia. In: Kliegman RM, Behrman RE, Jenson HB, Standon BF, eds. Neslon Textbook of Pediatrics. 19th ed. Philadelphia, PA: Suanders Elsevier; 2011: chapt 456.7. |
|
Percy MJ, Butt NN, McMullin MF, et al. Identification of high oxygen affinity hemoglobin variants in the investigation of patients with erythrocytosis. Hematologica. 2009; 94: 1321-22. |
|
Steinberg MH. Sickle hemoglobin polymer: Structure and functional properties. In: UpToDate, Mahoney DH (Ed), UpToDate, Waltham, MA. (Accessed May 18, 201 |