
Developmental Abnormalities of the Female Reproductive Organs
Authors
INTRODUCTION
An understanding of congenital anomalies as they are encountered in clinical practice is greatly enhanced by not only a knowledge of normal embryology and the mechanism of formation of normal infants, but also an insight into the processes that result in the development of anomalies.1, 2, 3, 4 An awareness of malformations and a systematic examination and appraisal of every neonate will greatly increase the number of such anomalies found. In some instances, e.g., congenital adrenal hyperplasia, imperforate anus, diaphragmatic hernia, and esophageal atresia, early detection and prompt intervention may be lifesaving. In adults, amenorrhea is an important clue and may suggest an imperforate hymen, vaginal septum or absence of the uterus. The finding of one anomaly should stimulate a careful gynecologist to carry out a complete study to detect renal and ureteral anomalies, particularly the solitary pelvic kidney which might be removed as a “pelvic mass”. Many anomalies occur infrequently so that only physicians in large medical centers may see them frequently enough to be aware of the possible anomalies and their causation, prognosis, and, in some cases, correction. The identification and interpretation of such abnormalities constitute a real challenge to the clinician. A knowledge of the problems and pitfalls in the management of these defects will benefit both the obstetrician and the gynecologic surgeon.
CAUSES OF MALFORMATIONS
The causes of congenital malformations or abnormalities present at birth may be either environmental or genetic (chromosomal abnormalities).5 It is not always easy to separate the two factors; both may be at work in the same embryo or fetus. Rapidly growing embryonic organs are the most sensitive to environmental influences. Millen6 has classified the mechanisms of anomaly production as follows:
- Developmental arrest—cessation of development before completion
- Agenesis or aplasia—failure of normal development
- Hyperplasia or local overgrowth
- Aberrant development
- Failure of normal resorption (either too much or too little) or resorption in the wrong locations
- Secondary degeneration of normally developed structures
Millen also emphasizes “that the period when environmental agents may affect the development of an embryo is very short, being nearly over by the end of the eighth week of pregnancy". Organogenesis occurs from day 13 to day 60; teratogenic (G. teras, monster) agents are most dangerous during this period. There is a time relationship between specific organ systems and sensitivity to environmental factors as well as a relationship between specific teratogens and specific organ systems. Examples are rubella infections occurring in the first trimester, with a high incidence of cataracts, deafness, and cardiac malformations, and use of thalidomide, with varied malformations of arms and legs.
Nugent7 has evaluated in detail the mechanisms of action of various environmental teratogenic factors. These include the following:
- Ionizing radiation
- Vital disease and related infections
- Chemical factors
- Immunologic disturbances
- Hormones
- Nutritional factors
Ionizing radiation is probably one of the best known damaging factors. Infections such as rubella virus, cytomegalovirus, and Toxoplasma gondii can cause severe damage to the eyes and central nervous system. Chemicals include aminopterin (causing skeletal defects and nervous system damage), methotrexate, and thalidomide. Immunologic disturbances include Rhesus incompatibilities. Hormone damage is particularly interesting: the administration of exogenous testosterone, synthetic progestogens, and similar preparations can cause iatrogenic deformities of the female genitalia. Pathologic hyperandrogenemia, as seen in luteomas of pregnancy, can result in virilization in the female newborn. Environmental factors, such as exposure to diesel fumes, have also been associated with virilization due to inhibition of aromatase and accumulation of excess testosterone.8 Nutritional factors apparently have little direct teratogenic effect on the fetus.
CYTOGENETICS
In 1923, Painter reported that there were 48 chromosomes in the normal human cell. The correct number of 46 was established in 1956 by Tijo and Levan.9 In 1956, Down syndrome, Turner syndrome, and Klinefelter syndrome were shown to be the result of chromosomal anomalies. Porter10 pointed out that we can now distinguish every individual chromosome and different regions of each chromosome. Normal variants can be defined reliably, extra chromosomes involved in abnormalities can be identified accurately, small defects previously missed can now be recognized, and structural defects can be mapped accurately by tracing exchange segments of chromosomes. A clear cytogenetic diagnosis can serve as a guide to a rational plan of management, to counseling with regard to prognosis and genetic problems, and in the monitoring of pregnancies in people with an increased risk of having children with defects.
There are hundreds of chromosomal abnormalities; most involve visible morphologic defects which should alert the clinician to the possibility of chromosomal anomalies. Disorders of gonadal development such as Klinefelter syndrome (47XXY) and Turner syndrome (45X) have been described with sex chromosome aneuploidy. Even deletions within regions of the X or Y chromosome can be deleterious to normal development. Gene deletions in the distal arms of the X chromosome (Xp22.3) cause short stature, mental retardation, X linked ichthyosis, and Kallman syndrome.11 The distal region of the Y chromosome contains the sex-determining region Y chromosome (SRY) that encodes the gene for testis-determining factor (TDF).12 Deletions in this region cause gonadal dysgenesis and streak gonads. Transfer of this region to the X chromosome causes an XX male. Another region of interest within the Y chromosome is the azoospermia factor region (AZF) that is related to spermatogenesis. It has been discovered that one subset of gene rearrangements on the Y chromosome, "micro-deletions", is a major cause of male infertility in some populations.13 With refined cytogenetic mapping, we will be able to better correlate phenotype with genotype.
The complexity of the rapidly changing field of cytogenetics is not only startling but also a fascinating promise of new knowledge, new discoveries, and new clinical tools. With these new tools, challenges emerge for the obstetrician/gynecologist, reproductive endocrinologist, and pediatrician. Genetic analysis may become routine for every embryo, fetus or neonate. Embryos can be biopsied and a single cell screened for aneuploidy prior to implantation.14 This technique has the potential to improve pregnancy outcomes and decrease miscarriage rates, especially in couples with multiple fetal losses.15 The public health implications are startling and indications for testing are increasing.
GAMETOGENESIS
The union of a spermatozoon and ovum marks the beginning of a new individual. The fertilization of an ovum is an amazingly complex act. Egg formation (oogenesis) and sperm formation (spermatogenesis) have many similarities, although they differ with regard to sex determination. There are 44 somatic chromosomes and two sex chromosomes in the normal human cell.9
Human oogenesis begins with the formation of a primary oocyte which contains 44 + XX chromosomes. The number of germ cells is fixed during fetal development and cannot be regenerated. The oocyte undergoes meiosis, a process that generates haploid gametes through a specialized process that consists of one round of DNA replication followed by two rounds of cell division. In humans, the oocyte begins meiosis during embryogenesis and remains arrested in prophase I until ovulation, when meiosis resumes. Completion of this first division resumes with ovulation and the normal, or diploid number of chromosomes, found in the body cells is reduced to the haploid (G. haplous, single) number. This occurs so that the normal chromosomal number of 44 somatic chromosomes and two sex chromosomes will be restored after fertilization. During this first maturation division, the secondary oocyte (22 + X) retains most of the cytoplasm, while the other half of the nucleus remains as a small first polar body. A second maturation division results in the formation of the mature ovum (22 + X) plus a second polar body. Meiosis is a requisite step in sexual reproduction and is critical for generating genetic diversity through recombination. In humans, meiotic errors lead to reproductive failure via aneuploidy, spontaneous abortion or infertility.16, 17
The progression through meiosis in the male is continuous, and in humans, begins with puberty. This is in sharp contrast to the female where meiosis is begun during embryogenesis, but is maintained in a state of prophase I arrest until after puberty when ovulation occurs. The oocyte can be maintained in prophase arrest for over 40 years. The spermatogonium is formed in the testis, with a chromosome complement of 44 + XY. This Y sex chromosome determines male development. The spermatogonium develops into a relatively large spermatocyte (44 + XY). This in turn undergoes a first maturation (meiotic) division to form secondary spermatocytes, half of which are 22 + X and half of which are 22 + Y. During a second maturation division, the secondary spermatocytes again divide into spermatids, half of which are 22 + X and half of which are 22 + Y. After 1 or 2 weeks the spermatids become mature spermatozoa. Unlike the female where a single primary oocyte yields a single mature oocyte, a single spermatogonium yields four mature spermatozoa. These spermatozoa must undergo further changes before they can fertilize an ovum. The first change, known as capacitation, is a physiologic change probably associated with removal of a protective coating.18 Following this, an acrosome reaction occurs at the anterior extremity of the spermatozoon, where small perforations develop in the wall. Enzymes passing through these openings digest a path for the sperm through the corona and the zona pellucida.
Ovum transport is the mechanism by which the nonmotile ovum is carried by a stream of peritoneal fluid into the infundibulum of the tube. This stream is produced by sweeping movements of the fimbriae. The ovum is carried into the tubal ampulla partially by muscular contractions of the tube but mostly as a result of ciliary action. At any given normal intercourse, over 300 million sperm are deposited in the vagina near the cervical os. Only a few thousand sperm reach the oviducts and only a few hundred reach the ampulla, where fertilization usually occurs. As the sperm head advances, it reaches the surface of the ovum and attaches so that its nucleus is within the membrane of the ovum. As a result, the zona pellucida changes and the entrance of other spermatozoa is inhibited. At the same time, the secondary oocyte is completing its second meiotic division and expelling the second polar body. The male and female pronuclei come into contact near the center of the ovum and mingle their chromosomes. During the process of fertilization, the chromosomes of father and mother mingle, the diploid number (46) of chromosomes is restored, and sex is determined by the presence or absence of the Y, or male, chromosome.
Cleavage
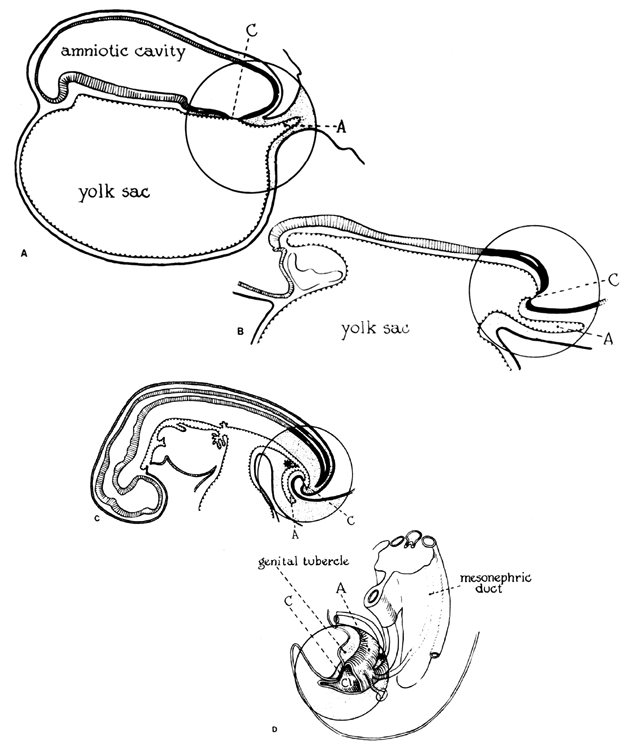
The fertilized egg undergoes a series of rapid mitotic divisions while passing down the tube. After several days, a ball of cells called the “morula” (L. morus, mulberry) is formed. After 5 days, a fluid-filled space appears in the morula, which is now called the “blastocyst” (G. blastos, germ) (Fig. 1.). An inner cell mass appears on one side of the blastocyst cavity at the site of the embryo. By 6 days the blastocyst has implanted and the trophoblast cells invade the succulent endometrium or decidua. During the second week, the embryo becomes a bilaminar disc (Fig. 2).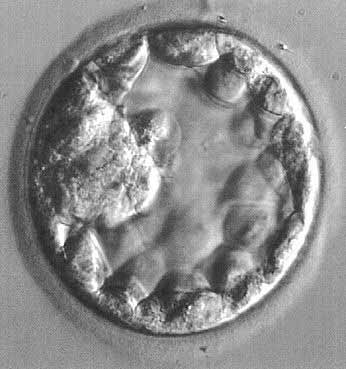
Fig. 1. Human blastocyst, showing inner cell mass at 9 o'clock position and trophectoderm lining periphery.
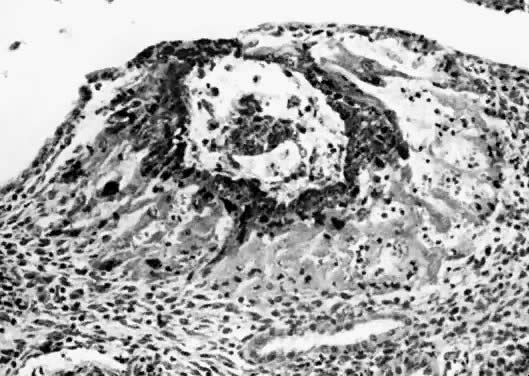
Fig. 2. Bilaminar embryonic disc. The cells of the embryonic disc flatter and differentiate into two layers, the epiblast and the hypoblast. The epiblast differentiates for forma all three layers of the trilaminar embryonic disc. Extraembryonic endoderm, including the yolk sac, is derived from the hypoblast. The bilaminar disc is sandwiched between the amniotic cavity and the yolk sac. The large circle of dark cells is the cytotrophoblast.
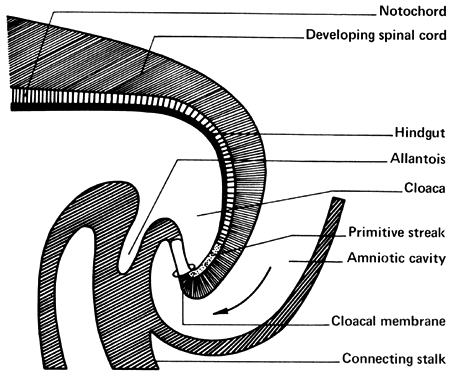
The amniotic cavity appears above the embryonic ectoderm while the blastocyst cavity develops into the yolk sac. In humans the yolk sac never develops yolk as such but makes important contributions to organogenesis. Around amnion, yolk sac, and embryo lies the extraembryonic mesoderm in which isolated lacunas appear. These celomic spaces fuse rapidly and form large areas of extraembryonic celom (G. koiloma, hollow). During the third week, a three-layered embryo develops, consisting of ectoderm, endoderm, and mesoderm. A notochord and the notochordal canal develop. The flat embryonic disk folds into a roughly cylindric shape, and the rapid growth of the embryo results in folding at both ends of the embryo. At the caudal end, a tail fold, which includes part of the yolk sac, develops and becomes a hindgut. Part of the hindgut becomes the cloaca, separated from the amniotic cavity by the cloacal membrane, while a ventral outpouching develops as the allantois (G. alias, sausage).
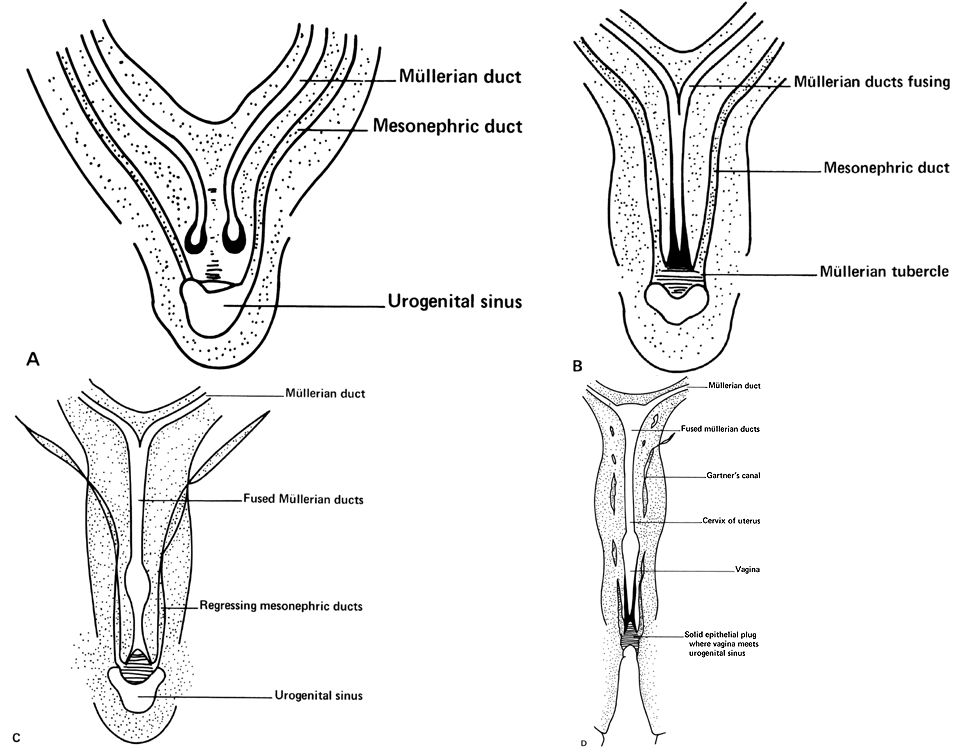
Very early in embryonic development, the primitive body cavity (an intraembryonic celom) appears. Running longitudinally along the sides of the cavity, the anlage of the genital ridges develops. Primordial cells aggregate along these ridges to form the earliest elements of the future gonads. Two sets of future duct systems develop. The mesonephric urinary ducts are formed and connect at the caudal end with the urogenital sinus and the cloaca. The paramesonephric or müllerian ducts develop lateral and parallel to the mesonephric ducts.19 These urogenital ridges become more prominent and fuse caudally to form the genital cord. During their development, the paramesonephric (müllerian) ducts migrate until they lie side by side in the midline. These paramesonephric ducts develop initially at the cranial ends where funnel-shaped openings communicate with the future peritoneal cavity. As these ducts develop caudally, they cross the ventral aspect of the mesonephric ducts to fuse in the midline, resulting in a Y-shaped tubular structure. The caudal portion of this fused paramesonephric duct forms an elevation known as the müllerian tubercle, which protrudes into the urogenital sinus. The caudal ends of the mesonephric ducts enter the urogenital sinus on either side of the müllerian tubercle. Under the influence of the ovarian hormones (and due to the lack of male hormones in females) the paramesonephric ducts continue their development to form the genital tract, while the mesonephric system regresses except for certain vestigial structures. The anterior or cranial portions of the paramesonephric duct system develop into the oviducts or fallopian tubes; the middle portion develops into the body of the uterus, while the caudal fused portions become the primordium of the cervix and the upper vagina. With further development they fuse to form a double-barreled organ which terminates in the müllerian tubercle located in the dorsal portion of the urogenital sinus. In the early embryo, sexual differentiation is not apparent until the tenth week of development. After this time the mesonephric or wolffian ducts regress in the female, leaving a few vestigial fragments, chiefly in the broad ligament. The paramesonephric or müllerian ducts continue their developmenl to form vagina, uterus, and tubes (Table 1).
Table 1. Timetable of development of the female genital tract
Size (mm) | Age (weeks)* | |
0.08 | 7.5 days | Bilaminar disk embryo |
0.21 | 13 days | Yolk sac forming, hindgut, foregut |
0.36 | 20 days | Allantois formed |
1.5 | 2+ | Gonocytes are located in yolk sac entoderm Urogenital ridge begins to form |
2 | 3 | Wolffian duct appears as coalescence of pronephric ducts Allantois well formed, joins hindgut at cloaca Cloacal membrane forms as cloaca contacts hind-end ectoderm |
4 | 4 | Wolffian ducts open into urogenital sinus (cloaca) |
5 | Primordium of gonad appears | |
6 | 4+ | Primordial germ cells in mesenchyme |
7 | 1 month + | Indifferent gonad developing |
5 | Wolffian ducts open to urogenital sinus Ureteric buds External genital tubercle forming | |
8 | Sex cords budding from celomic epithelium Anlage of adrenal cortex forms Urorectal septum starts partition of cloaca | |
10 | Free ends of sex cords form rete testis | |
11 | 5.5 | Müllerian funnel and duct begin to form Primary bifurcation of ureteric bud Sex difference in external genitalia: urethral groove shorter in female Urethral folds develop lateral to central groove |
15 | 5¾ | Differentiation of gonad into testis in male Ureter opens separately from wolffian duct into urogenital sinus |
16 | 6 | Cloaca subdividing; urorectal septum completed Phallus forming from genital tubercle |
18 | Secondary bifurcation of ureteric bud Urethral membrane (and cloacal membrane) perforate | |
20 | 7 | Gonad recognizable as ovary; cortex forming Labioscrotal folds developed (lateral genital swellings) |
23 | 8 | |
25 | Coronary sulcus of phallus appears Female phallus shows caudal curvature | |
30 | Interstitial cells recognizable | |
32 | 9 | Müllerian ducts reach urogenital sinus Müllerian tubercle formed |
42 | Lower müllerian ducts fused | |
45 | 10 | Wolffian ducts begin to regress in female Coronary sulcus of phallus forms groove (easily recognized) |
50 | Posterior commissure formed Great increase of Leydig cells from mesenchyme Prostatic buds first appear in male | |
56 | 11 | Sex differences in external genitals first noticeable Uterovaginal canal formed, fusion of müllerian ducts with sinovaginal bulbs |
65 | Wolffian ducts begin to disappear Longitudinal folds in urogenital sinus Sinovaginal bulbs formed, obliterate müllerian tubercle | |
70 | 12 | Beginning solidification of uterovaginal canal by “proliferation of both müllerian and sinovaginal” epithelium |
94 | Sinus upgrowth along vaginal plate | |
100 | 16 | Wolffian ducts have disappeared by this stage External genitalia of male fully formed |
125 | Fetal ovarian stroma differentiates | |
150 | Primordial follicles in central part of ovary Vagina canalizes from urogenital sinus forward Maximum development of Leydig cells | |
175 | 20 | Beginning shrinkage and degeneration of Leydig cells |
180 | Vagina fully canalized; glycogenated epithelium | |
187 | Seminiferous tubules coiled; lumina | |
200 | 24 | |
240 | 7 months (early) | Testes descend into scrotum |
250 | 28 | During cortical differentiation, medullary cords crowded centrally; some medullary tubules provided with germ cells persist in hilum |
286 | 32 | Reduction in Leydig cells (male) |
Disappearance of medullary cords in ovary (female) |
*Except as otherwise noted.
From Dougherty CM, Spencer R: Female Sex Anomalies. Hagerstown, MD: Harper & Row, 1972.
The cumbersome terms “mesonephric” (wolffian) and “paramesonephric” (müllerian) have always been difficult to keep clearly differentiated. Mesonephric means “middle-kidney” (pronephric elements are not a factor in human development). The eponym “müllerian duct” is deeply ingrained in the literature as relating to uterus, tubes, and vagina, while the term “paramesonephric”, meaning "alongside the middle kidney system" is less difficult to remember. The concept of the paramesonephric (müllerian) duct system as a double-barreled organ (suggesting a shotgun) is useful in understanding developmental anomalies, since various growth arrests and failures of development at this stage explain many of the anomalies encountered.
UTERINE ANOMALIES
The most frequent uterine anomalies (Fig. 3) are those resulting from varying degrees of failure of fusion of the müllerian ducts. This variability makes classification difficult and determination of the true incidence uncertain. Many of these malformations are detected by radiologic or sonographic studies. Estimates of incidence vary from 0.13% to 4.0%. The incidence of müllerian anomalies in patients with infertility has been reported to be as high as 6.3%.20 Pregnancy occurs in many women despite these anomalies. The complication rates with pregnancy are considerably increased; complications include abortion, prematurity, postpartum hemorrhage, retained placenta, and breech presentation.21 Not surprisingly, the rate of cesarean delivery is markedly higher.
|
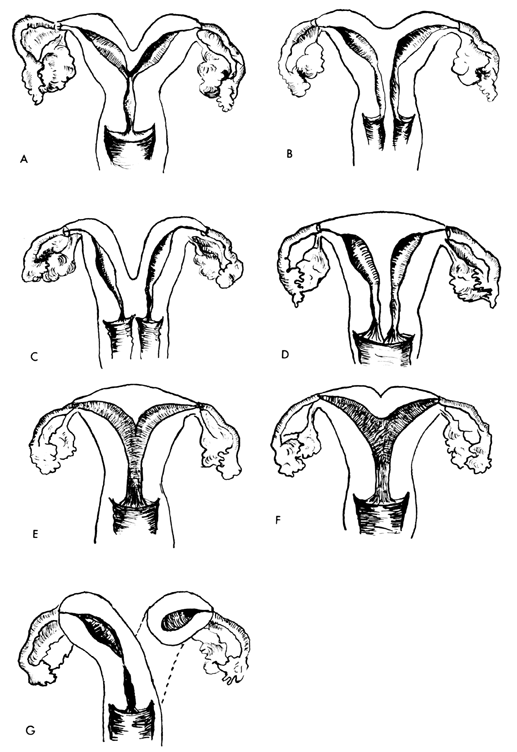
Uterus duplex, or the bicornuate uterus, is the most frequent uterine anomaly. The unicollis type in which there is a single cervix with a septum that does not reach the cervix is the most frequent type, occurring in over one third of all patients with uterine anomalies.
Uterus duplex bicollis, in which two cervices are present, is less frequent. Obstetric complications are frequent, but live births do occur.
Uterus didelphys, with completely separate uterine cavities, is also frequent. The cervices are externally united and the uterine fundi are externally separate. In most patients the vagina is septate, causing a double vagina. The halves of such a uterus are often of different sizes. If there is an asymmetric vaginal septum which occludes one vagina, mucocolpos or hematocolpos may result. Communicating uteri, involving an incomplete uterine septum with part of the fetus in each uterine cavity, occasionally occur.
Uterus septus is an essentially normal uterus with a septum reaching to the cervix.
Uterus subseptus involves a partial septum that does not reach the cervix. Twins apparently occur approximately three times more often in women with this condition than in women with normal uteri; the cause is not clear.
Uterus arcuatus is a normal uterus without a septum. The fundus, however, is notched or flattened. There is usually no interference with normal pregnancy.
Uterus unicornis is a uterus with a single horn. A normal vagina and a single normal tube are usually present. The other half of the uterus is usually absent or rudimentary. In most patients the kidney is missing on the side of the missing uterus. Successful pregnancy can occur.
Separate hemiuteri with separate vaginas is a rare condition that is usually associated with duplications of urethra and bladder or of the colon and anus. Pregnancy in each of the two hemiuteri in the same woman at different times has been reported.
These anomalies result from failure of fusion of the paired müllerian ducts, but in some instances there is a true duplication of the ducts on one or both sides. Such duplications result from splitting of the müllerian duct during the seventh week of development. Accessory tubes or ovaries may be present.
Semmens22 extensively reviewed the literature on genital tract anomalies and evaluated 56 personal patients as well as 500 cases from the literature. He employed a simplified classification, based entirely on the functional capacity of the uterine cavity, which divided genital tract anomalies into two groups: group I—hemiuterus of single müllerian origin, paired or otherwise, and group II—uterus of dual müllerian origin, associated with varying degrees of absorption. He concluded that if the entire functional component has been derived from a single müllerian duct and its vaginal outlet is a cervical canal of similar origin its capacity is smaller than that of the uterus resulting from fusion of bilateral ducts.
By contrast, if the uterus is derived from two müllerian ducts, its capacity is larger. Semmens concluded that the space available for the developing fetus as well as the variations in uterine circulation had a direct effect on the length of gestation, onset and behavior of labor, and overall fetal salvage. The diagnosis of uterine anomalies during pregnancy is always difficult. The most important factor is awareness of their existence and frequency and of the problems that frequently develop. Findings such as a floating head at term without apparent cause, notching and broadening of the uterine fundus, abnormal lie, recurrent breech presentations, prolonged third stage of labor, and trapped or retained placenta indicate the possibility of an anomaly. In abnormal uteri, triangular spasm and cornual pocketing of the placenta may occur and necessitate manual removal of the placenta. The administration of oxytocin under these circumstances usually increases the degree of pocketing in an abnormal uterus. Among antepartum patients, pyelitis (often associated with a urologic anomaly), passage of a decidual cast, hemorrhage, and premature rupture of the membranes are the most common complications. Intrapartum complications include septal dystocia, incarcerated nongravid horn, uterine inertia, and retained placenta. Fetal dystocia is considerably increased. Postpartum complications are few and include hemorrhage, retained placenta, and failure of uterine involution. Most complications of uterine anomalies in the pregnant woman can be anticipated and managed successfully if the clinician is alert to the possibility of anomalies and if the nature of the anomaly is known. The finding of an asymmetrically located cervix in the vaginal fornix, an excessively large cervix whether or not a septum is present, or a duplicated cervix suggests uterine abnormality. Abnormal configurations of the fundus of the uterus in the third trimester should suggest abnormalities to the clinician, as should abnormal presentations and failure of the presenting part to engage without apparent reason. An obstetric history of successive abortions, recurrent breech presentations, or recurrent pyelitis should alert the obstetrician to the possibility of anomalies and, in nonpregnant women, to the need for hysterosalpingogram, saline sonography, magnetic resonance imaging, and renal imaging.
RUDIMENTARY HORN PREGNANCY
O'Leary and O'Leary23 reviewed and analyzed 240 published cases of pregnancies that occurred in a rudimentary uterine horn. They found that 89% ruptured and 61% of these ruptured in the second trimester. Fetal death occurred in 98% of cases. There have been a handful of case reports describing fetal survival after rupture of a rudimentary horn24, 25, 26 and a handful of cases describing delivery of a term fetus in an unruptured horn.27, 28 Since most rudimentary uterine horns are thicker than the fallopian tube, rupture tends to occur later than in tubal ectopic pregnancy and precipitates more severe concealed hemorrhage. Expectant management is not an option for a pregnancy in a rudimentary horn.
With the advent of better imaging studies, it should become more common practice to detect pregnancies in the rudimentary horn earlier in pregnancy. In 2005, Jayasinghe et al published a review of 336 rudimentary horn presentations (210 gynecologic and 156 obstetric).29 Noncommunicating horns accounted for 92% of cases (95% confidence interval 88–95%, p <0.001). Diagnosis before clinical symptoms occurred in only 14% (95% confidence interval 7–23%). The majority of functional noncommunicating horns presented with acute obstetric uterine rupture. If a non-communicating horn is identified pre-conception, surgical removal before pregnancy is recommended. Removal of the rudimentary horn can be accomplished via laparoscopy.30, 31 Sadly, rates of diagnosis prior to catastrophic rupture remain disappointingly low.
In addition to the morbidity associated with uterine rupture, abnormal placentation is commonly encountered in these pregnancies and adds further complication. Many rudimentary horn pregnancies have an associated placenta accreta or placenta percreta.32, 33, 34 The obstetrician caring for the patient with a rudimentary horn pregnancy must be alerted to this and the operating room team must be prepared during delivery should life threatening hemorrhage occur.
TUBAL ANOMALIES
The fallopian tubes are formed from the most cephalic portions of the müllerian ducts. Late in the sixth week (crown rump length, 11 mm), a shallow depression can be observed near the mesonephric duct. Commencing as a groove, a tube forms, a lumen develops, and the paramesonephric ducts grow caudad, lying lateral to the wolffian ducts. The ostia at the cephalic ends remain open. Anomalies of the oviducts are not prominent and frequently pass unnoticed. Absence of one or both tubes may occur and is almost always associated with absence of the uterus as well as with other anomalies. Localized factors may result in an incomplete tube. Rudimentary tubes occur as fibromuscular cords without lumens. If the tube is a long, thin hypoplastic structure and responds to appropriate endocrine therapy, it is classified as infantile. Occasionally, ostia are duplicated or an accessory tube may be present. Most anomalies as such do not require treatment.
Acquired defects of the midportion of the fallopian tube can also occur. In 2007, Grover reported a case of torsion of a paratubal cyst that interrupted the ampullary portion of the fallopian tube.35 Had this interruption not been diagnosed at the time of emergency surgery for torsion, it may have later been mistaken for a congenital anomaly. Prior to the advent of assisted reproductive technology (ART), reproductive options for women with congenital anomalies of the fallopian tubes were limited. ART can bypass the fallopian tubes and enable women without fallopian tubes to achieve pregnancy.
OVARIAN ANOMALIES
The sex of each human is determined at the time of union of spermatozoon and ovum, but no morphologic sex differentiation in the ovaries can be demonstrated until the seventh week. The primordial germ cells are first found at the 24th day near the allantois. The germ cells proliferate and migrate to reach the genital ridge, which by the fifth week, becomes elevated and thickened. Sex differentiation becomes visible early in the eighth week. Proliferation of oogonia (germ cells) by mitosis continues until approximately the 15th week. By the fourth month primary follicles appear. In the meantime, the ovary has become recognizable as a discrete organ which “descends” to the level of the pelvic brim and undergoes lateral rotation.
Ovarian anomalies other than the streak ovaries of gonadal dysgenesis are quite rare. Complete absence of an ovary is extremely rare and is usually associated with renal agenesis and absence of the ipsilateral fallopian tube. True ovarian duplication is rarely reported; it occurs in conjunction with duplication of genital ridge and a duplicated müllerian duct. Excess ovarian tissue near the normal ovarian tissue which has developed from it (and may be connected with it) is classified as an accessory ovary. Lobulation of an ovary is not infrequent and is of little clinical importance. Supernumerary ovaries or the presence of ovarian tissue not connected to the tubes or uterus is very unusual.
Gonadal dysgenesis generally refers to a condition where gonadal development is abnormal, often only presenting streaks of connective tissue that are often called "streak gonads". Streak ovaries extend from the lateral pelvic wall to the attachment of the utero-ovarian ligaments. They vary considerably in size but are usually approximately 4 cm in length and 2–3 mm in width. Dysgenetic ovaries are characterized by absence of follicular structures and oocytes. In women, the most common cause of gonadal dysgenesis is Turner syndrome, 45X. Phenotypic females with streak gonads can also have XX gonadal dysgenesis, XY gonadal dysgenesis or mixed gonadal dysgenesis. In phenotypic females with a Y chromosome, there is a high risk of the development of a gonadoblastoma and removal of the gonads is usually indicated.36
MESONEPHRIC REMNANTS OR VESTIGES
At 28 days after fertilization, when the embryo is 4 mm long, the mesonephros and the mesonephric duct have begun to differentiate. By 49 days post-fertilization (20 mm) the mesonephric duct system has begun to degenerate. Degeneration is never complete, however, and a number of structures may persist to various degrees in the normal adult female (Fig. 4). Such structures include the following:
|
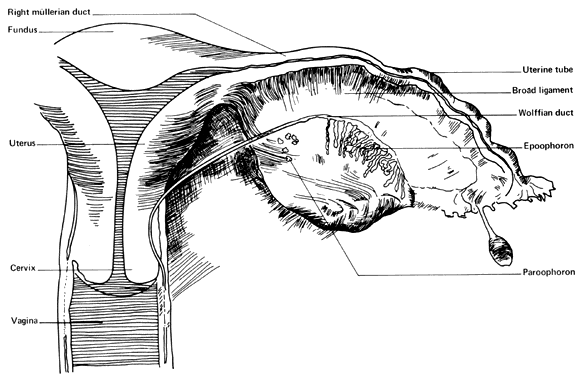
- Hydatid of Morgagni (probably of müllerian origin)
- "Vesicular appendage"
- Epoophoron (organ of Rosenmfiller)
- Paroophoron (Kobeit's tubules)
- Gartner's duct or canal (ductus epoophori longitudinalis)
The broad ligament contains a number of important structures. In addition to the normal tubes, ovaries, uterine and ovarian blood vessels, and the round ligaments, vestiges of the mesonephric duct system are usually present to varying degrees. The regressing mesonephric or wolffian duct system begins near the tubal ostium and runs parallel to the tube in the broad ligament, entering the wall of the uterus near the level of the internal os. It continues in the cervical and vaginal walls until it ends at or near the introitus. These vestiges are variable with respect to place and size. Occasionally, a small structure called the “appendix vesiculosa” (sessile hydatid) may be encountered below the tubal ostium at the edge of the broad ligament at the distal end of the mesonephric duct and is believed to represent its blind end.
In the lateral third of the mesovarium lies the epoophoron, consisting of eight to 13 tubules running from the mesonephric duct toward the ovary. These convoluted tubules are lined with a low cuboidal epithelium, but no secretory activity has been noted in them. They are of little clinical significance, although benign cysts are believed to occasionally arise in them.
Farther caudad along the regressing mesonephtic duct may be found a small group of mesonephric tubules called the “paroophoron”. They are usually found only in infants and are clinically unimportant.
Farther along the course of the vestiges of the mesonephric duct can be found remnants of the duct, here called “Gartner's duct” (ductus epoophori longitudinalis). Coiled tubes frequently occur in the lower part of the supravaginal cervical wall, where they are called the “ampulla”. In some cases, cystic remnants may occur in the vaginal walls, where their unsuspected presence may cause diagnostic confusion. In rare cases, Gartner's duct may form an ectopic ureter as a result of having maintained its ureteral connection. Although believed to be paramesonephric rather than mesonephric in origin, clear pedunculated hydatid or cystic structures arising at the ostium at the end of the tube are found frequently. These are called the “hydatids of Morgagni” (appendix vesiculosa) and are usually harmless but are removed when encountered since they can undergo torsion with or without gangrene and have been misdiagnosed as dysmenorrhea, appendicitis, and ectopic pregnancy.
UMBILICAL ANOMALIES
The umbilicus may contain vestiges of either the yolk sac or the allantoic stalk. Normally these structures have almost disappeared by birth.
Persistent yolk stalk (vitelline or omphalomesenteric duct) anomalies include Meckel's diverticulum (of which over one fifth are attached to the abdominal wall), omphaloileal fistula (characterized by a patent tube connecting umbilicus and ileum), remnants of umbilical mucosa (forming an umbilical polyp or sinus), remnants of vitelline blood vessels (a solid cord from ileum to umbilicus), and various cystic remains of the vitelline duct in the abdominal cavity or body wall. Prolapse of the vitelline duct or of the ileum through the umbilicus carries with it a high mortality. It is frequently associated with intestinal obstruction and is a complication which requires early diagnosis and prompt, skillful surgery.
The allantoic remnant, the urachus, is normally a fibrous cord from the anterior abdominal wall to the apex of the bladder. A number of anomalies can result if the urachus does not undergo complete regression. The urachus may be patent its entire length so that urine leaks from the umbilicus. Patency of the distal portion of the urachus may result in a urachal sinus at the umbilicus, with persistent discharge. However, if the proximal portion is patent, a urachal diverticulum of the bladder will be present. In some patients both ends may be closed, resulting in a urachal cyst. Infections, abscesses, stone formation, and carcinoma occur as complications of these urachal remnants.
VAGINAL ANOMALIES
As the paramesonephric (müllerian) ducts grow caudad, they reach the urogenital sinus by approximately the ninth week (32 mm) and fuse with it to form an elevation known as the müllerian tubercle, with the openings of the paramesonephric ducts on either side of it. A ribbon of epithelium replaces the uterovaginal canal and is the precursor of the vagina. The vagina is formed between the 16th and 20th weeks by the development of lacunas; complete canalization later occurs to form the vaginal lumen (Fig. 5).
|
The principal congenital anomalies of the vagina include the following:
Longitudinal septum (32)
Transverse septum (15)
Vaginal agenesis (11)
Mesonephric remnants (15)
The numbers represent the relative frequencies of these conditions, as described by Dougherty and Spencer.37
Longitudinal septums produce the “double vagina” which is always startling and suggests the appearance of a double-barreled shotgun. Such septums may be complete or incomplete. They are frequently associated with uterus didelphys, but all combinations have been encountered, including the presence of a normal uterus and cervix. Sometimes one cervix may be blocked off, with retention of mucus or blood. Because the external genitalia usually appear normal, such septums are frequently not diagnosed unless painful coitus or labor dystocia occurs. Simple incision with appropriate ligation of bleeding points is usually all that is required. Fistula formation or vaginal stenosis may result from incautious or extensive surgery. Longitudinal septums occur approximately twice as frequently as transverse septums. Transverse septums vary from complete occlusion to a mild constriction. Frequently, there is a small opening allowing secretions or blood to drain. Unlike the bulging membrane associated with imperforate hymen, there is no external sign of the blockage. In nearly all patients, the internal organs are normal and pregnancy is not infrequent. Such transverse septums are believed to represent failure of complete canalization of the vaginal epithelial mass. The stenosis may be caused by a constricting fibromuscular bond. If menstruation and coitus occur without trouble, the condition may not be detected until pelvic examination reveals its presence.
The symptoms depend on the presence of adequate uterine drainage. If there is complete vaginal atresia, a mass may develop in the lower abdomen, due to hematometrocolpos (Fig. 6). Pelvic abscesses may develop in such entrapped blood. Vaginal septums may cause dystocia and make cesarean section the safest method of delivery. Treatment depends on the degree of stenosis and the rigidity of the constricting band. No treatment may be required, or two or three longitudinal incisions may suffice. Attempts at complete excision of an annular segment of vaginal wall may result in postoperative scarring or fistula formation.
|
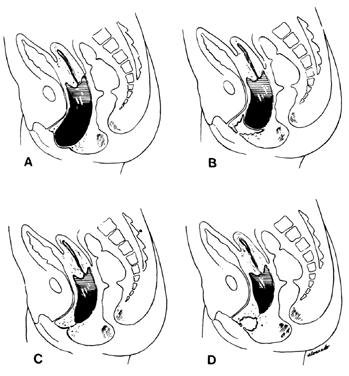
Agenesis of the vagina may only be detected on pelvic examination for amenorrhea. Absence of a vagina is always associated with absence of a hymen. Since agenesis of the vagina is usually misdiagnosed as imperforate hymen, this is of diagnostic importance. In nearly every case of vaginal agenesis there is an absent or very rudimentary uterus; ovarian agenesis may be present. Nearly all patients without a vagina have associated anomalies of the urinary tract, including renal agenesis, ectopic pelvic kidney, and ureteral anomalies. The mechanism of these vaginal anomalies is not entirely clear; they may result from failure of development of the epithelial vaginal mass or from failure of development of the urogenital sinus. The treatment of vaginal agenesis and the formation of an artificial vagina is described by Capraro.38 Operative results are usually excellent if molds are worn regularly or if there is regular coitus.
A congenital imperforate hymen is a result of failure of completion of the canalization or cavitation of the epithelial plug that fills the vagina. The condition is usually discovered at puberty, when the patient develops a lower abdominal mass, abdominal pain, and a bulging vaginal mass full of mucus or blood.39 This procedure should be performed using sterile technique in the operating room; after careful catheterization, a cruciate incision followed by excision of the four wedge-shaped tabs of hymen corrects the condition. Prophylactic antibiotics should be given postoperatively. A related condition is a microperforate hymen, in which no hymeneal opening can be found but in which a tiny opening allows passage of menstrual blood.
ABNORMALITIES OF EXTERNAL GENITALIA
The hindgut (Fig. 7 and Fig. 8) plays an exceedingly important part in the development of the infraumbilical abdominal wall, external genitalia, perineum, anus, and lower genitourinary tract. As early as 13 days after conception, the hindgut becomes active, and its importance and differentiation continue through the 12th week of gestation. A variety of malformations can occur, depending on the age of the embryo and whether agenesis, embryonic fissure, embryonic arrest, or duplication mechanisms are involved. The most damaging malformation is sympodia, or absence of the entire hind end of the body. Such damage occurs early and results in destruction of portions of the lower extremities, perineum, and cloaca. Absence of the perineum leaves no genital, urinary, or anal orifices; lower urinary and müllerian tract derivatives are absent. Damage occurring later may result in exstrophy of the cloaca and, later still, exstrophy of the bladder. Exstrophy of the bladder may be incomplete or complete. Complete exstrophy is the most common type and is associated with wide separation of the pelvic bones, complete epispadias, and protrusion of the entire posterior bladder wall. The failure is that of midline fusion rather than absence of abdominal wall musculature. Ureteral duplications, uterine and vaginal duplications, and imperforate anus are frequently associated with exstrophy of the bladder. This occurs in approximately 1 of 40,000 births. Unless adequately treated, ascending pyelonephritis results in early death. The results of surgery are usually good, with fair life expectancy. A number of women have delivered living infants after operative repair of the defect. Prolapse of the uterus usually follows vaginal delivery.
|
|
UROGENITAL SYSTEM
The early development of the paramesonephric and mesonephric duct systems has been described. As the paramesonephric (müllerian) duct develops, a solid wedge appears between the epithelial layer and the mesonephric ducts. A lumen develops and then the paramesonephric ducts become separate from the mesonephric ducts. The fused tips of the paramesonephric ducts grow caudad and reach the urogenital sinus. As the general elongation of the area develops, a solid epithelial cord is drawn out between the tubular portion of the paramesonephric duct and the müllerian tubercle on the wall of the urogenital sinus. This solid mass of cells constitutes the primordium of the solid vaginal cord. As development progresses, lacunas form in the center of this epithelial cord and the vagina becomes a hollow organ by the seventh month of fetal life. Although there is some controversy, it is generally accepted that the upper two thirds of the primitive vaginal plate is of paramesonephric origin, while the lower third is derived from the urogenital sinus. The hymen is formed as a partial septum at the level of the junction of the primitive urogenital sinus and the vagina. The caudal portion of the celomic cavity develops as the hindgut and becomes the cloaca, continuous with the yolk sac. The cloaca divides into a dorsally placed rectum and a ventrally placed urogenital sinus.
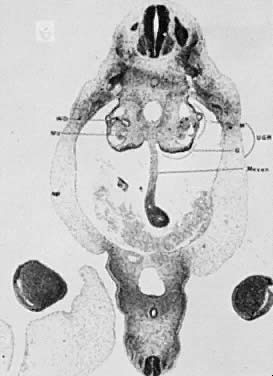
The mesonephric ducts connect with the urogenital sinus, but by the fifth week of fetal life the ureteric buds have appeared on either side of the müllerian tubercle and the metanephrogenic mass of cells (the future kidney) appears. At this time the primordial germ cells are migrating from the hindgut to form the primitive gonads along the urogenital ridge. The kidneys develop in the pelvis, beginning as a metanephrogenic mass of intermediate mesoderm around the ureteric bud. As the kidneys develop they migrate toward the head of the embryo, chiefly as a result of the growth of the embryo caudal to the kidneys. The kidneys are supplied by arteries and veins at successively higher levels as they move cephalad from the pelvis. The ureters are elongating at the same time and the kidneys rotate so that the hila come to face the midline. At the same time the mesonephric system is undergoing degeneration (Fig. 9, Fig. 10, Fig. 11, Fig. 12, and Fig. 13).
|
|

|
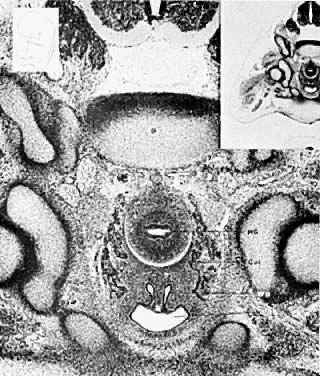
|
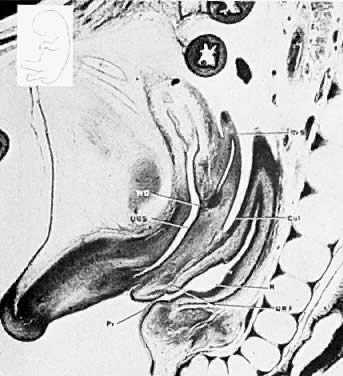
|
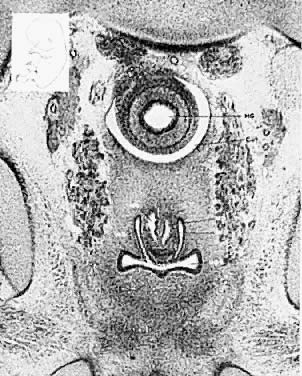
The relative frequencies of anomalies of the urogenital system, as described by Dougherty and Spencer37 are listed in Table 2.
Table 2. Anomalies of the genitourinary system
Type | No. |
Double ureter, branched, unilateral or bilateral | 15 |
Hydronephrosis and/or hydroureter | 10 |
Unilateral renal agenesis (all males) | 3 |
Pelvic or “unascended” kidney | 7 |
Horseshoe kidney | 3 |
Bilateral cystic kidneys (both males) | 2 |
Bilateral renal agenesis (both males) | 2 |
Malrotation of kidney (all females) | 3 |
Crossed renal ectopia | 1 |
Urachal cyst | 1 |
Bicornuate uterus (all types) | 1 |
Unicornuate uterus | 2 |
Clitoral hypertrophy | 2 |
Fused labia | 1 |
Absence of external genitalia (sirenomelia) | 2 |
Imperforate urogenital sinus | 2 |
Persistent urogenital sinus | 1 |
Urethra atresia (male) | 1 |
Renal hyperplasia | 1 |
Penile chordee | 2 |
From Dougherty CM, Spencer R: Female Sex Anomalies. Hagerstown, MD: Harper & Row, 1972, p. 52.
Because of the close association of mesonephric and paramesonephric ducts, urinary tract anomalies are frequently associated with malformations of the external genitalia and vagina. It is extremely important that thorough urologic studies be performed on all patients with müllerian anomalies. Ectopic kidneys and ureters, particularly the solitary pelvic kidney, present a real hazard for the gynecologic surgeon. Renal ectopia per se usually is not a problem, but if the kidney has failed to ascend to the normal level, complications can arise. The short ureter and short blood vessels preclude an attempt to "replace" the kidney at the normal level. The incidence of solitary kidneys is approximately 1 in 22,000 patients. Associated anomalies of the reproductive tract are present in nearly all women with a single kidney. Careful and thorough diagnosis is essential to avoid complications. There are at least two recorded cases in which solitary kidneys have been removed. Double ureters may come from a solitary kidney and do not prove that there are two kidneys.
CONGENITAL ADRENAL CORTICAL HYPERPLASIA
Congenital adrenal hyperplasia (CAH) is a fascinating developmental complication that is characterized by masculinized external genitalia and the diagnosis is made by demonstrating excess adrenal androgen production.40 The syndrome can appear in utero, or can be of late onset with symptoms of hyperandrogenism presenting in adolescence or early adulthood.
The most common cause of CAH is a block in the production of cortisol due to deficiency of the enzyme 21-hydroxylase (P450c21) (Fig. 14). 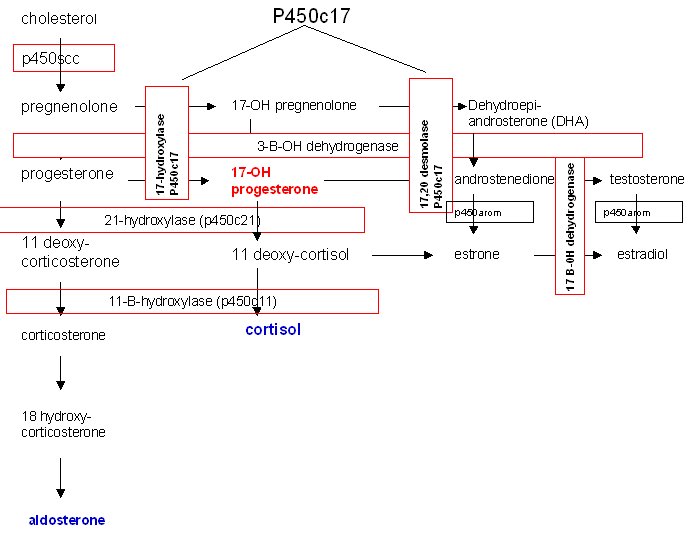
Fig. 14. Biosynthetic pathway of steroid production.
21-hydroxylase deficiency is responsible for 95% of all cases of CAH and has the potential, if undiagnosed, to be fatal. In the most severe form, production of both aldosterone and cortisol is halted and salt-wasting and shock accompany significant virilization.41 Two 21-hydroxylase genes have been identified, CYP21A and CYP21B. CYP21B is active in adrenal steroid production, while CYP21A is not involved and is a pseudogene. Various mutations in CYP21B are responsible for 21-hydroxylase deficiency and the severity of the disease is determined by the specific mutation within the gene.
The onset of production of excess androgens begins to occur between the 60-mm and 80-mm stages of fetal development. Due to the block in steroidogenesis at the level of 21-hydroxylase, the precursors progesterone and 17-hydroxyprogesterone are unable to be converted to aldosterone and cortisol, respectively. These precursors are then shunted to production of androstenedione and testosterone. Excess androgens in utero masculinize the external genitalia of a female infant. The degree of masculinization depends on the time of onset of the condition. Early in development, both vagina and urethra open into a persistent urogenital sinus. Clitoromegaly is present in all patients. Separate urethral and vaginal orifices may occur with only minimal labial fusion. A more advanced degree of labial fusion results in a persistent urogenital sinus containing urethra and cervix but with no urethra in the phallus. The greatest degree of masculinization presents a phallus with urethra and no visible vaginal opening since the vagina has a hidden connection with the urethra.42 The physician can easily miss the anomaly, thus endangering the infant. The gonads are, of course, undescended ovaries. The associated disturbance of metabolic processes requires prompt diagnosis and treatment, and demands immediate attention and neonatal treatment rather than surgery to correct the anatomic appearance.
Of cases of congenital adrenal hyperplasia 5–8% are due to 11ß-hydroxylase deficiency rather than 21-hydroxylase deficiency. In this enzyme defect, 11-deoxycortisol is not converted to cortisol and virilization will occur due to shunting of precursors into androgen biosynthesis, similar to that seen in 21-hydroxylase deficiency. In addition, 11-deoxycorticosterone may not be converted to corticosterone and to aldosterone, though the degree to which this block exists is variable.43
Prenatal diagnosis and in utero treatment of 21-hydroxylase deficiency and 11ß-hydroxylase deficiency are possible and can prevent virilization of a female fetus. Prenatal treatment beginning at 4–5 weeks' gestation can be empirically begun with dexamethasone and continued until diagnosis has been established or ruled out. Reduction in the degree of virilization or complete prevention of virilization has been reported in affected fetuses.44
3ß-hydroxysteroid dehydrogenase deficiency leads to decreased synthesis of glucocorticoids, mineralcorticoids, androgens, and estrogens. These neonates are severely ill and this enzyme deficiency is fatal. Females may be slightly virilized and males may be incompletely masculinized. Milder, late-onset cases that present with hyperandrogenemia have recently been described.45
Genetic females with excess androgen production are referred to as female pseudohermaphrodites while genetic males lacking in androgens are referred to as male pseudohermaphrodites. True hermaphrodites possess both testicular and ovarian tissue. Both types may be contained in one gonad, ovotestes, or one side may be an ovary and the other side a testis. The internal structures correlate to the adjacent gonad. External genitalia are ambiguous, but may be sufficiently developed to have a male gender assignment. However, 75% develop gynecomastia and over 50% menstruate at puberty, perhaps making female sex assignment easier.
ECTOPIC URETERS
The ureteric buds arise from the posterior portions of the mesonephric ducts and shortly meet the mass of nephrogenic mesoderm. The ureter elongates, forming primary calyces. A bifurcation of the growing ureter at this early stage produces two ureters and two renal pelves. Later divisions (in the fifth week) may only result in bifid pelves. Accessory ureteric buds may result in ureteral triplications. Duplicate ureters occur in approximately 1% of humans. Ectopic ureters may implant in a variety of places and commonly cause incontinence in females. Such ectopic ureteral openings may occur on the skin of the vulva, in the vagina, and the urethra, and, rarely, into the uterus or cervix. The majority of ectopic ureteral orifices are associated with a ureter that drains the upper portion of a double kidney and has complete duplication of the ureter. If the distal portion of a mesonephric duct is present as Gartner's duct, an ectopic ureter usually will connect with it. Incontinence and infection are the almost invariable complications of ectopic ureteral orifices outside the bladder in a female. In children the usual misdiagnosis is enuresis. Careful urologic study involving excretory and retrograde pyelograms and intravenous indigo carmine and intravesical methylene blue studies are often necessary to fully evaluate the urologic problem. It should be remembered that multiple ureteral orifices may be present and that finding a single ectopic ureter in no way excludes the presence of others. If feasible and if the kidney is healthy, the operation of choice is transplantation of the ectopic ureter into the bladder.
Persistence of the urogenital sinus with outlet obstruction is relatively uncommon. During the fifth week the urorectal folds grow into the cloaca, dividing it into two portions. The dorsal portion becomes the rectum and the ventral portion becomes the urogenital sinus. The cloacal membrane ordinarily ruptures during the seventh week, allowing the urogenital sinus to drain externally and the anus to become patent. Persistence of the cloacal membrane may result in urethral obstruction or imperforate anus. Tank et al46 reported three such patients with complete anuria and a lower abdominal mass. Hydronephrosis and hydroureter were present. A marked compression of the vagina was present in all patients. Two of the patients also had imperforate anus. This series included five cases of stenosis of the urogenital sinus, a much less serious condition. Persistent urogenital sinus membrane constitutes a real surgical emergency because of the urologic tract damage and mounting azotemia. Careful and methodical examination of the newborn should result in early diagnosis and prompt surgery. In all cases, associated congenital anomalies can be expected. Adrenogenital syndrome, imperforate anus, mixed gonadal dysgenesis, and renal agenesis are among the reported complications.
The genitourinary anomalies may be evaluated by careful examination of the lower abdominal wall for failures of closure, exstrophies of bladder and cloaca, and umbilical anomalies such as cysts, leakage of urine, and patent urachus. A single umbilical artery is frequently associated with genitourinary anomalies and chromosomal trisomies. In a male neonate, an erect voiding penis and the palpation of descended testicles is a reassuring sign that the urogenital system is intact. Imperforate anus can be ruled out with passage of a rectal thermometer or a No. 8 F catheter. Passage of meconium is an important sign, though intestinal atresia may be present even if meconium is encountered, though this is less likely. In females, careful inspection of the vaginal introitus normally reveals whitish cervical secretion; the easy passage of a No. 8 F catheter excludes imperforate hymen although it does not rule out longitudinal or transverse vaginal septums. The complete differential diagnosis in infants with ambiguous genitalia is more extensive and involves obtaining a karyotype, 17-ketosteroid determinations and, possibly, an MRI or laparotomy. In cases of ambiguous genitalia, the diagnosis of congenital adrenal hyperplasia should be suspected as this can potentially be a life-threatening disease and, when indicated, treatment must be promply initiated.
REFERENCES
Gray SW, Skandalakis JE: Embryology for Surgeons. Philadelphia: Saunders, 1972 |
|
Hughes EC: ObstetricGynecologic Terminology. Philadelphia: Davis, 1972 |
|
Moore KL: The Developing Human. Philadelphia: Saunders, 1973 |
|
Patten BM: Human Embryology. New York: McGraw Hill, 1968 |
|
Spitzer RF, Wherrett D, Chitayat D et al: Maternal luteoma of pregnancy presenting with virilization of the female infant. J Obstet Gynaecol Can 29: 835–40, 2007 |
|
Millen JW: Timing of congenital malformations: With a time table of human development. Dev Med Child Neurol 5: 343, 1963 |
|
Nugent FB: Environmental causes of fetal malformations. Pa Med 74: 63, 1971 |
|
Watanabe N, Kurita M: The masculinization of the fetus during pregnancy due to inhalation of diesel exhaust. Environ Health Perspect 109: 111–9, 2001 |
|
Tijo JH, Levan A: The chromosome number of man. Hereditas 42: l, 1956 |
|
Porter IH: Clinical side of cytogenetics. J Reprod Med 17: 3, 1976 |
|
James RS, Coppin B, Dalton P et al: A study of females with deletions of the short arm of the X chromosome. Hum Genet 102: 507–16, 1998 |
|
Tsutsumi O, Iida T, Nakahori Y et al: Analysis of the testis-determining gene SRY in patients with XY gonadaldysgenesis. Horm Res 46 Suppl 1: 6–10, 1996 |
|
Vogt P, Chandley AC, Hargreave TB et al: Microdeletions in interval 6 of the Y chromosome of males with idiopathic sterility point to disruption of AZF, a human spermatogenesis gene. Hum Genet 89: 491–6, 1992 |
|
Munne S, Lee A, Rosenwaks Z et al: Diagnosis of major chromosome aneuploidies in human preimplantation embryos. Hum Reprod 8: 2185–91, 1993 |
|
Munne S, Chen S, Fischer J et al: Preimplantation genetic diagnosis reduces pregnancy loss in women aged 35 years and older with a history of recurrent miscarriages. Fertil Steril 84: 331–5, 2005 |
|
Hassold T, Hunt P: To err (meiotically) is human: the genesis of human aneuploidy. Nat Rev Genet 2: 280–91, 2001 |
|
Judis L, Chan ER, Schwartz S et al: Meiosis I arrest and azoospermia in an infertile male explained by failure of formation of a component of the synaptonemal complex. Fertil Steril 81: 205–9, 2004 |
|
Austin CR: The egg and fertilization. Science Journal (Lond) 6: 37, 1970 |
|
Cosby WM, Hill EC: Embryology of the Müllerian duct system. Obstet Gynecol 20: 507, 1962 |
|
Raga F, Bauset C, Remohi J et al: Reproductive impact of congenital Mullerian anomalies. Hum Reprod 12: 2277–81, 1997 |
|
Blair RG: Pregnancy associated with congenital malformations of the reproductive tract. J Obstet Gynaecol Br Emp 67: 36, 1960 |
|
Semmens J P: Congenital anomalies of female genital tract. Obstet Gynecol 19: 328, 1962 |
|
O'Leary JL, O'Leary JA: Rudimentary horn pregnancy. Obstet Gynecol 22: 371, 1963 |
|
Pal K, Majumdar S, Mukhopadhyay S: Rupture of rudimentary uterine horn pregnancy at 37 weeks gestation with fetal survival. Arch Gynecol Obstet 274: 325–6, 2006 |
|
Zaidi J, Carr J: Rupture of pregnant rudimentary uterine horn with fetal salvage. Acta Obstet Gynecol Scand 73: 359–60, 1994 |
|
Pal K, Majumdar S, Mukhopadhyay S: Rupture of rudimentary uterine horn pregnancy at 37 weeks gestation with fetal survival. Arch Gynecol Obstet 274: 325–6, 2006 |
|
Patra S, Puri M, Trivedi SS et al: Unruptured term pregnancy with a live fetus with placenta percreta in a non-communicating rudimentary horn. Congenit Anom (Kyoto) 47: 156–7, 2007 |
|
Chou MM, Ho ES, Lin SK et al: Term pregnancy in a noncommunicating rudimentary horn of an unicornuate uterus: a case report. Zhonghua Yi Xue Za Zhi (Taipei) 62: 383–7, 1999 |
|
Jayasinghe Y, Rane A, Stalewski H et al: The presentation and early diagnosis of the rudimentary uterine horn. Obstet Gynecol 105: 1456–67, 2005 |
|
Sonmezer M, Taskin S, Atabekoglu C et al: Laparoscopic management of rudimentary uterine horn pregnancy: case report andliterature review. JSLS 10: 396–9, 2006 |
|
Fedele L, Bianchi S, Zanconato G et al: Laparoscopic removal of the cavitated noncommunicating rudimentary uterine horn:surgical aspects in 10 cases. Fertil Steril 83: 432–6, 2005 |
|
Oral B, Guney M, Ozsoy M et al: Placenta accreta associated with a ruptured pregnant rudimentary uterine horn.Case report and review of the literature. Arch Gynecol Obstet 265: 100–2, 2001 |
|
Henriet E, Roman H, Zanati J et al: Pregnant noncommunicating rudimentary uterine horn with placenta percreta. JSLS 12: 101–3, 2008 |
|
Oral B, Guney M, Ozsoy M et al: Placenta accreta associated with a ruptured pregnant rudimentary uterine horn. Case report and review of the literature. Arch Gynecol Obstet 265: 100–2, 2001 |
|
Grover S: Torsion causing interruption of the ampullary portion of the fallopian tube. Fertil Steril 88: 968.e13–4, 2007 |
|
Pena-Alonso R, Nieto K, Alvarez R et al: Distribution of Y-chromosome-bearing cells in gonadoblastoma and dysgenetictestis in 45,X/46,XY infants. Mod Pathol 18: 439–45, 2005 |
|
Dougherty CM, Spencer R: Female Sex Anomalies. Hagerstown, MD: Harper & Row, 1972 |
|
Capraro V J: Surgical correction of genital anomalies. In Sciarra JJ (ed): Gynecology and Obstetrics. Hagcrstown, MD: Harper & Row, 1977, vol 1, chap 53 |
|
Huffman JW: Gynecology of Childhood and Adolescence. Philadelphia: Saunders, 1968 |
|
Jones HW, Scott WW: Genital Anomalies and Related Endocrine Disorders. Baltimore: Williams & Wilkins, 1958 |
|
White PC, New MI, Dupont B: Congenital adrenal hyperplasia. (1). N Engl J Med 316: 1519–24, 1987 |
|
Van Leeuwen G, Glenn L: Screening for hidden congenital anomalies. Pediatrics 41: 147, 1968 |
|
White PC, Curnow KM, Pascoe L: Disorders of steroid 11 beta-hydroxylase isozymes. Endocr Rev 15: 421–38, 1994 |
|
Cerame BI, Newfield RS, Pascoe L et al: Prenatal diagnosis and treatment of 11beta-hydroxylase deficiency congenital adrenal hyperplasia resulting in normal female genitalia. J Clin Endocrinol Metab 84: 3129–34, 1999 |
|
Pang S: Congenital adrenal hyperplasia owing to 3 beta-hydroxysteroid dehydrogenase deficiency. Endocrinol Metab Clin North Am 30: 81–99, vi–vii, 2001 |
|
Tank ES, Konnak JW, Lapides J: Urogenital sinus outlet obstruction. J Urol 104: 769, 1970 |