
Endocrine Diseases in Pregnancy
Authors
INTRODUCTION
Pregnancy is a unique clinical scenario in which several endocrine disorders may be more frequent and/or have specific considerations for diagnosis and treatment. In this review, anterior pituitary insufficiency, adrenal, parathyroid, and thyroid disorders of pregnancy are discussed.
ANTERIOR PITUITARY INSUFFICIENCY
Anterior pituitary insufficiency is an uncommon disease. The etiology includes destruction of the anterior pituitary gland by tumors, infarction (postpartum necrosis or Sheehan's syndrome), idiopathic disease (Simmonds' disease), surgery, and radiotherapy to the pituitary gland. There have also been reports of pituitary necrosis in patients with elevated intracerebral pressure.1 Disease of the hypothalamus affecting the secretion of releasing hormones may produce a similar clinical picture; some cases of Sheehan's syndrome and idiopathic hypopituitarism are due to hypothalamic diseases.2 Finally, congenital hypopituitarism is a rare diagnosis among newborn infants.3
Sheehan’s syndrome
The most common cause of panhypopituitarism in women of childbearing age is postpartum necrosis, or Sheehan's syndrome.4 The pathogenesis is not clear, although Sheehan in his original description did associate it with severe postpartum hemorrhage.5 Although the classic clinical etiology of Sheehan's syndrome in about 90% of patients is severe bleeding of the anterior pituitary during delivery or immediately postpartum, no catastrophic event can be detected in more than 10% of patients.
Lack of lactation after delivery, amenorrhea, loss of pubic and axillary hair or failure of pubic hair to grow back, anorexia and nausea, lethargy and weakness, and weight loss are typical presenting signs and symptoms. On physical examination, the findings depend on the severity and duration of the disease. Commonly, the skin has a waxy character with fine wrinkles about the eyes and mouth. There is some periorbital edema, and a decrease in pigmentation is often seen. Axillary and pubic hair becomes increasingly sparse. Atrophy of the breast tissue may be present. Even in those patients losing weight, cachexia is not a feature of the disease. Hypotension may be present, and normocytic anemia is common. However, this full constellation of symptoms does not occur in every patient, and it is not unusual for the full-blown picture to take 10–20 years to develop. Occasionally, the diagnosis is made when the patient develops acute adrenal insufficiency secondary to a stressful situation (e.g. infection, trauma, surgery).
It was recognized by Sheehan that not all patients with pituitary apoplexy develop panhypopituitarism, and partial pituitary insufficiency is not uncommon. In one retrospective case series of 44 patients in France, only 88% had hypopituitarism, with adrenocorticotropic hormone (ACTH) deficiency most common (70%).6 A few patients with partial hypopituitarism may present with the classic syndrome of acute panhypopituitarism with deficiency of all pituitary hormones. However, after treatment with corticosteroids alone, there is a spontaneous normalization in the menstrual cycle, with a return of thyroid test results to normal limits.
Successful pregnancies following a diagnosis of Sheehan's syndrome have been reported.7, 8, 9 In a few patients, the diagnosis of partial hypopituitarism may occur upon the presentation of a pregnancy. Although several patients conceive after treatment with gonadotropin, others conceive spontaneously, an indication of partial pituitary failure. Placental function is not altered in patients with pituitary insufficiency.
Pituitary adenoma
During normal pregnancy, the pituitary enlarges by approximately one-third of its size.10 Pituitary insufficiency in women of childbearing age may result in the setting of a pituitary tumor, usually in association with increased production of prolactin. The most common symptom is secondary amenorrhea with galactorrhea, although cases of primary amenorrhea have been reported. When there is local expansion of the tumor, patients may have neurologic symptoms, such as headache or bilateral temporal hemianopia. In such cases, other pituitary hormones may become affected with growth hormone, ACTH, and thyrotropin-stimulating hormone (TSH) deficiencies.
The diagnosis is confirmed by the use of appropriate tests to investigate each of the pituitary hormones. Baseline or random determination of serum pituitary hormone concentrations is of no value in the diagnosis of the disease; dynamic tests to evaluate pituitary reserve must be used. The most practical tests are presented in Table 1. However, their use in pregnancy is limited because of the blunted response of many of these tests.
Table 1. Tests of anterior pituitary hormone reserve
Hormone | Test | Normal Response | Response in Pregnancy |
GH
| L-Dopa, 500 mg, GH levels at 0, 1, 2 hour | ↑ by 10 ng/dl | Blunted |
Insulin hypoglycemia 0.1 U regular IV/kg, then draw GH at 0, 20, 60, 90 min | ↑ by 10 ng/dl | Blunted | |
ACTH
| Insulin hypoglycemia (see above), then draw cortisol at 0, 20, 60, 90 min | ↑ by 10 μg/dl | Blunted |
Metyrapone 750 mg every 4 hour × 6 | ↑ Urinary 17-KGS | Blunted | |
TSH
| Free thyroxine index |
| Normal |
Serum TSH |
| Normal | |
Prolactin |
Can no longer be tested, given the inavailability of TRH |
|
|
LH-FSH |
Assess by regularity/presence of menses |
|
|
| Measurement of estradiol and/or administration of progesterone 10 mg daily for 10 days for withdrawal bleeding can also be used |
GH, growth hormone; ACTH, adrenocorticotropic hormone; TSH, thyrotropin-stimulating hormone; TRH, thyrotropin-releasing hormone; LH, luteinizing hormone; FSH, follicle-stimulating hormone; LHRH, luteinizing hormone-releasing hormone; 17-KGS,17-ketogenic steroids; substance S, 11-desoxycortisol
When anterior pituitary insufficiency develops in pregnancy, the clinical manifestations may be local signs, such as headaches and visual disturbances, which are the consequence of an acute enlargement of, or bleeding into, the pituitary gland.11 The initial manifestations also could be related to endocrine deficiency, mainly hypoglycemia, nausea, vomiting, and hypotension secondary to ACTH deficiency.
Isolated ACTH deficiency is rare and has been infrequently described.12, 13 Acute enlargement of the pituitary gland is characterized by severe, deep, midline headaches (lasting for 2–3 days) and visual field disturbances. Severe hypoglycemia with convulsions and coma, unresponsive to large doses of glucose, but rapidly reversible after the administration of hydrocortisone, can be seen.
Partial or total hypopituitarism developing in patients with diabetes mellitus has been reported.14 In a review of 31 cases (19 women), the episode was associated with pregnancy in 11 (during the postpartum period in seven and during the antepartum period in four, with three maternal deaths).15 The mean age of the patients in this case series was 27 years, and the mean duration of their diabetes mellitus was 6 years, which makes vascular complication an unlikely cause of pituitary insufficiency. Furthermore, no specific vascular changes were found in the examined pituitary glands. Characteristically, the patients developed severe headaches that lasted for a few days with or without visual field disturbances, and a decrease in insulin requirement was observed. There was a high proportion of women with fetal loss. Although the mechanism supporting the increased risk of hypopituitarism among individuals with diabetes mellitus remains unclear, an association between pituitary antibodies and type 1 diabetes mellitus has been described.16
Lymphocytic hypophysitis
Lymphocytic hypophysitis can be another cause of pituitary dysfunction,17 and in pregnant women, usually presents close to delivery or in the immediate postpartum period.18 Sheehan described lymphocytic infiltration of the pituitary gland in some women with postpartum pituitary insufficiency,19 and it is possible that many of the cases mentioned above were due to lymphocytic hypophysitis.
The clinical presentation may be characterized by headaches and visual disturbances related to pressure from the expanding lesion mimicking a pituitary tumor;20, 21 spontaneous regression of the lesion was seen in several cases.22 diabetes insipidus and galactorrhea. Report of a case and review of the literature) The differential diagnosis between pituitary tumor and hypophysitis can be made only by histologic examination.18 Conversely, the patient may present with signs and symptoms of hypopituitarism, such as protracted hypoglycemia responding to glucocorticoid therapy and hypotension. It can also present in the postpartum period as pituitary insufficiency, similar to Sheehan's syndrome without the history of profound bleeding.22, 23, 24, 25
Involvement of other endocrine glands has been recognized, consistent with the concept of an autoimmune disease,26 in addition to antibodies against pituitary cells.27 It is possible that these cases are typical of the autoimmune polyendocrine deficiency syndrome that may be exacerbated during pregnancy or in the immediate postpartum period.
Treatment
Patients with partial or total hypopituitarism who become pregnant spontaneously or after treatment with gonadotropins may carry a normal pregnancy with no increase in the dose of corticosteroid replacement therapy. The usual amount of hydrocortisone in patients with pituitary insufficiency is 20–30 mg/day (two-thirds of the total amount in the morning and one-third in the evening). In some instances, the amount of hydrocortisone can be decreased by one-third of the total dose because the effect of hydrocortisone is potentiated during pregnancy by estrogen.28 However, this potentiation does not occur when synthetic corticosteroids (i.e. prednisone, dexamethasone) are used. The equivalent amounts of prednisone and dexamethasone, respectively, are 5.0–7.5 mg daily and 0.5–0.75 mg daily. Because these patients have ACTH deficiency, aldosterone secretion is normal and there is no need for mineralocorticoid replacement therapy. If thyroid deficiency is present, the amount of levothyroxine needed for replacement is usually 0.1–0.2 µg daily.
Successful pregnancy in cases of isolated growth hormone deficiency has been reported.29, 30 In these patients, lactation was unimpaired and placental function studies and intrauterine growth were normal.
PROLACTINOMAS
The most common pituitary tumor diagnosed in women of childbearing age is a prolactinoma.31 It can be accompanied by amenorrhea, oligohypomenorrhea, and anovulation, and with or without galactorrhea. Hyperprolactinemia decreases gonadotropin-releasing hormone (GnRH) secretion, accounting for the infertility observed in these patients. Pituitary tumors are divided, according to size, into microadenomas (less than 10 mm in diameter) and macroadenomas (greater than 10 mm in diameter); the latter are further classified according to suprasellar extension and invasion of adjacent structures. Serum prolactin concentrations correlate fairly well with the size of the tumor. Hyperprolactinemia in the absence of a pituitary adenoma (idiopathic hyperprolactinemia) is a common finding.
Complications
Serum prolactin levels in women with prepregnancy hyperprolactinemia, with a few exceptions, remained unchanged during pregnancy. It was shown that prolactin levels did not change significantly in most women with baseline prolactin levels of over 60 pg/dL.32 However, in those patients with prolactin levels of less than 60 pg/dL, the mean level doubled at the end of pregnancy and returned to pretreatment levels at the end of lactation. Therefore, serum prolactin determination during pregnancy is not a predictor of tumor growth and is of no value in monitoring tumor growth.
The incidence of complications during pregnancy in patients with pituitary tumors varies according to tumor size. Due to the stimulatory effect of estrogen on lactotrophs, the size of the tumor increases in 2.7% of microprolactinomas and 22.9% of macroprolactinomas during pregnancy.31 In one study of 56 pregnant women with microprolactinomas, one developed headaches and five showed mild tumor growth.33 In studies of pregnant women with macroprolactinomas, the proportion of women developing neurologic symptoms and visual disturbances is significantly decreased upon treatment.31
Complications can occur at any stage of pregnancy. In patients with microadenomas, visual field examinations are indicated only if there are signs and symptoms of tumor enlargement, in which case an MRI is also indicated. If there is any objective evidence of tumor enlargement, bromocriptine is resumed and continued throughout pregnancy at up to 20 mg/day. If after a few days there is no improvement, dexamethasone 4 mg every 6 hours can be added. Surgery is indicated in those complicated cases not responding to the above therapies, but the recurrence rate is high among those with invasive prolactinomas even after surgery.34
Breastfeeding is not contraindicated in mothers with a diagnosis of prolactinoma. There is no difference in the remission rates of women with prolactinomas managed with dopamine-receptor agonists who breastfeed following delivery versus those who do not.35 It is advisable in patients with microadenomas to measure prolactin levels a few months after delivery and to reinstate bromocriptine therapy in the presence of persistent hyperprolactinemia. A pituitary MRI should be repeated in cases of macroprolactinoma soon after delivery because of the potential for tumor size increase.
Treatment
Once the diagnosis of prolactinoma is made, several types of therapy are available. The choice of therapy depends on tumor size, radiologic classification, local symptoms, and the patient's age and desire for pregnancy or current pregnancy.36
Medical therapy with dopamine-receptor agonists has been very effective in producing ovulation among hyperprolactinemic women37 and restores ovulation in approximately 90% of cases.31 Bromocriptine has historically been the preferred option, and no significant adverse effects have been observed in over 6000 pregnancies managed with bromocriptine.38 Most patients respond to doses of 2.5–5 mg/day, although occasionally a dose of 7.5 mg/day or more is needed. Bromocriptine is effective not only in normalizing prolactin levels but also in reducing the size of the tumor.31 It is advisable to use mechanical contraception during the first few months of bromocriptine therapy until the rhythm of the menstrual period is established. In those patients who have side effects such as nausea and vomiting, the oral bromocriptine tablet can be administered vaginally.39
Cabergoline is another dopamine-receptor agonist which can be considered.40 Although only 800 pregnancies have been reported with its use, there similarly does not appear to be any increased risks of preterm delivery or congenital malformations associated with this medication.38 In one 10-year observational study of 143 women, carbergoline therapy during pregnancy resulted in the ability of nearly 98% of the women to breastfeed following delivery.41 Once conception takes place, the dopamine-receptor agonist should be discontinued and the patient followed closely. For women in whom the macroprolactinoma is likely to increase, or in whom pressure symptoms occur, therapy during pregnancy should be continued.42
Radiation therapy as the initial and only therapy is seldom indicated, as medical therapy is usually very effective. The duration required for radiation therapy to normalize serum prolactin levels is lengthy and may produce hypopituitarism as a last sequela. Radiation therapy is indicated in those with prolactinomas refractory to conventional therapy.43
Surgical treatment, mainly transsphenoidal adenectomy, has been effective in restoring ovulation in patients with small tumors.32 The cure rate (i.e. sustained normalization of serum prolactin concentrations) is about approximately 70% at both 5 and 10 years of follow up; the associated proportion of successful pregnancy was similar.44 The best results are obtained in patients with microadenomas with low initial serum prolactin levels and lack of abnormal postoperative residual tissue.45
A recommended treatment approach in patients who wish to conceive is summarized in Table 2. It is suggested that treatment with bromocriptine be continued for at least 12 months before conception because it seems to reduce the risk of tumor enlargement during pregnancy.46
Table 2. Management of women with pre-conception hyperprolactinemia
| Tumor | Management | Pregnancy Follow-up |
| No tumor | Bromocriptine | Visual field (?) |
| Microadenoma | Bromocriptine* or surgery | Visual field every trimester |
| Macroadenoma | (A) Surgery + bromocriptine | Visual field monthly |
| (B) Radiotherapy + bromocriptine | Visual field monthly |
*Therapy for 1 year before conception
ACROMEGALY
Acromegaly is a chronic disease caused by hypersecretion of growth hormone by the adenohypophysis of the pituitary gland. It is almost always associated with a benign pituitary tumor and is characterized by slow and progressive enlargement of the acral parts. Facial changes are typical, but they usually develop so gradually that neither the family nor the patient recognizes the changes. As in other endocrine disorders, comparison of the patient's photographs taken over many years may be the only clue to the progression of the disease. Symptoms may be due to local expansion of the tumor (i.e. headaches and visual field disturbances), or they may be due to the somatic effects of chronic excess growth hormone, such as hyperhidrosis, weight gain, arthralgias, and acroparesthesia (carpal tunnel syndrome). Most women with acromegaly have been reported to suffer from oligohypomenorrhea or amenorrhea. In addition to the bony deformities, organomegaly (particularly enlargement of the heart, thyroid, and liver) is not uncommon on physical examination. The skin appears coarse and leathery. Galactorrhea with hyperprolactinemia is a common finding.
Diagnosis
The diagnosis is confirmed by an elevation in plasma insulin-like growth factor 1 (IGF-1) levels and a lack of suppression of growth hormone following the administration of a glucose load.47 However, IGF-1 levels may not be reliable during pregnancy, as they can be physiologically increased48 or decreased during pregnancy.49
Thus, suspected cases of acromegaly among pregnant women should be confirmed with a growth hormone suppression test, which requires determination of plasma growth hormone levels before and 1 and 2 hours after the administration of a solution of 100 g glucose orally. A normal response is characterized by growth hormone levels lower than 1 µg/L after glucose administration. Patients with acromegaly typically have elevated baseline IGF-1 levels and respond to the glucose load with no growth hormone suppression of growth hormone concentration or even occasionally with a paradoxical increase.
In patients with acromegaly, there are increased risks of several associated cormorbidities, including hypertension, diabetes mellitus, cardiovascular disease, osteoarthritis, and sleep apnea, which should be evaluated for upon the confirmed diagnosis of acromegaly.47
Treatment
Treatment is mandatory in patients with the disease because the long-term prognosis is poor; untreated individuals have an almost 3-fold increased mortality rate.48 Conventional pituitary irradiation, transsphenoidal hypophysectomy,50 and drug therapy with octreotide (or other somatostatin receptor analogues) or the growth receptor antagonist, pegvisomant51 are used most often and can improve disease survival.52
Acromegaly during pregnancy
There are limited data of successful pregnancies in women with acromegaly. In 1954, Abelove and colleagues reported two normal pregnancies in an acromegalic woman and reviewed 33 reported cases from the world literature.53 Since that time, several other cases have been published, including a recent report of ten pregnancies among eight acromegalic women in Brazil, in which plasma IGF-1 levels were not significantly changed during gestation.54 In most instances, the infants have been reported as being normal. However, in a case described by Fisch et al.,55 the infant was born with acromegalic features. In this infant, growth was above average during the neonatal period, but a normal growth pattern subsequently returned, although no serum laboratory measurements were obtained. The lack of acromegalic features in most cases is in accordance with the report by King and colleagues demonstrating no placental transfer of growth hormone from mother to fetus.56
Historically, bromocriptine has been used as a successful treatment to induce pregnancy in patients with acromegaly.57, 58 In each of these cases, pregnancy occurred in spite of persistent elevated serum growth hormone levels.
The current guidelines for management of acromegalic women during pregnancy have been summarized in the 2014 Endocrine Society guidelines for acromegaly.47 In general, discontinuation of long-acting medical therapy (somatostatin receptor analogues or pegvisomant) is recommended approximately 2 months prior to attempting to conceive; therapy can be replaced with short-acting octreotide instead during the pre-conception period. During gestation, medical therapy should only be administered only for tumor and headache control, and plasma growth hormone and IGF-1 levels should not be monitored.
DIABETES INSIPIDUS
Diabetes insipidus is an uncommon disease characterized by polyuria and polydipsia due to a deficiency of antidiuretic hormone (central or neurogenic diabetes insipidus) or the peripheral resistance to the antidiuretic hormone at the renal tubules (nephrogenic diabetes insipidus). Central diabetes insipidus may be a result of a lesion at the level of the hypothalamus or pituitary gland. It may arise following hypophysectomy, invasion of the neurohypophysis by tumors, malignant metastasis (i.e. breast cancer), trauma, granulomas, or infection. In 50% of cases, however, it is considered idiopathic, with some causes probably on an autoimmune basis. Nephrogenic diabetes insipidus is a hereditary disorder affecting males; therefore, symptomatic women carriers are extremely rare. Several cases of transient nephrogenic diabetes insipidus during pregnancy and/or postpartum have been reported. A third type of diabetes insipidus, called psychogenic, which is rarely reported in pregnancy,59 is differentiated from the other two in most cases by the results of the water deprivation test.
Diagnosis
The clinical presentation of idiopathic central diabetes insipidus is generally acute, with polyuria and polydipsia developing in a few days. Patients usually remember the day their symptoms began, they prefer cold water to drink, and their urinary output varies from 4 to 15 L per 24 hours.
The diagnosis of diabetes insipidus is confirmed by the results of a water deprivation test (Table 3). In patients with central diabetes insipidus, there is a decrease in urinary output and an increase in urinary osmolarity. No changes are seen in the nephrogenic subtype of diabetes insipidus. Patients with primary polydipsia or psychogenic diabetes insipidus respond to the administration of desmopressin with some increase in urine osmolarity, but the increase is not as high as it is in patients with central diabetes insipidus. In emergency situations with acute onset of symptoms, such as cases of acute fatty liver of pregnancy or preeclampsia, treatment cannot be delayed, and diagnostic tests to assess the etiology of diabetes insipidus must be postponed.
Table 3. Water deprivation test*
| 1. Nothing by mouth following the initiation of water deprivation (overnight fluid restriction should be advised against, given the potential for severe dehydration) |
| 2. Baseline serum sodium and osmolality measurements |
| 3. Baseline urine osmolality and specific gravity measurements |
| 4. Measurements of urine tests and body weight hourly |
| 5. Measurement of serum tests every 2 hours |
| 6.Water deprivation is continued until:
a. Two subsequent urine osmolalities vary by less than 10% or when 3–5% body weight is lost; b. Urine osmolality reaches a normal concentration (i.e. >600 mosmol/kg); c. Plasma osmolality is >295 mosmol/kg; or d. Plasma sodium >145 mEq/L |
| 7. Administration of desmopressin (10 µg intranasally or 4 µg subcutaneously or intravenously) |
| 8. Measurement of urine osmolality one hour after the injection |
*The test must be carried out under careful supervision in order to avoid either severe dehydration in patients with significant ADH deficiency, or water intoxication in patients with primary polydipsia due to continuous water ingestion after the administration of desmopressin.
In patients with newly diagnosed central diabetes insipidus, a complete evaluation to define the cause of the disease is imperative. Radiologic examination (i.e. pituitary MRI) of the hypothalamic–pituitary area and hormonal studies for anterior pituitary function assessment should be performed.
Treatment
Desmopressin (dDAVP), the synthetic analogue of vasopressin, is the standard treatment in patients with diabetes insipidus. Its antidiuretic effect last from 8 to 24 hours and is available in three forms: nasal insufflation, parenteral, and tablet form. The usual dose of the nasal spray is 10–25 μg once or twice daily as needed to control polyuria. In the presence of rhinitis or other conditions that would interfere with absorption, or in the postoperative period, the parenteral form is used. Two to 4 μg are injected subcutaneously or intravenously and repeated every 12–24 hours, according to urinary osmolality and output. Plasma electrolytes and water intake and urinary output should be followed closely to avoid water intoxication.
The experience of dDAVP use in pregnancy is extensive.60, 61 The fetus suffers no side effects, and it is safe to use in lactating mothers. In the rare case of nephrogenic diabetes insipidus, patients will be typically resistant to dDAVP therapy and thiazide diuretics are the only drug that can be used.62
Diabetes insipidus during pregnancy
Shortly after conception, plasma osmolality decreases by about 5–10 mOsm/kg and remains low throughout pregnancy.63 In accordance with this, basal levels of serum arginine vasopressin (antidiuretic hormone) also decrease during gestation,64 particular during the first and second trimesters.59 The ability to concentrate the urine remains within normal limits.65
Diabetes insipidus during the pre-pregnancy and pregnancy settings can occur as various clinical scenarios, as summarized in Table 4.66 The prevalence of diabetes in pregnancy is approximately 1:30,000.62
Table 4. Forms of diabetes insipidus in the pre-pregnant and pregnant woman
| Pregestational diabetes insipidus |
| Diabetes insipidus presenting for the first time in pregnancy and persisting thereafter |
| Transient diabetes insipidus (occurring during gestation or in the immediate postpartum period) associated with preeclampsia, liver disease, or the HELLP (hemolysis, elevated liver enzymes, and low platelet count) syndrome |
| Transient diabetes insipidus, due to the increase in the placental enzyme vasopressinase (tends to be recurrent in subsequent pregnancies) |
| Postpartum diabetes insipidus in patients with acute pituitary insufficiency, such as in Sheehan's syndrome or hypophysitis |
| An unusual transient form of diabetes insipidus resistant to desmopressin administration |
Transient vasopressin-resistant diabetes insipidus typically occurs during the last trimester of pregnancy. It is associated with preeclampsia, acute fatty liver of pregnancy, or HELLP syndrome. The lack of response to vasopressin is due to the significant increase in the plasma clearance of antidiuretic hormone (ADH) caused by the activity of the enzyme vasopressinase. This also explains the good response to dDAVP, which is not metabolized by vasopressinase.
In the transient recurrent form of diabetes insipidus, patients with a decreased ADH reserve may manifest symptoms of diabetes insipidus in pregnancy for the first time. This occurs because of the inability to increase the secretion of ADH in the presence of an increased metabolic clearance rate of ADH due to the normal increased secretion of vasopressinase in pregnancy, which may underscore mild, asymptomatic diabetes insipidus.
PARATHYROID DISEASES
Parathyroid diseases, although uncommon in pregnancy, are associated with significant perinatal and maternal morbidity and mortality if not diagnosed and properly managed.67 Calcium homeostasis during pregnancy, primary hyperparathyroidism, hypoparathyroidism, and osteoporosis are discussed.
Calcium homeostasis during pregnancy
Two hormones, parathyroid hormone (PTH) and 1,25-dihydroxyvitamin D (1,25-D), are responsible for maintaining calcium homeostasis. About 50% of serum calcium is protein bound (mostly to albumin), 10% is complexed to anions, and 40% circulates free as ionized calcium. During pregnancy, there is an active transfer of maternal calcium to the fetus. A full-term infant requires 25–30 g of calcium during the course of pregnancy for new bone mineralization, particularly during the last trimester of pregnancy.68 The World Health Organization recommends 1.5–2.0 g elemental calcium intake daily from 20 weeks' gestation until the end of pregnancy.69
Total serum calcium is slightly decreased during gestation due to physiologic hypoalbuminemia. The ionized calcium levels, however, remain unchanged throughout gestation. Normal total serum calcium is 8% below the postpartum level,70 with the upper limit of normal for total serum calcium during pregnancy approximately 9.5 mg/dL.
Maternal serum PTH concentrations have been shown to remain mostly unchanged throughout pregnancy when measured by an immunoradiometric assay. In contrast, blood levels of 1,25-dihydroxyvitamin D (calcitriol) increase after the 20th week of gestation as a result of stimulation of renal 1α-hydroxylase activity by estrogen, placental lactogen, and PTH, and as a result of synthesis of calcitriol by the placenta.71 Twenty-four-hour urinary calcium excretion also increases with each trimester of gestation and falls in the postpartum period.72 Similarly, parathyroid hormone-related protein (PTHrP), a peptide responsible for the hypercalcemia found in many malignant tumors, also increases during pregnancy due to its production by mammary and fetal tissue to facilitate transport of calcium to the developing fetus.73 Osteocalcin is a bone-specific protein released by osteoblasts into the circulation proportional to the rate of new bone formation and is slightly decreased during pregnancy, particularly during the first or second trimester, similar to other markers of bone formation (i.e. procollagen I carboxypeptides, bone-specific alkaline phosphatase).73
Differential diagnosis of hypercalcemia
Although the leading diagnosis of hypercalcemia in young women is primary hyperparathyroidism, other endocrine causes should be ruled out, including vitamin D or A overdose, the use of thiazide diuretics, and granulomatous diseases (Table 5).
Table 5. Causes of hypercalcemia in pregnancy and the puerperium
| Most common |
| Primary hyperparathyroidism |
| Rare causes related to pregnancy |
| Familial hypocalciuric hypercalcemia |
| Postpartum hypercalcemia in hypoparathyroidism |
| PTHrp (PTH-related protein)-induced hypercalcemia |
| Other causes unrelated to pregnancy |
| Malignancy |
| Endocrine-related Thyrotoxicosis Adrenal insufficiency |
| Vitamin overdoses Vitamin D overdose Vitamin A overdose |
| Medications Thiazide diuretics Lithium |
| Granulomatous diseases Sarcoidosis Tuberculosis Histoplasmosis Coccidiomycosis |
| Milk-alkali syndrome |
| Acute and chronic renal failure |
| Total parenteral nutrition |
Three uncommon syndromes associated with hypercalcemia during pregnancy are briefly discussed:
- Familial hypocalciuric hypercalcemia (FHH) is an autosomal dominant condition with a high penetrance of hypercalcemia. Patients present with mild hypercalcemia, a mild elevation in serum PTH concentrations, and low urinary calcium excretion. There is moderate enlargement of the four parathyroid glands, but total parathyroidectomy is seldom indicated because of the benign course of the disease. Specific clinical manifestations in the newborn have been described in association with this disease. First, asymptomatic hypercalcemia can develop in an affected offspring if the mother is a carrier for FHH. Second, severe neonatal hypocalcemia can occur in a mother with FHH syndrome. Although the neonatal hypocalcemia can be initially severe, neonatal parathyroid function returns to normal a few weeks after delivery. Third, in the autosomal recessive disease of severe neonatal hypercalcemia (also called neonatal severe hyperparathyroidism), biallelic inactivating mutations of the calcium sensing receptor (CASR) are inherited from parents with FHH who each have one mutated CASR allele.74 Some infants with neonatal severe hyperparathyroidism require parathyroidectomy soon after birth.75
- Postpartum hypercalcemia can occur in women with treated hypoparathyroidism. In this situation, a hypoparathyroid mother treated with vitamin D and calcium during pregnancy may develop significant hypercalcemia in the postpartum period.76 The mechanisms for these hypercalcemic changes include the increased serum levels of 1,25-dihydroxyvitamin D during the second half of pregnancy and the placental synthesis of calcitriol.77 Nausea and vomiting may develop a few days after delivery associated with significant hypercalcemia. Patients should be followed postpartum with serum calcium determinations, and vitamin D should be discontinued in cases of hypercalcemia. In severe cases, intravenous fluids and glucocorticoid therapy are required (Fig. 1).78 In contrast, in some of these women, there may be a small window of time in which calcitriol is required immediately postpartum to maintain normocalcemia.79, 80
Fig. 1. Serum calcium (closed circles) and creatinine (closed squares) levels during pregnancy and for 1 month after delivery in a woman with hypoparathyroidism who was treated with vitamin D and calcium (see text). Stippled area shows normal range. IV, intravenous. (Ficinski M, Mestman JH: Hyperparathyroidism in pregnancy. Endocr Prac 1996;2(5):362–7)
3. A third syndrome of hypercalcemia during pregnancy and in the postpartum period has been described in association with high levels of PTHrP,81 which may also be exacerbated by concurrent use of vitamin D supplements.82 In one case, the mother developed significant hypercalcemia in two successive pregnancies. In the second pregnancy, the PTHrP was elevated three times the normal level and the baby was born with mild hypercalcemia that returned to normal within 24 hours of delivery.83 In the case reported by Khosla and colleagues, a 25-year-old woman with massive bilateral breast enlargement at 24 weeks' gestation had a serum calcium level of 14.3 mg/dL.84 The serum PTH level was undetectable and she underwent bilateral mastectomy during pregnancy. The immunohistochemical studies demonstrated PTHrP antigenic activity in breast tissue.

Primary hyperparathyroidism
The first case of primary hyperparathyroidism during pregnancy was reported in 1931.85 Shortly thereafter, the first case of neonatal hypocalcemia causing tetany in a mother with undiagnosed hypercalcemia due to hyperparathyroidism was described by Friderichsen.86 The disease remains fairly uncommon, and less than 200 pregnant patients with primary hyperparathyroidism have been described.87
As in non-pregnant individuals, the most common cause of primary hyperparathyroidism in pregnancy is a single parathyroid adenoma, which constitutes about 80% of cases. Primary hyperplasia of the four parathyroid glands accounts for about 15% of the cases reported; 3% are due to multiple adenomas; and 2% are due to parathyroid carcinoma. In the first review of the literature in 1962, Ludwig found an incidence of fetal wasting of 27.5% in 40 pregnancies of 21 women with primary hyperparathyroidism.88 The incidence of neonatal tetany was 19%, which was the first indication of maternal hyperparathyroidism.
The majority of nonpregnant patients with primary hyperparathyroidism are asymptomatic, and the diagnosis is made through the routine use of biochemical screening. In pregnancy, however, manifestations of the disease may be more common and commonly overlap with the symptoms of normal pregnancy, including nausea and vomiting.89
There can be significant maternal complications associated with primary hyperparathyroidism, including nephrolithiasis, radiographic bone disease, hyperemesis gravidarum, muscle weakness, and confusion.90 One interesting aspect of primary hyperparathyroidism among pregnant women is the relative increased incidence of pancreatitis in this group, compared to their nonpregnancy counterparts (7–13% vs. 1–2%).87 It is more common in primipara than in women who have had multiple pregnancies and is most likely to occur during the first or last trimesters of pregnancy or the postpartum period.87 In addition, hyperparathyroid crisis has been reported during gestation91 and in the postpartum period, due to the loss of fetal shunting of calcium stores and an increase in placental production of 1,25-dihydroxyvitamin D toward the end of gestation. Patients present with severe nausea, vomiting, generalized weakness, changes in mental status, elevation in serum creatinine due to dehydration; serum calcium levels of over 14 mg/dL are commonly found. Morbidity can be significant, and maternal deaths due to hyperparathyroid crisis have been reported.92, 93, 94 In the neonate, maternal primary hyperparathyroidism can result in hypocalcemia due to the suppressed parathyroid hormone levels.95
Treatment of primary hyperparathyroidism during pregnancy
The only effective treatment for primary hyperparathyroidism is removal of the abnormal parathyroid gland(s). The high perinatal morbidity and mortality reported in the early series was related to significant maternal hypercalcemia.96 If the diagnosis is made during the first two trimesters of pregnancy, surgical treatment is preferred, particularly for those patients with symptoms or those with a persistent serum calcium of over 11 mg/dL. Some reports describe an increased risk of complicated births and adverse neonatal events among women who remain hypercalcemic until the end of pregnancy.97 For women in whom primary hyperparathyroidism is first diagnosed in late gestation, the optimal treatment strategy is unclear. The decision in such a situation should be based on the patient’s overall condition and severity of hypercalcemia.
Medical therapy is reserved for those patients with significant hypercalcemia who are not surgical candidates. Oral phosphate therapy (1.5–2.5 g/day) has been shown to be effective in controlling hypercalcemia, but side effects may include nausea, vomiting, and hypokalemia, which should be managed by decreasing the dose of the medication. The safety of calcitonin and cinacalet needs to be determined during pregnancy, and bisphosphonates should only be used when there are no other alternatives.97 Adequate hydration, early treatment of urinary tract infections, and avoidance of medications that may produce an elevation in serum calcium, such as vitamin D, vitamin A, and thiazide diuretics, are mandatory. Serum calcium should be checked regularly.
In patients undergoing surgical treatment, hypocalcemia, albeit transient, may occur postsurgery. Serum calcium should be checked every 6 hours. If the patient develops hypocalcemic symptoms, intravenous calcium in the form of calcium gluconate 10% solution 10–20 mL should be given during a period of 5–10 minutes. Intermittent infusions can be repeated, or calcium gluconate can be diluted in 5% dextrose or isotonic saline and infused continuously at 1 mg/kg/bw per hour. In patients with bone disease, postsurgical hypocalcemia may be profound, and aggressive treatment is needed. These patients may benefit from vitamin D supplementation in the form of calcitriol 0.25–0.5 μg/day.
Hypoparathyroidism
The most common cause of hypoparathyroidism is damage to or removal of the parathyroid glands in the course of an operation on the thyroid gland. The incidence of hypoparathyroidism after thyroid surgery has been estimated to be up to 10%, but the hypocalcemia in the majority of these cases are only transitory.98 Idiopathic hypoparathyroidism is a much less common cause of the disease.
The diagnosis of hypoparathyroidism is based on previous history (particularly a history of thyroid surgery) and on clinical, radiologic, and laboratory information. Symptoms of hypocalcemia include numbness and tingling of the fingers and toes and around the lips. Patients may complain of carpopedal spasm, laryngeal stridor, and dyspnea. Convulsions may be a manifestation of severe hypoparathyroidism. Symptoms of irritability, emotional lability, impairment of memory, and depression are common. On physical examination, papilledema and cataracts may be seen. In patients with idiopathic hypoparathyroidism, changes in the teeth, skin, nails, and hair are common. Chvostek's sign, which is a twitch of the facial muscles (notably those of the upper lip) when a sharp tap is given over the facial nerve, is seen in many patients with hypocalcemia, although it has been described in 10% of normal adults. Trousseau's is another sign of hypocalcemia, described as carpopedal spasm caused by reducing the circulation in the arm with a blood pressure cuff; the constriction should be maintained above the systolic blood pressure for 2 minutes before the test is considered negative. The diagnosis is confirmed by the presence of persistent low serum calcium and high serum phosphate levels. The plasma alkaline phosphatase level usually is normal. The differential diagnosis of hypocalcemia includes rickets, osteomalacia, and hypomagnesemia.
One of the first reports of hypoparathyroidism in pregnancy was in 1942, when Anderson and Musselman reviewed the literature on pregnancy and tetany and collected 240 cases.99 Of these, 26 were caused by post-thyroid surgery and 140 were the so-called idiopathic type. It is likely that in some of these cases tetany was not caused by hypoparathyroidism. Before specific therapy was available, fetal and maternal mortality was so high that therapeutic abortion was routinely recommended. With the availability of vitamin D and the use of calcium supplementation, the prognosis has improved and the infants reveal no unusual abnormalities.
Women with hypoparathyroidism during pregnancy can be treated with vitamin D therapy, with preference given to use small (i.e. once daily) doses of calcitriol to achieve and maintain serum calcium levels in the normal range.77 If the mother is not properly treated, hypoparathyroidism with hypocalcemia can be serious for the newborn. The infant can develop intrauterine hyperparathyroidism associated with radiologic bony changes. Loughead and colleagues reported 16 infants who were born with secondary hyperparathyroidism caused by severe maternal chronic hypocalcemia, in whom the abnormal bone mineral content was corrected by 1 month of age on no treatment.100
A common challenge in the treatment of hypoparathyroidism are the recurrent episodes of hypercalcemia and hypocalcemia, and thus, serum calcium determinations should be performed at regular intervals. The most common symptoms of vitamin D intoxication are nausea, constipation, fatigue, headaches, and, in more severe cases, vomiting and dehydration.
Pseudohypoparathyroidism
Pseudohypoparathyroidism encompasses several different disorders which feature varying degrees of target organ resistance to PTH. Somatic changes, such as short stature, obesity, round face, brachydactyly, and mental retardation, are present in some forms of the syndrome known as Albright's syndrome type 1a. Most of these patients demonstrate hypocalcemia caused by a derangement of renal 1α-hydroxylase and production of 1,25(OH)D2 (calcitriol).
A few cases of pseudohypoparathyroidism have been reported during pregnancy.101, 102 In the case presented by Singh and colleagues, a woman was found to have a low serum calcium of 7.8 mg/dL at 24 weeks' gestation; her previous pregnancy had been a miscarriage in the first trimester.102 Features of hypocalcemia included multiple calcifications in the brain shown by computed tomography scan. She was prescribed oral calcium (2.5 g/day) and calcitriol (1 µg/day), then upon delivery at 40 weeks' gestation, managed with IV calcium. Her delivery was uneventful to a healthy infant.
Osteoporosis
During pregnancy, maternal physiology adapts to meet the demands of the developing fetal skeleton, including increased maternal intestinal calcium absorption, skeletal resorption, and secretion of PTHrp by the placenta and breast tissues.103 Although this hypothesis suggests that maternal bone density is decreased in pregnancy, studies reporting the degree of impairment have not been consistent, which may in part be due to differences in sample size and methodological aspects of assessing bone density. Family history and genetic components of bone disease have also been suggested as contributors.104, 105, 106 Some investigators also hypothesize that heparin use may be an additional risk factor for pregnancy-associated osteoporosis,107 although this has not been consistently demonstrated.108
Although fairly uncommon, reports of osteoporosis diagnosed during late pregnancy and in the postpartum period have been described.104, 109 One of the earliest reports suggesting the syndrome of postpartum osteoporosis was published in 1955 by Nordin and Roper.110 Whether these are two different syndromes or the same entity is unclear, as the disease process may begin during pregnancy but the diagnosis may not be made until after delivery. Osteoporosis diagnosed during pregnancy may be localized in the hip(s) or lumbar spine, or both. Pain in one hip or back pain is the presenting symptom in most cases. Because radiographs cannot be performed during pregnancy, there should be a high index of suspicion and the diagnosis may have to be presumed. These women should have radiographic determinations and bone density studies after delivery.
Treatment of bone loss during pregnancy and lactation should include adequate calcium and vitamin D supplementation; one study suggested a vitamin D dose of 1200 IU/day.111 Other case reports have proposed the use of strontium in women with vertebral compression fractures during pregnancy.112 There may also be a beneficial effect of bisphosphonates, teriparatide,113, 114 or calcitonin.115
In a study by Phillips and colleagues, 13 women with pregnancy-associated osteoporosis (including some with vertebral collapse) were followed for up to 8 years.116 The mean bone mineral density at both the hip and spine increased significantly during this follow up period toward the lower end of the normal range, suggesting that some of the bone loss is reversible. In another report of 73 postpartum women, lumbar spine bone mineral density was 7.6 ± 0.1% lower than that of 55 age-matched controls, and further loss of 2.0 ± 1.0% was seen in the 1–6 months postpartum among the lactating subset.117 This study suggests that pregnancy is associated with low bone mass that is further exacerbated by the initial first few months of lactation, which stabilized in the following several months up to 1 year. The findings of a recent report in Denmark were consistent with this trend, and further follow up showed that bone mineral density returned to pre-pregnancy levels at 19 months postpartum.118
ADRENAL DISEASES
Adrenal gland physiology
Histologically, the adrenal gland is divided into three zones: zona glomerulosa, zona fasciculata, and zona reticularis. The glomerular zone produces aldosterone; the middle, or fascicular, zone produces mainly cortisol; and the inner, or reticular, zone produces androgens and a minimal amount of estrogen.
Cortisol is the chief secretory product of human adrenal glands. In addition to having glucocorticoid activity, it has a potent anti-inflammatory effect when used in pharmacologic doses. Cortisol is present in the circulation in at least three forms: bound to a specific binding protein (corticosteroid-binding globulin [CBG] or transcortin), bound to albumin, and in the free form. In the nonpregnant woman, 70–80% is bound to CBG, and only 10–15% is in the free or unbound fraction. In pregnancy, there is an increase in the plasma levels of CBG beginning at approximately 11 weeks of gestation,119 and by the end of the third trimester, there is an increase of serum levels of CBG, corresponding to a rise of total plasma cortisol, both by 3-fold.120 Free or unbound cortisol is the only biologically active fraction of cortisol, and its levels increase by 1.6-fold by the end of gestation.120
Aldosterone is the most potent mineralocorticoid; its daily secretion rate in nonpregnant women is about 50–250 μg during normal sodium intake. Its secretion is controlled by the renin–angiotensin system, potassium, and, to a lesser degree, ACTH. Aldosterone secretion rate and plasma concentration increase during pregnancy, despite the increases in plasma volume seen during this state.121
The adrenal cortex secretes dehydroepiandrosterone (DHEA) and dehydroepiandrosterone sulfate (DHEAS). The DHEA production rate is increased during pregnancy; it is then aromatized to estradiol and estrone by the term placenta and may contribute up to 9% of circulating estradiol.122
Cushing's syndrome
Cushing's syndrome is defined as a constellation of signs and symptoms due to chronic excessive production of glucocorticoids by the adrenal glands. As in many other endocrine conditions, women are more affected than men, with a ratio of about 3:1 and overall annual incidence of 1.2–1.7/million per year in one large population-based Danish study.123
Cushing's syndrome can be classified as ACTH dependent or ACTH nondependent (Table 6). ACTH-dependent Cushing's syndrome is caused by hyperproduction of glucocorticoids by the adrenal glands secondary to excessive or inappropriate secretion of ACTH or ACTH-releasing hormone by an ectopic tumor. ACTH-independent Cushing's syndrome is caused by an intrinsic disorder of the adrenal gland, such as a benign or malignant adrenal tumor. Alternatively, it may be iatrogenic, secondary to pharmacologic doses of glucocorticoids in the treatment of systemic disease. Plasma ACTH levels are helpful when interpreted in the light of urinary cortisol values. High-normal or elevated values in the presence of high urinary cortisol is suggestive of ACTH-dependent Cushing's syndrome (Cushing's disease). Low ACTH values are consistent with unilateral adrenal tumor.
Table 6. Classification of Cushing's syndrome
| ACTH-dependent |
| Chronic administration of ACTH |
| Excessive production of pituitary-ACTH (Cushing's disease), resulting in bilateral adrenal hyperplasia |
| Ectopic ACTH production |
| ACTH-independent |
| Chronic administration of synthetic corticosteroids |
| Adrenal adenoma |
| Adrenal carcinoma |
Given the adverse effect of increased cortisol levels in Cushing’s syndrome on ovulation, women of childbearing age with untreated Cushing’s syndrome rarely become pregnant. In the infrequent case, the most common cause is ACTH-independent Cushing’s syndrome, usually due to a benign adrenal adenoma124 and less commonly, Cushing’s disease due to a pituitary ACTH-secreting adenoma.125 Bilateral hyperplasia and adrenal carcinoma during pregnancy have also been described.126, 127 Similarly, ectopic ACTH syndrome is unusual in young people, but occasional cases have been reported in pregnancy.128
Cushing's syndrome is characterized clinically by the slow progression of symptoms. These manifest as muscle weakness, personality changes, oligomenorrhea or amenorrhea, hirsutism, weight change, and back pain due to osteoporosis. One of the common manifestations in women is oligomenorrhea, which progresses to amenorrhea. There is an increased incidence of thromboembolism. On physical examination, hypertension, truncal obesity, fat deposition in the supraclavicular areas and upper dorsal spine (buffalo hump), moon face, atrophy of skin that is easily bruised, and purple striae (over abdomen, axillae, and hip areas) are common clinical features. Weakness due to muscular atrophy can be very marked, particularly in the quadriceps muscles; the patient may be unable to rise from a deep knee bend without assistance. Headaches and visual field disturbances due to a large pituitary tumor are rare. Hirsutism, sometimes of the lanugo type, is common. The severity of virilization depends on the amount of androgen produced; in patients with adrenal adenoma, the secretion of androgens is low, whereas it is moderately high in patients with bilateral adrenal hyperplasia. Masculinization is rare, and when it occurs it is due to an adrenal carcinoma producing large amounts of androgens. Kidney stones due to hypercalciuria are reported to occur in some patients.
An increase in the hematocrit, leukocytosis with relative lymphopenia, and eosinopenia are usually seen. Abnormal glucose metabolism is present in approximately 50% of Cushing’s syndrome patients, of which overt diabetes mellitus comprises over two-thirds of cases.129 Mild hypokalemia with metabolic alkalosis is not uncommon. Radiologic examination of the chest may show an enlarged mediastinum secondary to fat deposit; osteoporosis is not uncommon.
The clinical diagnosis may be difficult during pregnancy because characteristically similar striae are seen in both pregnancy and Cushing's syndrome. Comparisons of close-up photographs from earlier years are helpful in detecting subtle changes. The diagnosis is confirmed by the proper use and interpretation of tests assessing the hypothalamic–pituitary–adrenal axis. A significant and persistent elevation in urinary free cortisol is the best indication of cortisol overproduction, although physiologic elevations occur in normal pregnancy. Random determinations of plasma cortisol are of little help, because values are normal in many patients with Cushing's syndrome. The overnight dexamethasone suppression test, although an excellent screening test in nonpregnant women, is of limited value in pregnancy, given its blunted response during this state and risk of false positive results. Lack of diurnal variation for cortisol is suggestive of the disease, but to properly assess the results of the test, specimens should be obtained early morning and late evening. A random plasma cortisol late afternoon value could be misleading. Hence, in pregnancy, the determination of free cortisol concentrations from a 24-hour urine specimen is the best screening test to rule out Cushing's syndrome. Concomitant determination of creatinine in the urine is necessary to ensure proper urinary collection.
Once the diagnosis of Cushing's syndrome is suggested by use of the urinary screening test in pregnancy, the cause should be investigated. Follow-up testing is not different from those recommended for the non-pregnant individual. The low-dose dexamethasone suppression test is the initial assessment, in which urinary cortisol is measured before and after dexamethasone (0.5 mg by mouth every 6 hours for eight doses); patients with Cushing's syndrome, regardless of the cause, fail to suppress urinary free cortisol with this test. To differentiate between Cushing’s disease and an ectopic source of cortisol excess, the high-dose dexamethasone (2 mg every 6 hours for eight doses) should be performed; patients with Cushing’s disease will demonstrate suppression, whereas nonpituitary/ectopic malignant tumors usually do not.
In patients diagnosed with Cushing's disease, an MRI of the pituitary gland may identify a tumor. However, as enlargement of the pituitary gland occurs during normal pregnancy, this finding may be difficult to assess. Selective petrosal sinus sampling, a procedure used in localization of the pituitary lesion, is not indicated in pregnancy because of radiation exposure. For cases in which the laboratory tests are suggestive of an ACTH-independent adrenal lesion, an MRI (preferred over CT due to ionizing radiation) may delineate the adrenal pathology.
Maternal and perinatal morbidity and mortality are significant in Cushing's syndrome. Increased maternal glucorticoid exposure has been associated with low birth weight and prematurity.130 Cushing’s syndrome in the mother has been associated with increased risks of hypertension, preeclampsia, gestational diabetes mellitus, myopathy, opportunistic infections, poor wound healing, osteoporotic fractures, psychiatric problems, congestive heart failure, and death.119 diagnosis and management strategies) In the fetus, there may be intrauterine growth restriction, and there is an increased risk of miscarriage and stillbirth.119
Medical treatment of Cushing's syndrome during pregnancy has been achieved primarily with the use of metyrapone or ketoconazole. Metyrapone acts primarily to inhibit steroid 11β-hydroxylation, therefore decreasing the secretion of cortisol by the adrenal glands. Its main use is as a diagnostic tool for assessing ACTH reserve, thus it is also used in the diagnosis of Cushing's syndrome. It has been used as a therapeutic agent in nonpregnant patients with Cushing's disease preoperatively or after radiation therapy to the pituitary gland. In a recent review, 15 patients with Cushing’s syndrome during pregnancy managed with metyrapone and ketoconazole were described.131 Within this series, 53% delivered close to term, 20% developed preeclampsia, and there was one stillborn and two neonatal deaths; hypercortisolemia was well-controlled overall. The use of octreotide and the dopamine-agonist, cabergoline, during pregnancy for the treatment of Cushing’s disease have also been described.132, 133
If medical management is not achievable or desired, adrenal surgery can be performed during the second trimester. One report described the successful use of laparascopic adrenalectomy at 32 weeks' gestation.134 Regarding Cushing’s disease, several reports of successful transsphenoidal pituitary surgery performed during pregnancy or shortly following delivery have been described.135 A recent report described the use of gamma-knife in treating Cushing’s disease among five pregnant women.136
In cases of Cushing’s disease in which pituitary surgery is not successful, bilateral adrenalectomy may be an option. However, following bilateral adrenalectomy, there is a concern for the development of Nelson’s syndrome, which is characterized by increased size of the corticotroph tumor. In one series of 11 such patients, there were no significant increases of the pituitary tumor during pregnancy, suggesting that this state does not contribute to tumor progression.137 However, these patients, as in patients with prolactinomas, should be followed carefully during pregnancy with visual field examinations to detect any potential enlargement of the pituitary gland.
Addison's disease
Addison's disease, or primary adrenal insufficiency, results from total destruction of the adrenal cortex. In cases of adrenal insufficiency secondary to ACTH deficiency, the zona glomerulosa is preserved; therefore, there is normal secretion of aldosterone. The most common cause of primary adrenal insufficiency is autoimmune disease of the adrenal glands, which occurs in about 70% of cases of Addison's disease, followed by tuberculosis. Other unusual causes include surgical bilateral adrenalectomy for systemic diseases, bilateral metastatic carcinoma, bilateral adrenal bleeding during anticoagulant therapy, and, rarely, fungal infections. Severe systemic infections by meningococcus (Waterhouse-Friderichsen syndrome) may also present as acute adrenal insufficiency characterized by vascular collapse and petechial hemorrhage in the skin and mucosa.
With the wide use of corticosteroids for the treatment of other systemic diseases, adrenal atrophy due to ACTH suppression is perhaps the most common cause of adrenal insufficiency. In these cases, adrenal insufficiency is caused by chronic suppression of pituitary ACTH. As a result, patients receiving corticosteroid therapy for various periods of time may develop relative adrenal insufficiency. This may manifest for the first time during periods of stress, which because of chronic ACTH suppression, the adrenal glands are unable to respond with an increase in cortisol secretion. The amount of time for the hypothalamic–ACTH–adrenal axis to recover completely after the discontinuation of corticosteroid therapy is reported to be quite variable and may not even be necessarily dependent on the dose and duration of the corticosteroid.138, 139 In one series, smaller doses of chronic corticosteroids (i.e. prednisone 5 mg by mouth daily) were not associated with impaired cortisol responses as assessed by a rapid adrenocorticotropic hormone stimulation test.140
The most common symptoms in patients with chronic primary adrenal insufficiency are weakness, fatigue, and weight loss. Characteristically, these patients feel relatively well in the morning and experience increased fatigue and asthenia as the day progresses. Blood pressure is low, and patients may complain of postural hypotension. Gastrointestinal symptoms, such as nausea, anorexia, and sometimes vomiting and diarrhea, are not uncommon and may result in weight loss. Although pigmentation of the skin and mucosa is characteristically described in patients with Addison's disease, it is not seen in all such patients. If present, the pigmentation is usually seen in body creases, such as palms of the hands, knuckles, knees, and elbows; in scars; and in the genital, gingival, and buccal areas. Almost half of all Addison’s disease patients will develop another autoimmune disease, including vitiligo, type 1 diabetes mellitus, or autoimmune thyroid disease.141 Vitiligo occurs as lesions ascribed to autoimmune destruction of the melanocyte in hyperpigmented areas. Gonadal function is disturbed, and in women, axillary and pubic hair may be reduced because of lack of adrenal androgens. Adrenal crisis is characterized by severe hypotension, gastrointestinal symptoms such as nausea, vomiting, and diarrhea, and severe dehydration. These patients may present with elevated body temperature, although infection is not always demonstrated.
Typical laboratory findings in patients with Addison's disease include mild anemia, hyponatremia, hyperkalemia, and an increase in serum blood urea nitrogen. Serum electrolytes may remain within normal limits in some cases. Hypercalcemia may be seen during the acute phase of adrenal insufficiency. Hypoglycemia may be found in patients in the fasting state and during adrenal crisis. Chest radiographs may demonstrate a small heart, and the electrocardiogram may show changes related to electrolyte disturbance.
The diagnosis is confirmed by a lack of cortisol response to ACTH administration or elevated serum ACTH levels in the presence of low serum cortisol concentrations, or both:
1. ACTH stimulation test – Baseline or random serum cortisol levels are low in most patients with Addison's disease. However, in some patients the adrenal glands are still able to respond to the high levels of circulating plasma ACTH, and these patients may have normal cortisol levels on a random specimen. On the other hand, low plasma cortisol levels are not diagnostic of adrenal insufficiency, because they are seen in many other situations in which the adrenal glands are not diseased. Furthermore, in pregnancy or in patients on estrogen therapy, plasma cortisol is elevated because of an increase in plasma CBG concentrations. Therefore, the diagnosis of adrenal insufficiency is confirmed by dynamic tests, namely, serum cortisol response to the administration of ACTH.
A. Low-dose ACTH stimulation test – This is the simplest test available to rule out the presence of Addison's disease. For the test, synthetic ACTH (cosyntropin) 0.25 mg is given as a rapid intravenous bolus, and plasma cortisol is measured before administration and 30 and 60 minutes after. A peak cortisol response of 18 μg/dL over the baseline values rules out Addison's disease. However, a lack of response does not confirm the diagnosis, and a more prolonged ACTH stimulation test may be needed.
B. Two-day ACTH infusion test – There are several modifications of the original short ACTH stimulation test. The basis for the test is the continuous administration of ACTH for several days along with measurement of plasma or urinary corticosteroids.142 For the test, ACTH in the form of cosyntropin, 0.25 mg in 500 mL 5% dextrose in normal saline, is given every 12 hours for 48 hours, and serum cortisol is measured before administration and every 12 hours thereafter. An increase in serum plasma cortisol of over 30 μg/dL rules out the presence of primary adrenal insufficiency. To prevent an acute adrenal crisis in patients with significant symptoms of adrenal insufficiency, dexamethasone, 0.5 mg, can be added to the continuous ACTH infusion. This small amount of corticosteroid does not interfere with the determination of serum cortisol and provides enough glucocorticoid to correct the patient's symptoms. This 2-day continuous ACTH infusion test can accurately distinguish between primary and secondary adrenal insufficiency.
2. Plasma ACTH determination – Patients with cortisol deficiency caused by primary adrenal disorders have elevated concentrations of serum ACTH. Normal values early in the morning are between 20 and 150 pg/mL. Baseline cortisol values less than 10 μg/dL and ACTH levels of greater than 250 pg/mL are diagnostic of primary adrenal insufficiency.
Patients with primary adrenal insufficiency require lifelong treatment with only occasional exceptions. Hydrocortisone is the treatment of choice, in doses of 20–30 mg/day, with two-thirds of the dose being given in the morning and one-third in the evening. In most patients, a mineralocorticoid is also added in the form of fludrocortisone; the daily amount has to be adjusted for each patient, and the total dose varies from 0.05 to 0.2 mg. Although cortisol is the natural hormone produced by the adrenal gland, and it is the drug of choice, other synthetic glucocorticoids may be used. Prednisone and prednisolone represent other options, and either one can be used in doses of 3–5 mg in the morning and 1–3 mg in the evening. In cases of moderate stress, such as concurrent illness, the dose of corticosteroids should be doubled for 3 days.
Adrenal crisis is an emergency which requires immediate treatment. Intravenous fluids in the form of normal saline and glucose should be given, and 100 mg hydrocortisone should be injected as a bolus intravenously. An additional 200 mg in divided doses should be given during the first 24 hours. A search for the cause of the acute crisis should be made, as in most cases, infection is the underlying precipitant.
Patients with known adrenal insufficiency and patients on corticosteroid therapy undergoing selective surgery should be given increased cortisol in total divided doses of 200–300 mg daily perioperatively. Many regimens have been proposed for the management of these patients, but the one that we recommend is the use of cortisol, 100 mg intravenously every 8 hours in continuous infusion for the first 24 hours and then decreased by 50 mg/day until the patient is able to take oral medications. The infusion should be started early in the morning on the day of surgery. In most cases, the patient is able to return to a routine daily dose by the fourth or fifth day postoperatively. Because the amount of cortisol given has enough mineralocorticoid action, no mineralocorticoid supplementation is needed during this time. Daily determinations of serum electrolytes and glucose should be obtained, because this amount of cortisol may produce hypokalemia and hyperglycemia. To supply enough corticosteroids, it is suggested that an injection of cortisone acetate, 50–100 mg intramuscularly, be given the day before surgery. The same regimen is applied to patients who received corticosteroid therapy in the past and discontinued it within 1 year. An alternative regimen involves cortisone acetate, 50 mg intramuscularly, every 6 hours the first day, every 8 hours the second day, and every 12 hours the fourth day after a surgical procedure. During surgery, 100 mg of hydrocortisone is added to the intravenous drip.
Pregnancy in patients with known Addison's disease been associated with uncomplicated deliveries and healthy infants.143 Early reports have indicated no increase in miscarriage rate, prematurity, or intrauterine death.144, 145 In general, the amount of glucocorticoid needed is the same as in nonpregnant patients, with the requirement increasing during labor, similar to patients in stressful situations. As fasting serum glucose is lower during pregnancy, patients with untreated adrenal insufficiency have a tendency to develop hypoglycemia. Severe hypoglycemia may occur due to ACTH deficiency during pregnancy if insufficient corticosteroids are given or if the patient omits a dose. Patients with chronic adrenal insufficiency should be instructed to be strictly compliant with the medications, particularly in cases of nausea and vomiting, and should have injectable corticosteroids at home for such cases. Dexamethasone phosphate in 4-mg injectable syringes is available. Patients should always carry with them identification stating their diagnosis and medication. It has been reported that patients may need an increased amount of mineralocorticoids during pregnancy because of the high levels of progesterone, which have a sodium-losing effect. Elevated levels of serum aldosterone are characteristic in normal pregnancy and have been attributed to the high levels of progesterone that are also present.
Infants of mothers who receive corticosteroid therapy rarely develop adrenal insufficiency. In a historical review of 260 pregnancies in which the mother took steroids for variable periods, only one infant developed adrenal insufficiency.146 However, if maternal Addison’s disease is unrecognized and/or inadequately treated, there is a high incidence of both maternal and fetal complications, including postpartum adrenal crises and fetal death in utero.147
Congenital adrenal hyperplasia
Congenital adrenal hyperplasia (CAH) is a hereditary disorder involving both adrenal glands in which a number of defects of corticosteroid biosynthesis result in decreased production of cortisol and a secondary increase in ACTH secretion. Adrenal hyperplasia can result and an accumulation of hormonal precursors can be detected on laboratory testing. Defects of several enzymes, such as cytochrome P-450 oxidase, may produce specific clinical scenarios (Figure 2).
|

The most common defect in CAH is the 21-hydroxylation defect, which is found in more than 95% of patients with congenital adrenal hyperplasia.148 It is an autosomal recessive disorder involving the CYP21A2 gene on the short arm of chromosome 6. The classical form affects about 1 in every 5000 live births and 1 in 15,000 live births in North America and Europe, respectively.149 The clinical manifestations depend on the severity of the abnormality in the 21-hydroxylase genes. Women with severe deficiency of the enzyme (the classical form of 21-hydroxylase deficiency) have a decreased fertility rate150 because of oligoanovulation, elevated progestin levels, and aberrant endometrial implantation.151 Abnormal genitalia and sodium wasting are recognized in the neonate at the time of delivery, as are signs of virilization. The milder form of the disease, termed the classic simple virilizing form of CAH, affects 1 in 600 live births.150 In this form, virilization is the only manifestation, because aldosterone secretion is not significantly affected. The third form of the disease, nonclassical CAH, is mild and may not be detected until childhood or adulthood. Other types of defects include the 11-hydroxylation defect and, rarely, the defect of the 3-β-hydroxysteroid-dehydrogenase enzyme.
The goals of treatment are to prevent adrenal insufficiency, provide salt-retaining corticosteroids in the salt-losing types of cases, and suppress androgen secretion to prevent virilization and abnormalities of growth in a way that balances the potential side effects of excess steroid treatment. This is achieved with doses of hydrocortisone of 5–10 mg with each meal.152 Prednisolone or dexamethasone, which have a longer half-life, can be used if control is not achieved with hydrocortisone alone; these agents are more effective but also are associated with the side effects of Cushingoid features, weight gain, and striae.152 The use of prednisone is discouraged, as its conversion to prednisolone is inconsistent when used in the low doses required for CAH patients.152 Parameters of adequate glucocorticoid dosage include determination of plasma 17-hydroxyprogesterone (17-OHP), androstenedione, and testosterone. Patients with the salt-losing variety also require mineralocorticoid therapy in the form of Florinef (fludrocortisone) 0.05–0.3 mg/day; the dose is adjusted to maintain plasma renin activity in the lower part of the reference range. No adjustment of any of the medications is needed in pregnancy.153
Prenatal evaluation of the partner of a woman who has the disease or is a carrier is important in evaluating for possible fetal involvement. In one European study, the frequency of being a carrier of CAH was 1:55.154 Determination of plasma 17-OHP at baseline and after the administration of ACTH allows, in most cases, detection of the partner's heterozygous state.155 Fetuses are considered at risk if: (1) both parents are heterozygous, (2) both parents are homozygous, or (3) one parent is homozygous and the other heterozygous.
Prenatal diagnosis of CAH due to 21-hydroxylase deficiency was historically achieved with the detection of elevated amniotic fluid 17-OHP and adrenal androgen concentrations at approximately 16–17 weeks' gestation. However, genetic analysis of extracted fetal DNA has now become the mainstay of prenatal diagnosis of CAH.156 This is usually performed through chorionic villus biopsy at 8–10 weeks' gestation, which carries a fetal loss rate of 1.1%.157
Early treatment (i.e. before 6 weeks' gestation) may prevent the development of masculinization of external genitalia in the affected female fetus. Dexamethasone, at 20 µg//kg/day,156 is the drug of choice because it readily crosses the placenta (compared with the other glucocorticoids) and suppresses fetal adrenal androgen synthesis, thus reducing masculinization. Mothers who are potentially affected should be treated until fetal sex and involvement are known. Maternal urinary estriol levels are an accurate index of maternal compliance with dexamethasone.156
The maternal side effects of this therapy include the development of Cushingoid facial features, facial hirsutism, excessive weight gain, permanent scarring of striae, hypertension, and gestational diabetes. Patients should be informed about potential side effects and those receiving treatment should be closely monitored, medical intervention should be provided when indicated, and the dose of dexamethasone should be reduced during the second half of pregnancy, to reduce side effects. Some studies have suggested that prenatal dexamethasone treatment of potentially affected fetuses is associated with neuropsychological impairments, including those of IQ, learning, and long-term memory,158 as well as behavioral problems.159
11-Hydroxylase deficiency is another form of CAH and is due to a mutation in the CYP11B1 gene.160 The production of cortisol is reduced, and there is an excessive production of 11-deoxycortisol, 11-deoxycorticosterone, and adrenal androgens. Hypertension, hypokalemia, and virilization are the most common form of presentation,161 along with infertility. The diagnosis may be made usually in adolescence or adulthood, when the patient presents with infertility or oligomenorrhea, hirsutism, and/or short stature. Treatment with glucocorticoids normalizes infertility, and successful pregnancies have been reported.153
Primary aldosteronism
Primary aldosteronism was described by Conn in 1955162 and is characterized by hypertension and hypokalemia due to excessive production of aldosterone by the adrenal glands. The first cases described were of patients with single adrenal adenomas, but now it is known that the disease in some patients is from bilateral adrenal hyperplasia or multiple microadenomas; rare cases of glucocorticosteroid remediable aldosteronism (GRA), adrenal carcinoma, and congenital primary aldosteronism have also been reported.
Primary hyperaldosteronism accounts for up to 18% of patients with hypertension.163, 164 Symptoms such as headaches and muscular weakness due to hypokalemia are present in some patients; serum potassium is decreased in the vast majority of patients. Metabolic alkalosis with hypochloremia and an elevation in serum bicarbonate are commonly seen. Urinary potassium excretion is elevated and inappropriate to the low serum potassium levels. Renal function is within normal limits, and the pH of the urine is neutral or alkaline.
The diagnosis is suspected in patients with concomitant hypertension and hypokalemia. All other causes of hypokalemia, particularly ingestion of diuretics, must be ruled out. High levels of urinary potassium in the presence of hypokalemia are very suggestive of primary hyperaldosteronism. The diagnosis is confirmed by (1) an elevation in serum or urine aldosterone, (2) suppressed levels of plasma renin, (3) a lack of plasma renin elevation on administration of a potent diuretic such as furosemide, and (4) failure of the aldosterone level to be suppressed by the administration of a sodium load.
In normal pregnancy there is an increase in aldosterone secretion and plasma aldosterone levels starting in the first trimester; this peaks at 36 weeks' gestation at a level eight- to ten-fold higher than that in nonpregnant women. This elevation in plasma aldosterone is normally suppressed in pregnancy by the administration of 9-α-fludrocortisone.155 Plasma renin levels increase early in pregnancy, reaching a peak at 12 weeks and remaining elevated throughout pregnancy. Metabolic alkalosis is present instead of the respiratory alkalosis that is characteristically seen in normal gestation. Serum bicarbonate in plasma is normal or elevated, in contrast to the low values observed in pregnant women with primary hyperaldosteronism.
Approximately 30 cases of primary hyperaldosteronism have been reported in pregnancy.165 Surgery is preferred166 if it can be done safely in either the late first or early second trimester. There are limited options for medical therapy, as spironolactone, angiotensin-converting enzyme inhibitors, angiotensin II receptor antagonists are contraindicated. Use of eplerenone, although similarly not recommended nor contraindicated, has been reported in one case.165
Pheochromocytoma
Pheochromocytoma is a rare syndrome accounting for between 0.01 and 0.10% of all cases of newly diagnosed hypertension.167 The prevalence is estimated at 1.5–4 per 10,000, and if including those undiagnosed cases which are only found at autopsy, may increase to 0.1%.168 It is equally distributed between males and females,169 and the mean age of detection is 43 years.170
In most cases (90%), the disease is characterized by a single catecholamine-secreting adrenal adenoma. Involvement of both adrenal glands is more commonly seen in cases with a strong family history of pheochromocytoma. Approximately 10–20% are associated with a genetic syndrome,169 such as multiple endocrine neoplasia type II (MEN type IIA and IIB), in which medullary carcinoma of the thyroid and parathyroid adenoma (Sipple's syndrome) may also be found. Pheochromocytoma may also be associated with type 1 neurofibromatosis (NF) or von Hippel-Lindau (VHL) syndrome. In 15–20% of cases, the chromaffin tumors initiate from outside the adrenal gland and are termed paragangliomas or extra-adrenal pheochromocytomas.171 Approximately 10–20% of these tumors are malignant, with a higher incidence among paragangliomas.172
Hypertension is the most common symptom, which can be persistent or paroxysmal. Some patients may have markedly labile blood pressure. In addition to hypertension, headaches, sweating, palpitations, nervousness, tremor, weakness, abdominal pain, and warm flashes are not uncommon. The attacks can be precipitated by induction of anesthesia, examination of the abdomen, or changes in position. The patient with pheochromocytoma may be hypermetabolic and display weight loss, nervousness, and elevation of the basal metabolic rate. Glycosuria and mild intolerance to glucose are not unusual.
Pheochromocytoma associated with pregnancy is life-threatening for mother and fetus.173 It is exceedingly rare in pregnancy and carries an estimated incidence of 0.007%.119 Maternal and fetal mortality were as high as 50% prior to the 1970s, and due to advances in detection and appropriate treatment, fetal mortality has improved to 10% and maternal mortality to 2%.174 Survival is improved in cases in which the diagnosis is made antenatally.175
In the pregnant woman with a pheochromocytoma, symptoms are similarly variable and nonspecific, as is the case with a non-pregnant patient. Hypertension (paroxysmal or sustained), headaches, palpitations, anxiety, and convulsions may be present (Table 7).176 In several cases, sudden shock appeared spontaneously or was induced by anesthesia or vaginal delivery in patients without previous symptoms. In 22 patients, the diagnosis was made for the first time during pregnancy. Of 67 patients in whom pheochromocytoma remained undiagnosed during pregnancy, acute toxemia was diagnosed in 29 (43%) and essential hypertension was diagnosed in 11 (16.4%).
Table 7. Symptoms of pheochromocytoma in pregnancy
| Cases (%) | |
| Hypertension | 90 |
| Headaches | 70 |
| Palpitations | 40 |
| Sweating | 35 |
| Anxiety | 30 |
| Blurred vision | 20 |
| Convulsions | 10 |
| Dyspnea | 10 |
* Adapted from 176
The diagnosis of pheochromocytoma is confirmed by the biochemical demonstration of elevated levels of catecholamines or their metabolites (norepinephrine and epinephrine), methylated metabolites (normetanephrine and metanephrine), and the methylated deaminated metabolite vanillylmandelic acid (VMA), which are not affected by pregnancy.174 An elevation in VMA, catecholamines, or metanephrine in plasma and/or a 24-hour urine specimen is suggestive of the diagnosis. This should be confirmed, however, by a repeat collection. The following recommendations are suggested for the proper interpretation of urinary catecholamine and its metabolites:
- 24-hour urine collections are preferable to random ones; urinary creatinine should be measured in each 24-hour collection to assess its adequacy.
- It is preferable to collect the urine when the patient is at rest, is on no medication, and has not had recent exposure to radiographic contrast media. Among the medications for hypertension, diuretics, adrenergic blocking agents, and hydralazine do not interfere appreciably.
- Urine collection must be acidified (pH below 3.0) and kept cold during and after collection.
- The diagnostic information is greater in a patient with paroxysmal symptoms if a 24-hour collection is done when he or she experiences the crisis.
Plasma and urinary free metanephrines are elevated in most patients with pheochromocytoma,177 although the levels may be slightly elevated in preeclampsia.178 Specific quantitation of epinephrine and norepinephrine may assist in the localization of a lesion. Increased epinephrine excretion in excess of 50 μg/day is suggestive of an adrenal lesion. Excessive secretion of noradrenaline is suspicious, but not diagnostic, of extra-adrenal tumor. False-positive elevations may occur in patients who are taking methyldopa, are undergoing strenuous exercise, have increased intracranial pressure, have hypoglycemia, are undergoing clonidine withdrawal, or are undergoing aminophylline administration. Plasma levels of catecholamines can be measured, particularly during a paroxysmal episode. It is important to adhere strictly to the technique of blood collection. Result fluctuations are common under routine stressful situations, such as exercise, venipuncture, and anxiety. It is recommended that the patient rest in the supine position in a quiet environment for 30 minutes before blood is drawn. Blood is obtained through an indwelling catheter. The plasma should be centrifuged and the serum stored at temperatures lower than 4°C until assayed.
As soon as the diagnosis is confirmed, an imaging study should be performed to localize the lesion; given the contraindication of a CT scan during pregnancy, pregnant women should receive an MRI.177 Genetic testing should be considered if there are suspected features of a familial component to the pheochromocytoma.
Treatment consists of α-adrenergic blocking agents to normalize blood pressure and pulse. The drug most often used is phenoxybenzamine, which is safe in pregnancy179 and can be started at 10 mg/day; the dose can be increased by 10 mg every 2–3 days until blood pressure and symptoms are controlled. Although the dose may need to be adjusted because it has a half-life of about 24 hours and hence is cumulative, the usual dose of phenoxybenzamine is 40–80 mg/day in divided doses. The blocking dose of phenoxybenzamine is 1 mg/kg/day. Prazosin and doxazosin have also been used as selective α1-antagonists in pregnancy.179, 180 In addition, α-adrenergic receptor blockade increases the blood volume and may improve congestive heart failure as well as angina pectoris, if present. A 7–14 day course of an α-adrenergic receptor blockade is the cornerstone of preoperative management,177 β-blockers should not be used without concurrent α-adrenergic receptor blockade treatment given the risk of unopposed α-adrenergic effects by catecholamines, but can be added if tachycardia develops after the administration of an α-adrenergic blocking agent. Hypertensive crises can be treated with intravenous phentolamine.
Surgical intervention is the treatment of choice in cases of pheochromocytoma and should optimally be performed at the beginning of the second trimester. If the diagnosis is not made until the third trimester, surgery should be delayed until the time of delivery if possible. Postpartum resection of the pheochromocytoma is not recommended due to the potential further release of catecholamines induced by uterine fundal massaging.181 There have been some case reports of successful laparascopic adrenal resection of the pheochromocytoma during pregnancy.182 Cesarean section rather than vaginal delivery is strongly recommended as the safest procedure for both mother and fetus.
Virilization
Virilization (Table 8) in pregnancy is exceedingly rare. Several cases have been described with normal fetal outcome. The most common cause is luteoma of pregnancy, a benign human chorionic gonadotropin (hCG)-dependent ovarian tumor that develops during pregnancy in which virilization occurs in approximately 25–35% of such patients. As high levels of testosterone and androstenedione are the etiology of virilizing symptoms in these patients, hirsutism, acne, deepening of the voice, and clitoral enlargement can be seen in the mother, which may begin as early as the second trimester. Fetal virilization is seen in 50–67% of females born to patients whose virilization began during pregnancy.183 Regression with normalization of the elevated androgen levels occurs in the first few weeks of postpartum period.183
Table 8. Causes of fetal and maternal virilization in pregnancy
| Drugs |
| Dilantin |
| Danazol* |
| Progestogens* |
| Stilbestrol* (large doses) |
| Ovarian lesions |
| Arrhenoblastomas* |
| Luteoma of pregnancy* |
| Krukenberg tumor* |
| Mucinous cystadenoma |
| Leydig cell tumor |
| Lipoid cell tumor |
| Granulosa-theca-cell tumor |
| Dermoid cyst |
| Hyperreactio luteinalis |
| Polycystic ovarian syndrome |
| Adrenal lesions |
| Virilizing adenoma* |
| Virilizing carcinoma* |
| Aldosterone producing tumor* |
*Lesions and drugs producing fetal virilization
THYROID DISEASES
Thyroid disorders in pregnancy present a unique opportunity for health care professionals to use a team approach, similar to that of the care of diabetic women during gestation. Obstetricians, endocrinologists, and perinatologists are often called upon to interact in the management of complex thyroid problems. In addition to changes in thyroid function tests, hypermetabolic symptoms often seen in pregnancy present a challenge in the diagnosis of thyroid disease during pregnancy.
Thyroid function and antibody testing in pregnancy
Thyroid hormone concentrations, total thyroxine (TT4) and total triiodothyronine (TT3), increase during pregnancy as the result of both an elevation in thyroxine-binding globulin (TBG) secreted by the liver184 and a reduced peripheral TBG degradation rate. In spite of these changes in total hormone concentration, the serum free fractions of both T4 and T3 remain within normal limits. The serum levels of TSH, produced by the pituitary gland, have a tendency to decrease in the first trimester of pregnancy due to the mild thyroid stimulatory effect of hCG. High levels of hCG, as in cases of hydatidiform mole, hyperemesis gravidarum, and multiple gestation pregnancies, may cause elevations in serum T4 concentrations and even greater suppression of serum TSH concentrations, as is seen in thyrotoxicosis.185
Despite the limitations in the interpretation of serum TSH in the first trimester, it remains the most practical test to screen for thyroid dysfunction. High TSH values are consistent with the diagnosis of primary hypothyroidism, whereas suppressed values are suggestive of hyperthyroidism, with few exceptions (Fig. 3). In the presence of an abnormal serum TSH value, the determination of FT4 or its equivalent, free thyroxine index (FT4I), will help in the assessment of thyroid function. A low TSH value and high concentrations of FT4 or FT4I are diagnostic of hyperthyroidism; if the FT4 or FT4I is normal, a serum FT3 or FT3I determination is obtained. High values are diagnostic of hyperthyroidism, the so-called T3-toxicosis syndrome, which is sometimes seen in patients with an autonomous or “hot” thyroid nodule. Therefore, a few thyroid tests, if properly ordered and interpreted, allow the physician to assess thyroid function in pregnancy.
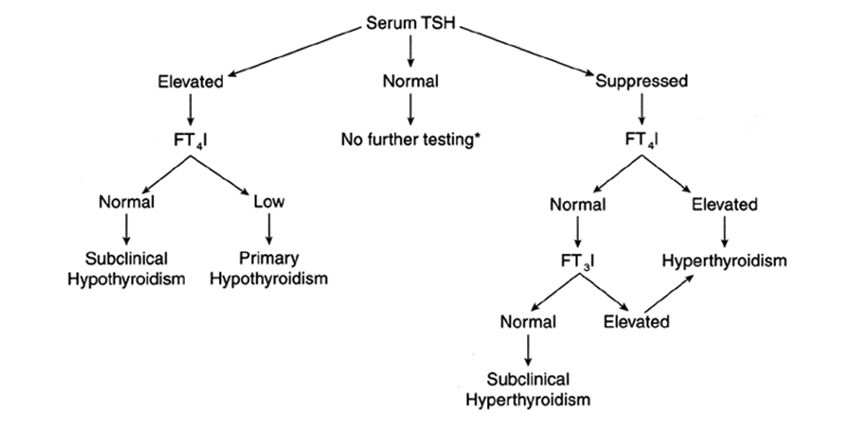 Fig. 3. Algorithm for the diagnosis of thyroid diseases. If there is clinical suspicion of secondary hypothyroidism, a determination of FT4 I is indicated. In this situation, the serum TSH is normal in the presence of low FT4 I. FT4 I, free thyroxine index or its equivalent, free thyroxine; FT3 I, free triiodothyronine index or its equivalent, free triiodothyronine; TSH, thyroid-stimulating hormone.
Fig. 3. Algorithm for the diagnosis of thyroid diseases. If there is clinical suspicion of secondary hypothyroidism, a determination of FT4 I is indicated. In this situation, the serum TSH is normal in the presence of low FT4 I. FT4 I, free thyroxine index or its equivalent, free thyroxine; FT3 I, free triiodothyronine index or its equivalent, free triiodothyronine; TSH, thyroid-stimulating hormone.
In geographical areas of iodine deficiency, goiter is commonly seen in pregnancy. However, in the United States and other areas of the world with sufficient iodine intake, the thyroid gland does not clinically increase in size during pregnancy. Therefore, the detection of a goiter in pregnancy is an abnormal finding that requires careful evaluation. One of the most common causes of diffuse goiter is chronic autoimmune thyroiditis or Hashimoto's thyroiditis. Most patients are euthyroid, and the diagnosis is made by the determination of thyroid antibodies (antimicrosomal or anti-thyroid peroxidase [anti-TPO]). Antibody concentration decreases during pregnancy and increases in the postpartum period. High values in the first trimester of pregnancy are predictors of the syndrome of postpartum thyroid dysfunction.
In some patients with a history of autoimmune thyroid disease, whether it is active or inactive, the determination of TSH receptor antibodies is indicated to assess the possibility of fetal or neonatal hyperthyroidism. These antibodies, mainly of the immunoglobulin G (IgG) class, have different functional activities, mainly stimulating or blocking antibodies on the TSH receptor of the thyroid gland. When high titers are present in the serum of pregnant women, they cross the placenta and may affect fetal thyroid function.186 Thyroid-stimulating immunoglobulin (TSI), formerly known as long-acting thyroid-stimulating antibody, may cause fetal or neonatal hyperthyroidism.187 Thyroid-blocking antibodies present in women with chronic thyroiditis, particularly those with atrophic thyroiditis, may produce neonatal hypothyroidism, a condition that is transient because of the short half-life of these antibodies.188
Titers of these antibodies decrease with progression of the pregnancy. This could explain, in the case of Graves' disease, the decrease in antithyroid drug requirement in many cases of hyperthyroidism in the second half of pregnancy. A TSH receptor antibody value three times greater than normal is considered predictive of neonatal or fetal hyperthyroidism.189 The test is relatively expensive and should be ordered only in very special circumstances (Table 10). The American Thyroid Association guidelines recommend ordering the test between 24 and 28 weeks' gestation because of the gradual decrease in titer concentration with progression of pregnancy.189
Table 10. Indications for serum thyrotropin receptor antibody determination in pregnant women
| Graves' disease |
| Fetal or neonatal hyperthyroidism in previous pregnancies |
| Hyperthyroidism, on treatment with antithyroid drugs |
Postablative euthyroidism in the presence of: |
| Chronic thyroiditis without goiter |
| Incidental fetal goiter on ultrasound |
| Infant born with congenital hypothyroidism |
Maternal–placental–fetal interactions
From animal and human studies, there is placental passage of small amounts of thyroid hormones from mother to fetus in the first few weeks of pregnancy, confirming an important role of thyroid hormones in embryogenesis.190 Maternal TSH does not cross the placenta, but TRH does and exerts a stimulatory effect on the fetal pituitary.191
Methimazole (MMI) and propylthiouracil (PTU), drugs used for the treatment of hyperthyroidism, also cross the placenta. If given in large doses, they may produce fetal goiter and hypothyroidism.192 Supraphysiologic iodine therapy is contraindicated in pregnancy because iodine is accumulated by the fetal thyroid and can potentially induce goiter and hypothyroidism.193
Thyrotoxicosis
The prevalence of thyrotoxicosis diagnosed in pregnancy is approximately 4.7 per 1000.194 In most cases, the etiology is Graves' disease; other causes are much less common (Table 11). In the authors' experience, single toxic adenoma and multinodular toxic goiter are seen in less than 10% of cases, and subacute thyroiditis is rarely seen during gestation. Increased secretion of hCG may also cause biochemical thyrotoxicosis, as in cases of hydatidiform mole and hyperemesis gravidarum.195
Table 11. Causes of hyperthyroidism in pregnancy
| Graves' disease* |
| Multinodular toxic goiter |
| Toxic adenoma |
| Subacute thyroiditis |
| Iatrogenic hyperthyroidism |
| Hydatidiform mole |
| Hyperemesis gravidarum |
*Accounts for 85–90% of all cases
Graves' disease
Graves' disease is the most common cause of hyperthyroidism in pregnancy and accounts for 75% of all cases.196 The natural course of the disease is characterized by an exacerbation of symptoms in the first trimester and an amelioration of symptoms in the second half of pregnancy; the disease may flare during the postpartum period. When Graves' disease is properly treated, the outcome for mother and fetus is good; however, untreated or poorly controlled disease may be catastrophic for both the woman and the neonate.197
In most patients whose diagnosis is made for the first time during pregnancy, symptoms antedate conception. The clinical diagnosis of thyrotoxicosis may be difficult during gestation because many hypermetabolic symptoms are commonly seen in normal pregnancy, such as palpitations, heat intolerance, and warm skin. There are a few important clues to guide the physician toward the diagnosis of hyperthyroidism: presence of goiter, ophthalmopathy, proximal muscle weakness, tachycardia, and weight loss or an inability to gain weight in spite of a good appetite.198 Occasionally the patient is seen for the first time in congestive heart failure. In patients with toxemia, the physician should suspect hyperthyroidism when there is systolic hypertension and an inappropriately low diastolic blood pressure (i.e. wide pulse pressure).
Classical symptoms of hyperthyroidism include nervousness, increased sweating, increased appetite, heat intolerance, insomnia, proximal muscle weakness, irritability, changes in personality, frequent bowel movements, a decreased tolerance to exercise (sometimes manifested as shortness of breath), pruritus, and weight loss. Not all symptoms are present in a given patient; the physician should be aware of subtle complaints, particularly in the presence of weight loss or the inability to gain weight. Differentiating Graves' disease from transient hyperthyroidism of pregnancy in the first trimester of pregnancy may be difficult.198
On physical examination, the thyroid gland is enlarged in almost every patient with Graves' disease. Indeed, the absence of a goiter makes the diagnosis very unlikely. The gland is diffusely enlarged (between two and six times its normal size) and varies from soft to firm; sometimes it is irregular to palpation, with one lobe more prominent than the other one. A thrill may be felt or a bruit may be heard; these are indications of hyperdynamic circulation. Examination of the eyes may reveal obvious ophthalmopathy, but in most cases exophthalmos is absent or mild, with one eye slightly more prominent than the other one. Stare is common, as well as injection or edema of the conjunctiva. Pretibial myxedema is rare; it is seen in less than 10% of women. A hyperdynamic heart and a loud systolic murmur are common findings. Proximal muscle weakness, fine tremor, and hyperkinetic symptoms are seen often. The skin is warm and moist, and palmar erythema is accentuated.
Cardiovascular manifestations include left ventricular dysfunction. Although these changes are reversible, they may persist for several weeks after euthyroidism is achieved. In one study, a reduction in peripheral vascular resistance and a high cardiac output were still present despite normalization of thyroxine levels.199 This is an important finding with significant clinical implications. Left ventricular decompensation in hyperthyroid pregnant women occurs in the presence of superimposed preeclampsia, at the time of delivery, or with intercurrent complications such as anemia or infection. Careful monitoring of fluid administration is imperative in these situations. Thyroid crisis or storm is rarely reported in pregnancy or the postpartum period.200
Biochemical diagnosis is the mainstay in cases of suspected hyperthyroidism. FT4 determination or calculation of the FT4I (using TT4 levels and a test for assessment of thyroxine-binding globulin, such as resin T3 uptake [RT3U]) are standard tests in most clinical laboratories. Almost every patient with Graves' disease has an elevated FT4 concentration. Serum TSH should be measured with a sensitive assay; this should be ordered at the time of the free T4 determination. A suppressed TSH value in the presence of a high free T4 or FT4 index confirms the diagnosis of hyperthyroidism.It must be kept in mind, however, that a suppressed serum TSH level is present in some pregnant women in the first trimester of pregnancy, and that trimester-specific reference ranges should be used.201, 202 In some unusual situations, the serum FT4 is in the upper limit of normal or is slightly elevated, in which case the determination of total T3 will confirm the diagnosis of hyperthyroidism.202 Thyroid peroxidase antibodies (antithyroid peroxidase) or thyroid antimicrosomal antibodies, markers of thyroid autoimmune disease, are elevated in most patients with Graves' disease. Determination of these antibodies is indicated for patients in whom the etiology of hyperthyroidism is in doubt.
Treatment of hyperthyroidism is essential to prevent maternal, fetal, and neonatal complications. The goal of treatment is to normalize the thyroid tests with the minimum amount of antithyroid medication.203 As antithyroid medications crossing the placenta may affect fetal thyroid function, patients should be monitored at regular intervals, and the amount of medication should be adjusted to keep the FT4 in the upper limits of the normal range. In patients with small goiters and a short duration of symptoms who are able to maintain euthyroidism on minimal amounts of antithyroid medication, the drug can be discontinued during the last few weeks of pregnancy. In the United States, the two antithyroid drugs available are propylthiouracil and methimazole. Although both drugs are effective in controlling the disease, propylthiouracil is recommended for treatment during the first trimester and methimazole for the second and third trimesters, due to the concerns of the rare risk of congenital malformations associated more so with methimazole and maternal hepatotoxicity with propylthiouracil.202
Resistance to drug therapy is very unusual; when it occurs, it is most likely due to poor compliance.204 Side effects of antithyroid drugs occur in 3–5% of treated patients.205 Patients should be advised to discontinue medication and contact their physician in case of pruritus or skin rash. This is the most common complication and resolves by switching to the other antithyroid drug; in general, this occurs 2–6 weeks after initiation of therapy, but in one case was reported after 11 years of use.206 Other much less common side effects include migratory polyarthritis, lupus-like syndrome, and cholestatic jaundice.
Agranulocytosis, a serious but unusual complication, has been reported in 0.35–0.37% of patients receiving an antithyroid drug.207 It is manifested by a high fever, malaise, and severe sore throat and usually occurs in the first 3 months of therapy. Patients should be made aware of the symptoms, and a leukocyte count should be immediately obtained in such scenarios. Checking a routine blood count in patients on antithyroid therapy is generally not needed. At the time of the diagnosis, β-adrenergic blocking agents may also be added for up to a week if symptoms of hyperthyroidism are very severe.
Subtotal thyroidectomy in pregnancy is effective in controlling the disease, but the indications for surgical treatment are few: allergy to both antithyroid drugs; some cases of very large goiters; and the exceptional case of resistance to drug therapy. Noncompliance could be another indication, although it does not solve the problem of noncompliance. Therapy with 131I is contraindicated in pregnancy because it produces fetal hypothyroidism when given after 10 weeks' gestation.208 A pregnancy test is mandatory in any woman of childbearing age before a therapeutic dose of 131I is administered.
Iodine crosses the placenta and in large doses, may produce fetal goiter and hypothyroidism.193, 209, 210 Although it has been used in some case reports of pregnant women with either mild hyperthyroidism211 or intolerance to antithyroid therapy,212 its use is contraindicated in pregnancy. Excessive amounts of antithyroid drug may also induce fetal hypothyroidism and goiter. The diagnosis of goiter has been made by ultrasonography, which can show hyperextension of the neck. If needed and the benefits outweigh the risk of this test, fetal hypothyroidism can be confirmed by measuring thyroxine and TSH in fetal blood obtained by cordocentesis.213, 214 Treatment with intra-amniotic injection of levothyroxine is able to result in resolution of the fetal goiter.
Following delivery, breastfeeding should be permitted if the daily dose of antithyroid drug is low194 (i.e. less than 300 mg/day of PTU or less than 20–30 mg/day of methimazole); methimazole is the preferred drug during breastfeeding given the concerns of hepatotoxicity with propylthiouracil.189 It is prudent to give the total dose in divided doses after each feeding, and the infant should be followed closely with thyroid function tests.
There may be associated risks of maternal and perinatal morbidity and mortality associated with hyperthyroidism in pregnancy, including fetal loss and preterm birth.215, 216 One study reported a higher risk of pregnancy-induced hypertension and preeclampsia,217 among untreated hyperthyroid pregnant women, compared to pregnant women with normal thyroid function. Mannisto et al. reported in a large retrospective cohort study that maternal hyperthyroidism was associated with neonatal sepsis, respiratory distress syndrome, transient tachypnea, and apnea.215 In a large national cohort registry study, Ohrling and colleagues described lower birth weights and smaller head circumference among neonates born to women with Graves’ disease or toxic nodular goiter treated with radioiodine or surgery pre-pregnancy, compared to women who had thyroid surgery for nontoxic goiters, thus suggesting that hyperthyroidism even prior to pregnancy may have effects of subsequent birth characteristics.218 length, and head circumference in children borne by women years after treatment for hyperthyroidism) Additionally, it has been hypothesized that maternal thyroid autoimmunity may independently play a role in adverse pregnancy outcomes.219, 220 A fetal well-being assessment with ultrasonography, a nonstress test, and a biophysical profile is indicated in women with poor metabolic thyroid dysfunction or when fetal tachycardia and/or intrauterine growth retardation is noted.
Transient hyperthyroidism of hyperemesis gravidarum
The presentation of transient hyperthyroidism of hyperemesis gravidarum (THHG) is characterized by severe nausea and vomiting requiring hospitalization (sometimes repeated hospitalizations) for intravenous hydration. Weight loss of at least 5 kg, ketonuria, and hypokalemia are common findings and thyroid function tests are in the hyperthyroid range. FT4 levels are elevated at sometimes two to three times the normal values. FT3 is also elevated, but not to the extent that FT4 may be. Serum TSH measured by a sensitive assay is consistently suppressed. The degree of thyroid function abnormality is directly related to the severity of vomiting and weight loss. In the study by Goodwin and colleagues, 67 patients with hyperemesis gravidarum were evaluated.221 In those with severe vomiting, weight loss of at least 5 kg, and significant dehydration, liver and electrolyte function abnormalities were often found. These patients had significant elevations in FT4 levels with suppression of serum TSH values. Those with lesser degrees of hyperemesis had less severe thyroid function abnormalities.
In spite of the significant biochemical hyperthyroidism, signs and symptoms of hypermetabolism are mild or absent. Patients may complain of mild palpitations, but heat intolerance, perspiration, and proximal muscle weakness are extremely rare. On physical examination, ophthalmopathy and goiter are absent, a mild tremor of the outstretched fingers is occasionally seen, and tachycardia may be present, mainly a result of dehydration. Significant in the medical history is the lack of hyperthyroid symptoms before conception, as patients with Graves' disease diagnosed for the first time during gestation should be symptomatic prior to the pregnancy. THHG should also be suspected when thyroid antibodies are negative.
Normalization of hyperthyroxinemia parallels the improvement in vomiting and weight gain, with most cases resolving spontaneously in 2–10 weeks. However, suppressed serum TSH may last for a few additional weeks (Fig. 4). No antithyroid medication is needed; furthermore, because of the severity of the vomiting, drug therapy is poorly tolerated. In most cases, total resolution occurred by midgestation, although in some series, persistence of hyperthyroidism beyond 20 weeks' gestation was reported in 15–25% of cases.222, 223 Occasionally, severe vomiting and hyperthyroidism require parenteral nutrition.
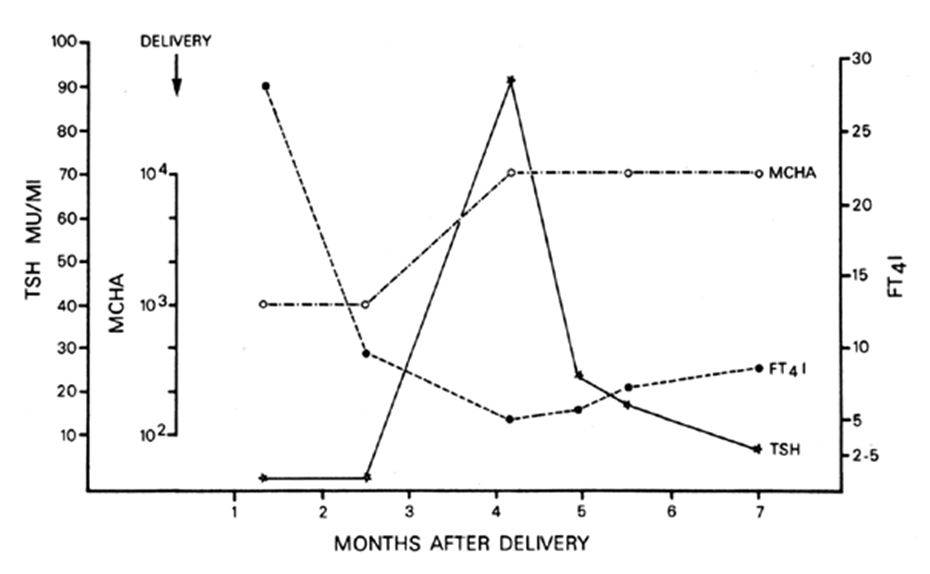
Fig. 4. Transient hyperthyroidism and hypothyroidism in the postpartum period in a patient with chronic autoimmune thyroiditis with spontaneous recovery.224 TSH, thyroid stimulating hormone; MCHA, microsomal hemagglutination antibodies; FT4 I, free thyroxine index.
The cause of biochemical hyperthyroidism in patients with hyperemesis gravidarum is related to high levels of hCG, a known stimulator of the TSH receptor. There is a significant, albeit weak, correlation between the degree of thyroid stimulation and total hCG levels in normal and hyperemetic women.221 It seems likely, however, that certain hCG fractions may be more important than the total hCG as thyroid stimulators. The thyroid-stimulating activity of early pregnancy and of molar gestations correlates best with the percentage of basic, partially desialylated hCG in serum.225
Fetal and neonatal hyperthyroidism
In mothers with a history of Graves' disease who were previously treated with thyroid surgery and/or radioiodine ablation therapy, concentrations of thyroid stimulating antibodies may remain elevated in spite of maternal euthyroidism. As such, fetal/neonatal Graves’ disease can still result even years later from placental passage of the maternal antibodies.226 A high titer of maternal TSH receptor antibody is a predictor of neonatal hyperthyroidism.227 Fetal hyperthyroidism is characterized by fetal tachycardia and intrauterine growth retardation. The diagnosis can be confirmed by measuring thyroid hormone levels in cord blood obtained by cordocentesis. Treatment consists of antithyroid medication given to the mother. Fetal tachycardia resolution and normal fetal growth are indications of a good therapeutic response. Because the half-life of the thyroid receptor antibody is only a few weeks, complete resolution of neonatal hyperthyroidism is expected following delivery.
Neonatal hyperthyroidism is rare and occurs in less than 1% of infants born to mothers with Graves' disease.228 If the mother with Graves’ disease is being treated with antithyroid medication during pregnancy, these medications pass through the placenta, and thus, the fetus should be adequately treated. However, as the protective effect of the antithyroid drug is lost after delivery, there is the potential for the neonate to develop clinical hyperthyroidism a few days after birth. If neonatal hyperthyroidism is not recognized and treated properly, neonatal mortality could be as high as 25%.229
Hypothyroidism
The prevalence of overt hypothyroidism in pregnancy is approximately 0.1–0.2% in the US,230 and likely higher in iodine-deficient regions, given the importance of adequate iodine nutrition required for normal thyroid hormone production. Subclinical hypothyroidism is more often encountered and has a prevalence which ranges up to 2.5%,189 of which many cases resolve following delivery.231
The spectrum of women with hypothyroidism in pregnancy includes the following:
- Women diagnosed with newly diagnosed overt or subclinical hypothyroidism during pregnancy
- Women who discontinue thyroid hormone therapy at the time of conception because of poor medical advice or because of the misconception that thyroid medication adversely affects the fetus
- Women on pre-existing thyroid hormone therapy who require but fail to receive larger doses during pregnancy
Most patients with subclinical hypothyroidism are asymptomatic. Patients with overt hypothyroidism may complain of tiredness, cold intolerance, fatigue, muscle cramps, constipation, and deepening of the voice. On physical examination the skin is dry and cold, deep tendon reflexes are delayed, bradycardia may be detected, and there may be periorbital edema.
Hypothyroidism in pregnancy has been associated with adverse obstetric and neonatal complications. In a retrospective study of over 9000 women, those with a serum TSH ≥6 mIU/L had an almost 4-fold higher risk of miscarriage, compared to those with a serum TSH <6 mIU/L.232 In a large retrospective cohort of 223,512 US women, maternal hypothyroidism was associated with preeclampsia, gestational diabetes, preterm birth, induction, cesarean section, ICU admission, placental abruption, and breech presentation.233 Casey et al. demonstrated that a maternal serum TSH greater than the 97.5th centile in a cohort of over 25,000 women was associated with increased risks of placental abruption (RR 3.0) and preterm delivery (RR 1.8), suggesting that even subclinical hypothyroidism can have some deleterious effects.234 Others have reported increased risks of gestational hypertension (i.e. eclampsia, preeclampsia, and pregnancy-induced hypertension) with insufficient maternal thyroid hormone levels.235, 236 Treatment with thyroid hormone replacement therapy decreases the proportion of women with miscarriages and increases the proportion of women with term deliveries.237 Other studies have not demonstrated a relationship between maternal hypothyroidism during pregnancy and adverse obstetric outcomes.238, 239, 240, 241
Adequate thyroid hormone for the developing fetus is crucial for normal neurodevelopment of the fetus throughout gestation and in the first few years after birth,242 and several investigations have demonstrated adverse neonatal outcomes associated with hypothyroidism during pregnancy. Maternal hypothyroidism has been associated with decreased IQ,243, 244 verbal and nonverbal cognitive delays,245 and attention deficit/hyperactivity disorder (ADHD).246
The diagnosis of hypothyroidism is confirmed by the determination of serum TSH using trimester-specific laboratory reference ranges or the following upper limits if unavailable: 2.5 mIU/L during the first trimester and 3.0 mIU/L for the second and third trimesters.189, 202 If the TSH is abnormal, a FT4 concentration or FT4I would be needed to differentiate between overt and subclinical hypothyroidism. In patients with overt hypothyroidism, the FT4 is low and the serum TSH is elevated, whereas in patients with subclinical hypothyroidism the FT4 is normal and the serum TSH is elevated. The degree of biochemical thyroid abnormality varies with the severity of the clinical symptoms; however, there is not always a good correlation between clinical and chemical parameters. Serum thyroid antibodies (thyroid peroxidase antibodies) are elevated in patients with autoimmune thyroiditis and are associated with an increased risk of hypothyroidism during pregnancy.247
Levothyroxine is mainstay of the treatment of hypothyroidism. In view of the complications mentioned above, it is important to normalize the thyroid function tests as soon as possible. Hypothyroid women on thyroid hormone replacement therapy require an increase in the dose of levothyroxine after conception to meet the demands of the developing fetus.248 Patients on thyroid therapy before conception should have their TSH checked on the first visit and the amount of levothyroxine adjusted accordingly, which is usually an increase of 2 tablets weekly.249 The serum TSH should be repeated every 4 weeks during the first half of pregnancy and once again between 26 and 32 weeks' gestation.189 Immediately after delivery, they should return to their prepregnancy dosage.
In patients with severe hypothyroidism, there may be a delay in the normalization of serum TSH, but normal serum FT4 values are achieved in the first 2 weeks of levothyroxine therapy. Higher doses may be required in patients who have had total thyroidectomy for thyroid carcinoma, because the goal in these cases is suppression of serum TSH.250 Finally, it should be noted that certain common medication/supplements taken during pregnancy, including iron and calcium, when given simultaneously with levothyroxine, may reduce the efficacy of thyroid hormone, and thus these should be ingested 1–2 hours prior to the administration of levothyroxine.251
Finally, adequate maternal iodine nutrition is recommended for to optimize normal thyroid function during the perinatal period. The American Thyroid Association,189 Endocrine Society,202 Teratology Society,252 American Association of Clinical Endocrinologists,251 the International Council for the Control of Iodine Deficiency Disorders Global Network,253 and the American Academy of Pediatrics254 all recommend the supplementation of 150 µg of iodine daily during preconception, pregnancy, and lactation. It is important to note that excess iodine (i.e. >1100 µg daily) can also induce thyroid dysfunction and should be avoided.254
Thyroid nodule
The prevalence of thyroid nodules is approximately 5% among women living in iodine-sufficient regions, of which 5–15% will be malignant based on such risk factors as previous radiation therapy to the head and neck, age, gender, and family history of thyroid cancer.250 Approximately 10% of women are diagnosed during pregnancy or the early postpartum period.255
Pregnancy does not appear to be a risk factor for more aggressive disease. A US study of 61 pregnant women (77% of whom received thyroidectomy after delivery) and 528 age- and gender-matched controls reported no difference in the prognosis of differentiated thyroid cancer between the two groups.256 However, one recent Italian study reported the diagnosis of differentiated thyroid cancer during pregnancy and up to 2 years postpartum was associated with a higher risks of tumor persistence/recurrence, compared to women who were diagnosed more than 2 years postpartum.257
Careful examination of the neck enables the physician to define and characterize the lesion. In addition to the size of the nodule, consistency, tenderness, fixation to the skin, and presence of metastasis should be noted. A hard, painless nodule measuring more than 2 cm in diameter is suspicious of malignancy. The following approach is that recommended at the LA-USC Medical Center (Los Angeles):
1. A suppressed serum TSH may indicate the presence of an autonomous nodule, which rarely is malignant. In such a case, thyroid function tests are performed to rule out hyperthyroidism. It should be kept in mind that serum TSH is suppressed in a minority of normal first-trimester pregnancies.
2. If the serum TSH is within normal limits, the next step is thyroid ultrasonography, which will distinguish a solid from a cystic lesion.
3. In the presence of a solid lesion less than 2 cm or a cystic lesion less than 4 cm in diameter, observation with or without thyroxine suppression therapy is recommended. If there is an increase in the size of the lesion or if cervical adenopathy is present, a fine needle aspiration biopsy (FNAB) of the lesion is indicated.
4. If ultrasound shows a solid or mixed lesion greater than 2 cm or a cystic nodule greater than 4 cm, the diagnostic approach differs according to gestational age.
5. Before 20 weeks' gestation, a FNAB is performed. The most important component of the FNAB is the cytopathologist's interpretation. A specific diagnosis is required: malignant, benign, follicular lesion, or inadequate specimen. In the latter, a repeat FNAB is recommended.
6. If the lesion is malignant, surgery is indicated. For follicular lesions, the decision about surgery is a personal one, because there is a 15–20% chance that the lesion is malignant. A similar approach is used with Hürthle cell lesions.
7. For lesions diagnosed after 24 weeks' gestation, the FNAB may be postponed until after delivery, unless there is a strong suspicion of malignancy. Suppressive therapy with thyroxine to prevent further growth of the lesion, although believed to be controversial by some, is the recommendation of the author's group. If there is further growth of the lesion in spite of suppression therapy, FNAB is recommended.
8. For lesions diagnosed between 20 and 24 weeks of gestation, the decision to wait until after delivery or to complete the workup is made by the patient and her physician. The patient's anxiety and fear about having a potentially malignant lesion should be considered when the advice is given. Most malignant lesions of the thyroid gland are slow growing, and the long-term prognosis is good in most patients.
During pregnancy, the American Thyroid Association recommends that a suspicious thyroid nodule undergo FNAB upon detection, and if indicated, thyroidectomy should be performed during the second trimester or following delivery.250 Women with persistently suppressed serum TSH concentrations (i.e. determined to not be transient hyperthyroidism of hyperemesis gravidarum) should have management deferred until after pregnancy and lactation in order to receive a radioactive uptake and scan prior to consideration of a thyroid nodule FNAB.
Postpartum thyroid dysfunction
Thyroid dysfunction (hyperthyroidism and hypothyroidism) is recognized with increasing frequency in the postpartum period and usually manifests within 12 months of delivery. It occurs in up to 18.2% of women without preexisting autoimmune disease.258 There are some features that may predict the development of postpartum thyroiditis: the presence of goiter, high titers of thyroid antibodies in the first half of pregnancy, episodes of postpartum thyroiditis in previous pregnancies, and personal or family history of autoimmune thyroid or other disease.Women with type 1 diabetes mellitus are at increased risk of developing postpartum thyroiditis.
Most of the cases are due to intrinsic thyroid disease, and a few cases are due to hypothalamic or pituitary lesions (Table 12). The differential diagnosis of patients who appear to be presenting with postpartum thyroiditis is important because the treatment is different. If not contraindicated because of breastfeeding, a thyroid radioactive iodine uptake is helpful. It will be decreased in patients with postpartum thyroiditis, and elevated in patients with recurrent hyperthyroidism due to Graves' disease. When it is due to recurrent Graves' disease, treatment with antithyroid medications is indicated, or the physician may advise ablation therapy or thyroid surgery. Hypothyroidism may also be secondary to hypothalamic or pituitary lesions, such as those seen in Sheehan's disease or lymphocytic hypophysitis. In such cases, there may also be clinical symptoms related to the deficiencies of other pituitary hormones present.
Table 12. Etiologies of postpartum thyroid dysfunction
| Chronic thyroiditis: Transient hyperthyroidism (RAIU will be decreased) Transient hypothyroidism Permanent hypothyroidism |
| Graves' disease |
| Exacerbation of hyperthyroidism (RAIU will be elevated) |
| Transient hyperthyroidism of chronic thyroiditis (RAIU will be decreased) |
| Hypothalamic–pituitary disease |
| Sheehan's syndrome |
| Lymphocytic hypophysitis |
RAIU, radioactive iodine uptake
The clinical diagnosis is not always clear, and the clinician should be concerned about nonspecific symptoms such as fatigue, depression, palpitations, and irritability in women after delivery. In about half of the cases, mild symptoms develop between 1 and 4 months postpartum. On physical examination, a goiter is felt in most cases; it is firm and nontender to palpation, and tachycardia may be detected. The goiter may be discovered for the first time, or the patient may notice an increase in the size of a previously diagnosed goiter. Serum thyroid function tests are in the hyperthyroid range, and thyroid antibody (antiperoxidase thyroid antibody or antimicrosomal antibody) titers are elevated. This is followed in a few months by hypothyroidism with spontaneous recovery a few months later. The antibody titer has a tendency to increase during this process, and in most cases, the size of the goiter increases. Other patients have a different cause of postpartum thyroiditis, characterized by an initial episode of hypothyroidism between 3 and 7 months postpartum without the initial hyperthyroid phase. In others the initial episode of hyperthyroidism is followed by a return to normal thyroid function.
Because most cases of postpartum thyroid dysfunction recover spontaneously, treatment is indicated mostly for symptomatic patients. In the presence of hyperthyroid symptoms, β-adrenergic blocking drugs (propranolol 20–40 mg every 6 hours or atenolol 25–50 mg every 24 hours) are effective in controlling the symptoms. Antithyroidal medications are not indicated, as the underlying etiology in postpartum thyroiditis is not increased thyroid hormone production, but rather leakage of preformed thyroid hormone stores. For hypothyroid symptoms, small amounts of levothyroxine (0.050 mg/day) will control symptoms and allow for a spontaneous recovery of thyroid function after discontinuation of the drug.
Postpartum thyroiditis is a predictor of permanent hypothyroidism.259 From several studies, there is a wide range (4–54%) of patients with postpartum thyroiditis who develop permanent hypothyroidism.258 The risks of recurrent postpartum thyroid dysfunction in a future pregnancy can be as high as 70%.260
Funding
This work was supported by NIH K23HD068552 (AML).
REFERENCES
Barber SG. Hypopituitarism and artificial ventilation. Acta Endocrinologica. 1979;90(2):211. |
|
Singer PA, Mestman JH, Manning PR, Nicoloff JT. Hypothalamic-hypothyroidism secondary to Sheehan's syndrome. The Western journal of medicine. 1974;120(5):416. |
|
Cavarzere P, Biban P, Gaudino R, Perlini S, Sartore L, Chini L, et al. Diagnostic pitfalls in the assessment of congenital hypopituitarism. Journal of endocrinological investigation. 2014. |
|
Kelestimur F. Sheehan's syndrome. Pituitary. 2003;6(4):181. |
|
Sheehan SL. Postpartum necrosis of the anterior pituitary. 1937;45:189. |
|
Leyer C, Castinetti F, Morange I, Gueydan M, Oliver C, Conte-Devolx B, et al. A conservative management is preferable in milder forms of pituitary tumor apoplexy. Journal of endocrinological investigation. 2011;34(7):502. |
|
Laway BA, Mir SA, Zargar AH. Recovery of prolactin function following spontaneous pregnancy in a woman with Sheehan's syndrome. Indian J Endocrinol Metab. 2013;17(Suppl 3):S696-9. |
|
Huang X, Liang J, Huang Y, Huang J. Complete hydatidiform mole and a coexistent fetus following ovulation induction in a patient with Sheehan's syndrome: a first case report and review of literature. Arch Gynecol Obstet. 2014;289(5):1145-50. |
|
Al-Sharafi BA, Nassar OH. Successful pregnancy in a female with a large prolactinoma after pituitary tumor apoplexy. Case Rep Obstet Gynecol. 2013;2013:817603. |
|
Khoo CM, Lee KO. Endocrine emergencies in pregnancy. Best Pract Res Clin Obstet Gynaecol. 2013;27(6):885-91. |
|
Piantanida E, Gallo D, Lombardi V, Tanda ML, Lai A, Ghezzi F, et al. Pituitary apoplexy during pregnancy: a rare, but dangerous headache. Journal of endocrinological investigation. 2014. |
|
Guo Q, Lu J, Mu Y, Chen K, Pan C. Adult idiopathic isolated ACTH deficiency: a short series and literature review. Neuro Endocrinol Lett. 2013;34(7):693-700. |
|
Pham LL, Garot C, Brue T, Brauner R. Clinical, biological and genetic analysis of 8 cases of congenital isolated adrenocorticotrophic hormone (ACTH) deficiency. PLoS One. 2011;6(10):e26516. |
|
Naha K, Vivek G, Dasari S, Prabhu M. Ketosis-prone type 2 diabetes mellitus in a patient with Sheehan's syndrome: a rare convergence of two distinct endocrine entities. BMJ Case Rep. 2012;2012. |
|
Schalch DS, Burday SZ. Antepartum pituitary insufficiency in diabetes mellitus. Ann Intern Med. 1971;74(3):357-60. |
|
Lupi I, Raffaelli V, Di Cianni G, Caturegli P, Manetti L, Ciccarone AM, et al. Pituitary autoimmunity in patients with diabetes mellitus and other endocrine disorders. J Endocrinol Invest. 2013;36(2):127-31. |
|
Rivera JA. Lymphocytic hypophysitis: disease spectrum and approach to diagnosis and therapy. Pituitary. 2006;9(1):35-45. |
|
Laway BA, Mir SA. Pregnancy and pituitary disorders: Challenges in diagnosis and management. Indian J Endocrinol Metab. 2013;17(6):996-1004. |
|
SHEEHAN HL. Post-partum necrosis of the anterior pituitary. Ir J Med Sci. 1948(270):241-55. |
|
Rumana M, Kirmani A, Khursheed N, Besina S, Khalil M. Lymphocytic hypophysitis with normal pituitary function mimicking a pituitary adenoma: a case report and review of literature. Clin Neuropathol. 2010;29(1):26-31. |
|
Mayfield RK, Levine JH, Gordon L, Powers J, Galbraith RM, Rawe SE. Lymphoid adenohypophysitis presenting as a pituitary tumor. Am J Med. 1980;69(4):619-23 |
|
Merker E, Futterweit W. Postpartum amenorrhea, diabetes insipidus and galactorrhea. Report of a case and review of the literature. Am J Med. 1974;56(4):554-8. |
|
Karaca Z, Kelestimur F. Pregnancy and other pituitary disorders (including GH deficiency). Best Pract Res Clin Endocrinol Metab. 2011;25(6):897-910. |
|
Leiba S, Schindel B, Weinstein R, Lidor I, Friedman S, Matz S. Spontaneous postpartum regression of pituitary mass with return of function. JAMA. 1986;255(2):230-2. |
|
Stacpoole PW, Kandell TW, Fisher WR. Primary empty sella, hyperprolactinemia, and isolated ACTH deficiency after postpartum hemorrhage. Am J Med. 1983;74(5):905-8. |
|
Ng WH, Gonzales M, Kaye AH. Lymphocytic hypophysitis. J Clin Neurosci. 2003;10(4):409-13. |
|
Caturegli P, Lupi I, Landek-Salgado M, Kimura H, Rose NR. Pituitary autoimmunity: 30 years later. Autoimmun Rev. 2008;7(8):631-7. |
|
NELSON DH, TANNEY H, MESTMAN G, GIESCHEN VW, WILSON LD. Potentiation of the biologic effect of administered cortisol by estrogen treatment. J Clin Endocrinol Metab. 1963;23:261-5. |
|
Cassar J, Verco CJ, Joplin GF. Successful pregnancy induced by human menopausal gonadotrophin in a patient with growth hormone deficiency and primary amenorrhoea: case report. Br J Obstet Gynaecol. 1980;87(4):337-9. |
|
Tyson JE, Barnes AC, Merimee TJ, McKusick VA. Isolated growth hormone deficiency: studies in pregnancy. J Clin Endocrinol Metab. 1970;31(2):147-52. |
|
Molitch ME. Prolactinoma in pregnancy. Best Pract Res Clin Endocrinol Metab. 2011;25(6):885-96. |
|
Babey M, Sahli R, Vajtai I, Andres RH, Seiler RW. Pituitary surgery for small prolactinomas as an alternative to treatment with dopamine agonists. Pituitary. 2011;14(3):222-30. |
|
Bronstein MD. Prolactinomas and pregnancy. Pituitary. 2005;8(1):31-8. |
|
Cho KR, Jo KI, Shin HJ. Bromocriptine therapy for the treatment of invasive prolactinoma: the single institute experience. Brain Tumor Res Treat. 2013;1(2):71-7. |
|
Domingue ME, Devuyst F, Alexopoulou O, Corvilain B, Maiter D. Outcome of prolactinoma after pregnancy and lactation: a study on 73 patients. Clin Endocrinol (Oxf). 2014;80(5):642-8. |
|
Witek P, Zieliński G. Management of prolactinomas during pregnancy. Minerva Endocrinol. 2013;38(4):351-63. |
|
Christin-Maître S, Delemer B, Touraine P, Young J. Prolactinoma and estrogens: pregnancy, contraception and hormonal replacement therapy. Ann Endocrinol (Paris). 2007;68(2-3):106-12. |
|
Glezer A, Bronstein MD. Prolactinomas, cabergoline, and pregnancy. Endocrine. 2014;47(1):64-9. |
|
Darwish AM, Farah E, Gadallah WA, Mohammad II. Superiority of newly developed vaginal suppositories over vaginal use of commercial bromocriptine tablets: a randomized controlled clinical trial. Reprod Sci. 2007;14(3):280-5. |
|
Bajwa SK, Bajwa SJ, Mohan P, Singh A. Management of prolactinoma with cabergoline treatment in a pregnant woman during her entire pregnancy. Indian J Endocrinol Metab. 2011;15 Suppl 3:S267-70. |
|
Auriemma RS, Perone Y, Di Sarno A, Grasso LF, Guerra E, Gasperi M, et al. Results of a single-center observational 10-year survey study on recurrence of hyperprolactinemia after pregnancy and lactation. J Clin Endocrinol Metab. 2013;98(1):372-9. |
|
Verhelst J, Abs R. Hyperprolactinemia: pathophysiology and management. Treat Endocrinol. 2003;2(1):23-32. |
|
Kim W, Clelland C, Yang I, Pouratian N. Comprehensive review of stereotactic radiosurgery for medically and surgically refractory pituitary adenomas. Surg Neurol Int. 2012;3(Suppl 2):S79-89. |
|
Thomson JA, Davies DL, McLaren EH, Teasdale GM. Ten year follow up of microprolactinoma treated by transsphenoidal surgery. BMJ. 1994;309(6966):1409-10. |
|
Primeau V, Raftopoulos C, Maiter D. Outcomes of transsphenoidal surgery in prolactinomas: improvement of hormonal control in dopamine agonist-resistant patients. Eur J Endocrinol. 2012;166(5):779-86. |
|
Holmgren U, Bergstrand G, Hagenfeldt K, Werner S. Women with prolactinoma--effect of pregnancy and lactation on serum prolactin and on tumour growth. Acta Endocrinol (Copenh). 1986;111(4):452-9. |
|
Katznelson L, Laws ER, Melmed S, Molitch ME, Murad MH, Utz A, et al. Acromegaly: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(11):3933-51. |
|
Melmed S, Kleinberg DL, Bonert V, Fleseriu M. Acromegaly: assessing the disorder and navigating therapeutic options for treatment. Endocr Pract. 2014;20(0):7-17. |
|
Persechini ML, Gennero I, Grunenwald S, Vezzosi D, Bennet A, Caron P. Decreased IGF-1 concentration during the first trimester of pregnancy in women with normal somatotroph function. Pituitary. 2014. |
|
Schwyzer L, Starke RM, Jane JA, Oldfield EH. Percent reduction of growth hormone levels correlates closely with percent resected tumor volume in acromegaly. J Neurosurg. 2014:1-5. |
|
Freda PU, Gordon MB, Kelepouris N, Jonsson P, Koltowska-Haggstrom M, van der Lely AJ. Long-Term Treatment with Pegvisomant as Monotherapy in Patients with Acromegaly: Experience from Acrostudy. Endocr Pract. 2014:1-32. |
|
Biermasz NR. Pituitary gland: Mortality in acromegaly reduced with multimodal therapy. Nat Rev Endocrinol. 2014;10(12):708-10. |
|
ABELOVE WA, RUPP JJ, PASCHKIS KE. Acromegaly and pregnancy. J Clin Endocrinol Metab. 1954;14(1):32-44. |
|
Dias M, Boguszewski C, Gadelha M, Kasuki L, Musolino N, Vieira JG, et al. Acromegaly and pregnancy: a prospective study. Eur J Endocrinol. 2014;170(2):301-10. |
|
Fisch RO, Prem KA, Feinberg SB, Gehrz RC. Acromegaly in a gravida and her infant. Obstet Gynecol. 1974;43(6):861-6. |
|
King KC, Adam PA, Schwartz R, Teramo K. Human placental transfer of human growth hormone-I 125. Pediatrics. 1971;48(4):534-9. |
|
Bigazzi M, Ronga R, Lancranjan I, Ferraro S, Branconi F, Buzzoni P, et al. A pregnancy in an acromegalic woman during bromocriptine treatment: effects on growth hormone and prolactin in the maternal, fetal, and amniotic compartments. J Clin Endocrinol Metab. 1979;48(1):9-12. |
|
Aono T, Shioji T, Kohno M, Ueda G, Kurachi K. Pregnancy following 2-bromo-alpha-ergocryptine (CB-154)-induced ovulation in an acromegalic patient with galactorrhea and amenorrhea. Fertil Steril. 1976;27(3):341-4. |
|
DeVane GW. Vasopressin levels during pregnancy and labor. J Reprod Med. 1985;30(4):324-7. |
|
Källén BA, Carlsson SS, Bengtsson BK. Diabetes insipidus and use of desmopressin (Minirin) during pregnancy. Eur J Endocrinol. 1995;132(2):144-6. |
|
Adonakis G, Kyriazopoulou V, Androutsopoulos G, Papadopoulos V, Decavalas G, Georgopoulos NA. Diabetes insipidus and two consecutive pregnancies: a case report and review of the literature. Clin Exp Obstet Gynecol. 2011;38(3):301-2. |
|
Ananthakrishnan S. Diabetes insipidus in pregnancy: etiology, evaluation, and management. Endocr Pract. 2009;15(4):377-82. |
|
Harris K, Shankar R, Black K, Rochelson B. Reset osmostat in pregnancy: a case report. J Matern Fetal Neonatal Med. 2014;27(5):530-3. |
|
van der Post JA, van Buul BJ, Hart AA, van Heerikhuize JJ, Pesman G, Legros JJ, et al. Vasopressin and oxytocin levels during normal pregnancy: effects of chronic dietary sodium restriction. J Endocrinol. 1997;152(3):345-54. |
|
Monson JP, Williams DJ. Osmoregulatory adaptation in pregnancy and its disorders. J Endocrinol. 1992;132(1):7-9. |
|
Durr JA, Lindheimer MD. Diagnosis and management of diabetes insipidus during pregnancy. Endocr Pract. 1996;2(5):353-61. |
|
Parkes I, Schenker JG, Shufaro Y. Parathyroid and calcium metabolism disorders during pregnancy. Gynecol Endocrinol. 2013;29(6):515-9. |
|
Riccardi D, Kemp PJ. The calcium-sensing receptor beyond extracellular calcium homeostasis: conception, development, adult physiology, and disease. Annu Rev Physiol. 2012;74:271-97. |
|
Organization WH. Calcium supplementation during pregnancy for the prevention of pre-eclampsia. Guidance Summary. 2014 [December 1, 2014]. Available from: http://www.who.int/elena/titles/guidance_summaries/calcium_pregnancy/en/. |
|
Pitkin RM. Calcium metabolism in pregnancy and the perinatal period: a review. Am J Obstet Gynecol. 1985;151(1):99-109. |
|
Seki K, Makimura N, Mitsui C, Hirata J, Nagata I. Calcium-regulating hormones and osteocalcin levels during pregnancy: a longitudinal study. Am J Obstet Gynecol. 1991;164(5 Pt 1):1248-52. |
|
Hacker AN, Fung EB, King JC. Role of calcium during pregnancy: maternal and fetal needs. Nutr Rev. 2012;70(7):397-409. |
|
Kovacs CS, Kronenberg HM. Maternal-fetal calcium and bone metabolism during pregnancy, puerperium, and lactation. Endocr Rev. 1997;18(6):832-72. |
|
Diaz-Thomas A, Cannon J, Iyer P, Al-Maawali A, Fazalullah M, Diamond F, et al. A novel CASR mutation associated with neonatal severe hyperparathyroidism transmitted as an autosomal recessive disorder. J Pediatr Endocrinol Metab. 2014;27(9-10):851-6. |
|
García-García E, Domínguez-Pascual I, Requena-Díaz M, Cabello-Laureano R, Fernández-Pineda I, Sánchez-Martín MJ. Intraoperative parathyroid hormone monitoring in neonatal severe primary hyperparathyroidism. Pediatrics. 2014;134(4):e1203-5. |
|
Caplan RH, Beguin EA. Hypercalcemia in a calcitriol-treated hypoparathyroid woman during lactation. Obstet Gynecol. 1990;76(3 Pt 2):485-9. |
|
Krysiak R, Kobielusz-Gembala I, Okopien B. Hypoparathyroidism in pregnancy. Gynecol Endocrinol. 2011;27(8):529-32. |
|
Ficinski ML, Mestman JH. Primary hyperparathyroidism during pregnancy. Endocr Pract. 1996;2(5):362-7. |
|
Sweeney LL, Malabanan AO, Rosen H. Decreased calcitriol requirement during pregnancy and lactation with a window of increased requirement immediately post partum. Endocr Pract. 2010;16(3):459-62. |
|
Durst R, Meirovitz A, Gross D, Kolker O, Muszkat M. Post-partum hypocalcemia: idiopatic hypoparathyroidism manifested early in lactation. J Endocrinol Invest. 2002;25(6):561-3. |
|
Sato K. Hypercalcemia during pregnancy, puerperium, and lactation: review and a case report of hypercalcemic crisis after delivery due to excessive production of PTH-related protein (PTHrP) without malignancy (humoral hypercalcemia of pregnancy). Endocr J. 2008;55(6):959-66. |
|
Shomali ME, Ross DS. Hypercalcemia in a woman with hypoparathyroidism associated with increased parathyroid hormone-related protein during lactation. Endocr Pract. 1999;5(4):198-200. |
|
Lepre F, Grill V, Ho PW, Martin TJ. Hypercalcemia in pregnancy and lactation associated with parathyroid hormone-related protein. N Engl J Med. 1993;328(9):666-7. |
|
Khosla S, van Heerden JA, Gharib H, Jackson IT, Danks J, Hayman JA, et al. Parathyroid hormone-related protein and hypercalcemia secondary to massive mammary hyperplasia. N Engl J Med. 1990;322(16):1157. |
|
Hunter D, Turnbull H. Hyperparathyroidism: Generalized osteitis fibrosa with observations upon bones, parathyroid tumors and the normal parathyroid glands. Br J Surg. 1931;19:203. |
|
Friderichsen D. Tetany in a suckling with latent osteitis fibrosis in the mother. Lancet. 1939;1:85. |
|
Krysiak R, Wilk M, Okopien B. Recurrent pancreatitis induced by hyperparathyroidism in pregnancy. Arch Gynecol Obstet. 2011;284(3):531-4. |
|
LUDWIG GD. Hyperparathyroidism in relation to pregnancy. N Engl J Med. 1962;267:637-42. |
|
Beattie GC, Ravi NR, Lewis M, Williams H, Blair AW, Campbell IW, et al. Rare presentation of maternal primary hyperparathyroidism. BMJ. 2000;321(7255):223-4. |
|
Som M, Stroup JS. Primary hyperparathyroidism and pregnancy. Proc (Bayl Univ Med Cent). 2011;24(3):220-3. |
|
Yilmaz BA, Altay M, Değertekin CK, Çimen AR, Iyidir Ö, Biri A, et al. Hyperparathyroid crisis presenting with hyperemesis gravidarum. Arch Gynecol Obstet. 2014;290(4):811-4. |
|
SCHENKER JG, KALLNER B. fatal postpartum hyperarathyroid crisis due to primary chief cell hyperplasia of parathyroids. Report of a case. Obstet Gynecol. 1965;25:705-9. |
|
Soyannwo MA, Bell M, McGeown MG, Milliken TG. A case of acute hyperparathyroidism, with thyrotoxicosis and pancreatitis, presenting as hyperemesis gravidarum. Postgrad Med J. 1968;44(517):861-6. |
|
Matthias GS, Helliwell TR, Williams A. Postpartum hyperparathyroid crisis. Case report. Br J Obstet Gynaecol. 1987;94(8):807-10. |
|
Çakır U, Alan S, Erdeve Ö, Atasay B, Şıklar Z, Berberoğlu M, et al. Late neonatal hypocalcemic tetany as a manifestation of unrecognized maternal primary hyperparathyroidism. Turk J Pediatr. 2013;55(4):438-40. |
|
Pothiwala P, Levine SN. Parathyroid surgery in pregnancy: review of the literature and localization by aspiration for parathyroid hormone levels. J Perinatol. 2009;29(12):779-84. |
|
Dochez V, Ducarme G. Primary hyperparathyroidism during pregnancy. Arch Gynecol Obstet. 2014. |
|
Khan MI, Waguespack SG, Hu MI. Medical management of postsurgical hypoparathyroidism. Endocr Pract. 2011;17 Suppl 1:18-25. |
|
Anderson G, Musselman L. The treatment of tetany in pregnancy. Am J Obstet Gynecol. 1942;43:547. |
|
Loughead JL, Mughal Z, Mimouni F, Tsang RC, Oestreich AE. Spectrum and natural history of congenital hyperparathyroidism secondary to maternal hypocalcemia. Am J Perinatol. 1990;7(4):350-5. |
|
Ngai YF, Chijiwa C, Mercimek-Mahmutoglu S, Stewart L, Yong SL, Robinson WP, et al. Pseudohypoparathyroidism type 1a and the GNAS p.R231H mutation: Somatic mosaicism in a mother with two affected sons. Am J Med Genet A. 2010;152A(11):2784-90. |
|
Singh A, Agarwal N, Chopra S, Sikka P, Suri V, Kumar B, et al. Management of Pseudohypoparathyroidism Type 1a during Pregnancy and Labor: A Case Report. Case Rep Obstet Gynecol. 2012;2012:629583. |
|
Kovacs CS. Osteoporosis presenting in pregnancy, puerperium, and lactation. Curr Opin Endocrinol Diabetes Obes. 2014;21(6):468-75. |
|
Tanriover MD, Oz SG, Sozen T, Kilicarslan A, Guven GS. Pregnancy- and lactation-associated osteoporosis with severe vertebral deformities: can strontium ranelate be a new alternative for the treatment? Spine J. 2009;9(4):e20-4. |
|
Campos-Obando N, Oei L, Hoefsloot LH, Kiewiet RM, Klaver CC, Simon ME, et al. Osteoporotic vertebral fractures during pregnancy: be aware of a potential underlying genetic cause. J Clin Endocrinol Metab. 2014;99(4):1107-11. |
|
Cook FJ, Mumm S, Whyte MP, Wenkert D. Pregnancy-associated osteoporosis with a heterozygous deactivating LDL receptor-related protein 5 (LRP5) mutation and a homozygous methylenetetrahydrofolate reductase (MTHFR) polymorphism. J Bone Miner Res. 2014;29(4):922-8. |
|
Ozdemir D, Tam AA, Dirikoc A, Ersoy R, Cakir B. Postpartum osteoporosis and vertebral fractures in two patients treated with enoxaparin during pregnancy. Osteoporos Int. 2014. |
|
Le Templier G, Rodger MA. Heparin-induced osteoporosis and pregnancy. Curr Opin Pulm Med. 2008;14(5):403-7. |
|
Ofluoglu O, Ofluoglu D. A case report: pregnancy-induced severe osteoporosis with eight vertebral fractures. Rheumatol Int. 2008;29(2):197-201. |
|
NORDIN BE, ROPER A. Post-pregnancy osteoporosis; a syndrome? Lancet. 1955;268(6861):431-4. |
|
Czech-Kowalska J, Latka-Grot J, Bulsiewicz D, Jaworski M, Pludowski P, Wygledowska G, et al. Impact of vitamin D supplementation during lactation on vitamin D status and body composition of mother-infant pairs: a MAVID randomized controlled trial. PLoS One. 2014;9(9):e107708. |
|
Zarattini G, Buffoli P, Isabelli G, Marchese M. Pregnancy-associated osteoporosis with seven vertebral compression fractures, a case treated with strontium ranelate. Clin Cases Miner Bone Metab. 2014;11(2):139-41. |
|
Takahashi N, Arai I, Kayama S, Ichiji K, Fukuda H, Handa JI, et al. Four-year follow-up of pregnancy-associated osteoporosis: a case report. Fukushima J Med Sci. 2014. |
|
Winarno AS, Kyvernitakis I, Hadji P. Successful treatment of 1-34 parathyroid hormone (PTH) after failure of bisphosphonate therapy in a complex case of pregnancy associated osteoporosis and multiple fractures. Z Geburtshilfe Neonatol. 2014;218(4):171-3. |
|
Ozturk C, Atamaz FC, Akkurt H, Akkoc Y. Pregnancy-associated osteoporosis presenting severe vertebral fractures. J Obstet Gynaecol Res. 2014;40(1):288-92. |
|
Phillips AJ, Ostlere SJ, Smith R. Pregnancy-associated osteoporosis: does the skeleton recover? Osteoporos Int. 2000;11(5):449-54. |
|
Karlsson C, Obrant KJ, Karlsson M. Pregnancy and lactation confer reversible bone loss in humans. Osteoporos Int. 2001;12(10):828-34. |
|
Møller UK, Við Streym S, Mosekilde L, Rejnmark L. Changes in bone mineral density and body composition during pregnancy and postpartum. A controlled cohort study. Osteoporos Int. 2012;23(4):1213-23. |
|
Kamoun M, Mnif MF, Charfi N, Kacem FH, Naceur BB, Mnif F, et al. Adrenal diseases during pregnancy: pathophysiology, diagnosis and management strategies. Am J Med Sci. 2014;347(1):64-73. |
|
Jung C, Ho JT, Torpy DJ, Rogers A, Doogue M, Lewis JG, et al. A longitudinal study of plasma and urinary cortisol in pregnancy and postpartum. J Clin Endocrinol Metab. 2011;96(5):1533-40. |
|
Gennari-Moser C, Escher G, Kramer S, Dick B, Eisele N, Baumann M, et al. Normotensive blood pressure in pregnancy: the role of salt and aldosterone. Hypertension. 2014;63(2):362-8. |
|
Gibson M, Tulchinsky D. The maternal adrenal. In: Tulchinsky D, Ryan K, editors. Maternal Fetal Endocrinology. Philadelphia: WB Saunders; 1980. p. 129-43. |
|
Steffensen C, Bak AM, Rubeck KZ, Jørgensen JO. Epidemiology of Cushing's syndrome. Neuroendocrinology. 2010;92 Suppl 1:1-5. |
|
Nassi R, Ladu C, Vezzosi C, Mannelli M. Cushing's syndrome in pregnancy. Gynecol Endocrinol. 2014:1-3. |
|
Lekarev O, New MI. Adrenal disease in pregnancy. Best Pract Res Clin Endocrinol Metab. 2011;25(6):959-73. |
|
Close CF, Mann MC, Watts JF, Taylor KG. ACTH-independent Cushing's syndrome in pregnancy with spontaneous resolution after delivery: control of the hypercortisolism with metyrapone. Clin Endocrinol (Oxf). 1993;39(3):375-9. |
|
Jairath A, Aulakh BS. Adrenocortical carcinoma in pregnancy: A diagnostic dilemma. Indian J Urol. 2014;30(3):342-4. |
|
Cohade C, Broussaud S, Louiset E, Bennet A, Huyghe E, Caron P. Ectopic Cushing's syndrome due to a pheochromocytoma: a new case in the post-partum and review of literature. Gynecol Endocrinol. 2009;25(9):624-7. |
|
Mazziotti G, Gazzaruso C, Giustina A. Diabetes in Cushing syndrome: basic and clinical aspects. Trends Endocrinol Metab. 2011;22(12):499-506. |
|
Duthie L, Reynolds RM. Changes in the maternal hypothalamic-pituitary-adrenal axis in pregnancy and postpartum: influences on maternal and fetal outcomes. Neuroendocrinology. 2013;98(2):106-15. |
|
Lim WH, Torpy DJ, Jeffries WS. The medical management of Cushing's syndrome during pregnancy. Eur J Obstet Gynecol Reprod Biol. 2013;168(1):1-6. |
|
Woo I, Ehsanipoor RM. Cabergoline therapy for Cushing disease throughout pregnancy. Obstet Gynecol. 2013;122(2 Pt 2):485-7. |
|
Naccache DD, Zaina A, Shen-Or Z, Armoni M, Kontogeorgos G, Yahia A. Uneventful octreotide LAR therapy throughout three pregnancies, with favorable delivery and anthropometric measures for each newborn: a case report. J Med Case Rep. 2011;5:386. |
|
Sammour RN, Saiegh L, Matter I, Gonen R, Shechner C, Cohen M, et al. Adrenalectomy for adrenocortical adenoma causing Cushing's syndrome in pregnancy: a case report and review of literature. Eur J Obstet Gynecol Reprod Biol. 2012;165(1):1-7. |
|
Ross RJ, Chew SL, Perry L, Erskine K, Medbak S, Afshar F. Diagnosis and selective cure of Cushing's disease during pregnancy by transsphenoidal surgery. Eur J Endocrinol. 1995;132(6):722-6. |
|
Ferraù F, Losa M, Cotta OR, Torre ML, Ragonese M, Trimarchi F, et al. Course of pregnancies in women with Cushing's disease treated by gamma-knife. Gynecol Endocrinol. 2012;28(10):827-9. |
|
Jornayvaz FR, Assie G, Bienvenu-Perrard M, Coste J, Guignat L, Bertherat J, et al. Pregnancy does not accelerate corticotroph tumor progression in Nelson's syndrome. J Clin Endocrinol Metab. 2011;96(4):E658-62. |
|
Ng AC, Kumar SK, Russell SJ, Rajkumar SV, Drake MT. Dexamethasone and the risk for adrenal suppression in multiple myeloma. Leukemia. 2009;23(5):1009-11. |
|
Krasner AS. Glucocorticoid-induced adrenal insufficiency. JAMA. 1999;282(7):671-6. |
|
LaRochelle GE, LaRochelle AG, Ratner RE, Borenstein DG. Recovery of the hypothalamic-pituitary-adrenal (HPA) axis in patients with rheumatic diseases receiving low-dose prednisone. Am J Med. 1993;95(3):258-64. |
|
Michels AW, Eisenbarth GS. Immunologic endocrine disorders. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S226-37. |
|
Rose LI, Williams GH, Jagger PI, Lauler DP. The 48-hour adrenocorticotrophin infusion test for adrenocortical insufficiency. Ann Intern Med. 1970;73(1):49-54. |
|
Cosimo C, Franco C. Addison's disease and pregnancy: case report. J Prenat Med. 2009;3(4):53-4. |
|
BRENT F. Addison's disease and pregnancy. Am J Surg. 1950;79(5):645-52. |
|
OSLER M. Addison's disease and pregnancy. Acta Endocrinol (Copenh). 1962;41:67-78. |
|
BONGIOVANNI AM, McPADDEN AJ. Steroids during pregnancy and possible fetal consequences. Fertil Steril. 1960;11:181-6. |
|
Ambrosi B, Barbetta L, Morricone L. Diagnosis and management of Addison's disease during pregnancy. J Endocrinol Invest. 2003;26(7):698-702. |
|
Marumudi E, Khadgawat R, Surana V, Shabir I, Joseph A, Ammini AC. Diagnosis and management of classical congenital adrenal hyperplasia. Steroids. 2013;78(8):741-6. |
|
Trakakis E, Loghis C, Kassanos D. Congenital adrenal hyperplasia because of 21-hydroxylase deficiency. A genetic disorder of interest to obstetricians and gynecologists. Obstet Gynecol Surv. 2009;64(3):177-89. |
|
Reichman DE, White PC, New MI, Rosenwaks Z. Fertility in patients with congenital adrenal hyperplasia. Fertil Steril. 2014;101(2):301-9. |
|
Witchel SF. Management of CAH during pregnancy: optimizing outcomes. Curr Opin Endocrinol Diabetes Obes. 2012;19(6):489-96. |
|
Auchus RJ, Arlt W. Approach to the patient: the adult with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2013;98(7):2645-55. |
|
Garner PR. Management of congenital adrenal hyperplasia during pregnancy. Endocr Pract. 1996;2(6):397-405. |
|
Baumgartner-Parzer SM, Nowotny P, Heinze G, Waldhäusl W, Vierhapper H. Carrier frequency of congenital adrenal hyperplasia (21-hydroxylase deficiency) in a middle European population. J Clin Endocrinol Metab. 2005;90(2):775-8. |
|
Ehrlich EN, Lindheimer MD. Effect of administered mineralocorticoids or ACTH in pregnant women. Attenuation of kaliuretic influence of mineralocorticoids during pregnancy. J Clin Invest. 1972;51(6):1301-9. |
|
Nimkarn S, New MI. Prenatal diagnosis and treatment of congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Nat Clin Pract Endocrinol Metab. 2007;3(5):405-13. |
|
Enzensberger C, Pulvermacher C, Degenhardt J, Kawacki A, Germer U, Gembruch U, et al. Fetal loss rate and associated risk factors after amniocentesis, chorionic villus sampling and fetal blood sampling. Ultraschall Med. 2012;33(7):E75-9. |
|
Lajic S, Nordenström A, Hirvikoski T. Long-term outcome of prenatal dexamethasone treatment of 21-hydroxylase deficiency. Endocr Dev. 2011;20:96-105. |
|
Hirvikoski T, Nordenström A, Lindholm T, Lindblad F, Ritzén EM, Lajic S. Long-term follow-up of prenatally treated children at risk for congenital adrenal hyperplasia: does dexamethasone cause behavioural problems? Eur J Endocrinol. 2008;159(3):309-16. |
|
Peter M, Dubuis JM, Sippell WG. Disorders of the aldosterone synthase and steroid 11beta-hydroxylase deficiencies. Horm Res. 1999;51(5):211-22. |
|
White PC. Steroid 11 beta-hydroxylase deficiency and related disorders. Endocrinol Metab Clin North Am. 2001;30(1):61-79, vi. |
|
CONN JW, LOUIS LH. Primary aldosteronism: a new clinical entity. Trans Assoc Am Physicians. 1955;68:215-31; discussion, 31-3. |
|
Rossi GP. Prevalence and diagnosis of primary aldosteronism. Curr Hypertens Rep. 2010;12(5):342-8. |
|
Aronova A, Iii TJ, Zarnegar R. Management of hypertension in primary aldosteronism. World J Cardiol. 2014;6(5):227-33. |
|
Cabassi A, Rocco R, Berretta R, Regolisti G, Bacchi-Modena A. Eplerenone use in primary aldosteronism during pregnancy. Hypertension. 2012;59(2):e18-9. |
|
Kosaka K, Onoda N, Ishikawa T, Iwanaga N, Yamamasu S, Tahara H, et al. Laparoscopic adrenalectomy on a patient with primary aldosteronism during pregnancy. Endocr J. 2006;53(4):461-6. |
|
Velasco A, Vongpatanasin W. The evaluation and treatment of endocrine forms of hypertension. Curr Cardiol Rep. 2014;16(9):528. |
|
Eisenhofer G, Pacak K, Maher ER, Young WF, de Krijger RR. Pheochromocytoma. Clin Chem. 2013;59(3):466-72. |
|
Hodin R, Lubitz C, Phitayakorn R, Stephen A. Diagnosis and management of pheochromocytoma. Curr Probl Surg. 2014;51(4):151-87. |
|
Martucci VL, Pacak K. Pheochromocytoma and paraganglioma: diagnosis, genetics, management, and treatment. Curr Probl Cancer. 2014;38(1):7-41. |
|
Tsirlin A, Oo Y, Sharma R, Kansara A, Gliwa A, Banerji MA. Pheochromocytoma: a review. Maturitas. 2014;77(3):229-38. |
|
Baudin E, Habra MA, Deschamps F, Cote G, Dumont F, Cabanillas M, et al. Therapy of endocrine disease: treatment of malignant pheochromocytoma and paraganglioma. Eur J Endocrinol. 2014;171(3):R111-22. |
|
Mannelli M, Bemporad D. Diagnosis and management of pheochromocytoma during pregnancy. J Endocrinol Invest. 2002;25(6):567-71. |
|
Plu I, Sec I, Barrès D, Lecomte D. Pregnancy, cesarean, and pheochromocytoma: a case report and literature review. J Forensic Sci. 2013;58(4):1075-9. |
|
Biggar MA, Lennard TW. Systematic review of phaeochromocytoma in pregnancy. Br J Surg. 2013;100(2):182-90. |
|
Schenker JG, Granat M. Phaeochromocytoma and pregnancy--an updated appraisal. Aust N Z J Obstet Gynaecol. 1982;22(1):1-10. |
|
Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915-42. |
|
Lenders JW. Pheochromocytoma and pregnancy: a deceptive connection. Eur J Endocrinol. 2012;166(2):143-50. |
|
Oliva R, Angelos P, Kaplan E, Bakris G. Pheochromocytoma in pregnancy: a case series and review. Hypertension. 2010;55(3):600-6. |
|
Lata I, Sahu S. Management of paroxysmal hypertension due to incidental pheochromocytoma in pregnancy. J Emerg Trauma Shock. 2011;4(3):415-7. |
|
Song Y, Liu J, Li H, Zeng Z, Bian X, Wang S. Outcomes of concurrent Caesarean delivery and pheochromocytoma resection in late pregnancy. Intern Med J. 2013;43(5):588-91. |
|
Zuluaga-Gómez A, Arrabal-Polo M, Arrabal-Martín M, Lahoz-García C, Escobar-Jiménez F, Torres-Vela E, et al. Management of pheochromocytoma during pregnancy: laparoscopic adrenalectomy. Am Surg. 2012;78(3):E156-8. |
|
Ugaki H, Enomoto T, Tokugawa Y, Kimura T. Luteoma-induced fetal virilization. J Obstet Gynaecol Res. 2009;35(5):991-3. |
|
Glinoer D. Increased TBG during pregnancy and increased hormonal requirements. Thyroid. 2004;14(6):479-80; author reply 80-1. |
|
Moleti M, Trimarchi F, Vermiglio F. Thyroid physiology in pregnancy. Endocr Pract. 2014;20(6):589-96. |
|
Barbesino G, Tomer Y. Clinical review: Clinical utility of TSH receptor antibodies. J Clin Endocrinol Metab. 2013;98(6):2247-55. |
|
Abeillon-du Payrat J, Chikh K, Bossard N, Bretones P, Gaucherand P, Claris O, et al. Predictive value of maternal second-generation thyroid-binding inhibitory immunoglobulin assay for neonatal autoimmune hyperthyroidism. Eur J Endocrinol. 2014;171(4):451-60. |
|
Evans C, Gregory JW, Barton J, Bidder C, Gibbs J, Pryce R, et al. Transient congenital hypothyroidism due to thyroid-stimulating hormone receptor blocking antibodies: a case series. Ann Clin Biochem. 2011;48(Pt 4):386-90. |
|
Stagnaro-Green A, Abalovich M, Alexander E, Azizi F, Mestman J, Negro R, et al. Guidelines of the American Thyroid Association for the diagnosis and management of thyroid disease during pregnancy and postpartum. Thyroid. 2011;21(10):1081-125. |
|
Emerson CH, Braverman LE. Transfer and metabolism of thyroid-related substances in the placenta. Adv Exp Med Biol. 1991;299:181-96. |
|
Roti E, Gnudi A, Braverman LE, Robuschi G, Emanuele R, Bandini P, et al. Human cord blood concentrations of thyrotropin, thyroglobulin, and iodothyronines after maternal administration of thyrotropin-releasing hormone. J Clin Endocrinol Metab. 1981;53(4):813-7. |
|
Mortimer RH, Cannell GR, Addison RS, Johnson LP, Roberts MS, Bernus I. Methimazole and propylthiouracil equally cross the perfused human term placental lobule. The Journal of clinical endocrinology and metabolism. 1997;82(9):3099. |
|
Shumer DE, Mehringer JE, Braverman LE, Dauber A. Acquired Hypothyroidism in an Infant Related to Excessive Maternal Iodine Intake: Food for Thought. Endocrine practice : official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists. 2013:1. |
|
Lazarus JH. Management of hyperthyroidism in pregnancy. Endocrine. 2014;45(2):190-4. |
|
Chiniwala NU, Woolf PD, Bruno CP, Kaur S, Spector H, Yacono K. Thyroid storm caused by a partial hydatidiform mole. Thyroid. 2008;18(4):479-81. |
|
Vaidya B, Pearce SH. Diagnosis and management of thyrotoxicosis. BMJ. 2014;349:g5128. |
|
Cooper DS, Laurberg P. Hyperthyroidism in pregnancy. Lancet Diabetes Endocrinol. 2013;1(3):238-49. |
|
Labadzhyan A, Brent GA, Hershman JM, Leung AM. Thyrotoxicosis of Pregnancy. J Clin Transl Endocrinol. 2014;1(4):140-4. |
|
Easterling TR, Schmucker BC, Carlson KL, Millard SP, Benedetti TJ. Maternal hemodynamics in pregnancies complicated by hyperthyroidism. Obstet Gynecol. 1991;78(3 Pt 1):348-52. |
|
Fitzpatrick DL, Russell MA. Diagnosis and management of thyroid disease in pregnancy. Obstet Gynecol Clin North Am. 2010;37(2):173-93. |
|
Panesar NS, Li CY, Rogers MS. Reference intervals for thyroid hormones in pregnant Chinese women. Annals of Clinical Biochemistry. 2001;38(Pt 4):329. |
|
De Groot L, Abalovich M, Alexander EK, Amino N, Barbour L, Cobin RH, et al. Management of Thyroid Dysfunction during Pregnancy and Postpartum: An Endocrine Society Clinical Practice Guideline. The Journal of clinical endocrinology and metabolism. 2012;97(8):2543. |
|
Marino M, Chiovato L, Pinchera A. In: DeGroot LJ, Jameson JL, editors. Endocrinology Adult and Pediatric: The Thyroid Gland, Volume 2. Philadelphia: Elsevier Saunders; 2010. |
|
Cooper DS. Propylthiouracil levels in hyperthyroid patients unresponsive to large doses. Evidence of poor patient compliance. Ann Intern Med. 1985;102(3):328-31. |
|
Mandel SJ, Cooper DS. The use of antithyroid drugs in pregnancy and lactation. The Journal of clinical endocrinology and metabolism. 2001;86(6):2354. |
|
Mutharasan P, Oatis W, Kwaan H, Molitch M. Delayed anithyroid drug-induced agranulocytosis. Endocr Pract. 2012;18(4):e69-72. |
|
Cooper DS. Antithyroid drugs. The New England journal of medicine. 2005;352(9):905. |
|
Stoffer SS, Hamburger JI. Inadvertent 131I therapy for hyperthyroidism in the first trimester of pregnancy. J Nucl Med. 1976;17(02):146-9. |
|
Omoto A, Kurimoto C, Minagawa M, Shozu M. A case of fetal goiter that resolved spontaneously after birth. J Clin Endocrinol Metab. 2013;98(10):3910-1. |
|
Connelly KJ, Boston BA, Pearce EN, Sesser D, Snyder D, Braverman LE, et al. Congenital hypothyroidism caused by excess prenatal maternal iodine ingestion. The Journal of pediatrics. 2012;161(4):760. |
|
Burrow GN. Neonatal goiter after maternal propylthiouracil therapy. J Clin Endocrinol Metab. 1965;25:403-8. |
|
Jamieson A, Semple CG. Successful treatment of Graves disease in pregnancy with Lugol's iodine. Scott Med J. 2000;45(1):20-1. |
|
Khamisi S, Lindgren P, Karlsson FA. A rare case of dyshormonogenetic fetal goiter responding to intra-amniotic thyroxine injections. Eur Thyroid J. 2014;3(1):51-6. |
|
Saini A, Reddy MM, Panchani R, Varma T, Gupta N, Tripathi S. Two cases of fetal goiter. Indian J Endocrinol Metab. 2012;16(Suppl 2):S358-60. |
|
Männistö T, Mendola P, Reddy U, Laughon SK. Neonatal outcomes and birth weight in pregnancies complicated by maternal thyroid disease. Am J Epidemiol. 2013;178(5):731-40. |
|
Mestman JH. Hyperthyroidism in pregnancy. Curr Opin Endocrinol Diabetes Obes. 2012;19(5):394-401. |
|
Medici M, Korevaar TI, Schalekamp-Timmermans S, Gaillard R, de Rijke YB, Visser WE, et al. Maternal early-pregnancy thyroid function is associated with subsequent hypertensive disorders of pregnancy: the generation R study. J Clin Endocrinol Metab. 2014;99(12):E2591-8. |
|
Ohrling H, Törring O, Yin L, Iliadou AN, Tullgren O, Abraham-Nordling M, et al. Decreased birth weight, length, and head circumference in children born by women years after treatment for hyperthyroidism. J Clin Endocrinol Metab. 2014;99(9):3217-23. |
|
Gleicher N. Maternal autoimmunity and adverse pregnancy outcomes. J Autoimmun. 2014;50:83-6. |
|
Nor Azlin MI, Bakin YD, Mustafa N, Wahab NA, Johari MJ, Kamarudin NA, et al. Thyroid autoantibodies and associated complications during pregnancy. J Obstet Gynaecol. 2010;30(7):675-8. |
|
Goodwin TM, Montoro M, Mestman JH. Transient hyperthyroidism and hyperemesis gravidarum: clinical aspects. American Journal of Obstetrics and Gynecology. 1992;167(3):648. |
|
Bouillon R, Naesens M, Van Assche FA, De Keyser L, De Moor P, Renaer M, et al. Thyroid function in patients with hyperemesis gravidarum. Am J Obstet Gynecol. 1982;143(8):922-6. |
|
Lao TT, Chin RK, Chang AM. The outcome of hyperemetic pregnancies complicated by transient hyperthyroidism. Aust N Z J Obstet Gynaecol. 1987;27(2):99-101. |
|
Mestman J. Management of thyroid disease in pregnancy. In: Berkowitz R, editor. Clinics of perinatology. Philadelphia: WB Saunders; 1980. |
|
Pekary AE, Jackson IM, Goodwin TM, Pang XP, Hein MD, Hershman JM. Increased in vitro thyrotropic activity of partially sialated human chorionic gonadotropin extracted from hydatidiform moles of patients with hyperthyroidism. J Clin Endocrinol Metab. 1993;76(1):70-4 |
|
Dierickx I, Decallonne B, Billen J, Vanhole C, Lewi L, De Catte L, et al. Severe fetal and neonatal hyperthyroidism years after surgical treatment of maternal Graves' disease. J Obstet Gynaecol. 2014;34(2):117-22. |
|
Besançon A, Beltrand J, Le Gac I, Luton D, Polak M. Management of neonates born to women with Graves' disease: a cohort study. Eur J Endocrinol. 2014;170(6):855-62 |
|
Polak M. Hyperthyroidism in early infancy: pathogenesis, clinical features and diagnosis with a focus on neonatal hyperthyroidism. Thyroid. 1998;8(12):1171-7. |
|
Skuza KA, Sills IN, Stene M, Rapaport R. Prediction of neonatal hyperthyroidism in infants born to mothers with Graves disease. J Pediatr. 1996;128(2):264-8. |
|
Casey B, de Veciana M. Thyroid screening in pregnancy. Am J Obstet Gynecol. 2014;211(4):351-3.e1. |
|
Shields BM, Knight BA, Hill AV, Hattersley AT, Vaidya B. Five-year follow-up for women with subclinical hypothyroidism in pregnancy. J Clin Endocrinol Metab. 2013;98(12):E1941-5. |
|
Allan WC, Haddow JE, Palomaki GE, Williams JR, Mitchell ML, Hermos RJ, et al. Maternal thyroid deficiency and pregnancy complications: implications for population screening. J Med Screen. 2000;7(3):127. |
|
Männistö T, Mendola P, Grewal J, Xie Y, Chen Z, Laughon SK. Thyroid diseases and adverse pregnancy outcomes in a contemporary US cohort. J Clin Endocrinol Metab. 2013;98(7):2725-33. |
|
Casey BM, Dashe JS, Wells CE, McIntire DD, Byrd W, Leveno KJ, Cunningham FG. Obstet Gynecol 2005;105(2):239-45. PMID: 15684146. |
|
Leung AS, Millar LK, Koonings PP, Montoro M, Mestman JH. Perinatal outcome in hypothyroid pregnancies. Obstet Gynecol. 1993;81(3):349. |
|
Millar LK, Wing DA, Leung AS, Koonings PP, Montoro MN, Mestman JH. Low birth weight and preeclampsia in pregnancies complicated by hyperthyroidism. Obstetrics and gynecology. 1994;84(6):946. |
|
Abalovich M, Gutierrez S, Alcaraz G, Maccallini G, Garcia A, Levalle O. Overt and subclinical hypothyroidism complicating pregnancy. Thyroid. 2002;12(1):63. |
|
Cleary-Goldman J, Malone FD, Lambert-Messerlian G, Sullivan L, Canick J, Porter TF, et al. Maternal thyroid hypofunction and pregnancy outcome. Obstet Gynecol. 2008;112(1):85. |
|
Mannisto T, Vaarasmaki M, Pouta A, Hartikainen AL, Ruokonen A, Surcel HM, et al. Perinatal outcome of children born to mothers with thyroid dysfunction or antibodies: a prospective population-based cohort study. J Clin Endocrinol Metab. 2009;94(3):772. |
|
Mannisto T, Vaarasmaki M, Pouta A, Hartikainen AL, Ruokonen A, Surcel HM, et al. Thyroid dysfunction and autoantibodies during pregnancy as predictive factors of pregnancy complications and maternal morbidity in later life. The Journal of clinical endocrinology and metabolism. 2010;95(3):1084. |
|
Ong GS, Hadlow NC, Brown SJ, Lim EM, Walsh JP. Does the thyroid-stimulating hormone measured concurrently with first trimester biochemical screening tests predict adverse pregnancy outcomes occurring after 20 weeks gestation? J Clin Endocrinol Metab. 2014;99(12):E2668-72. |
|
Bernal J. Thyroid hormone receptors in brain development and function. Nat Clin Pract Endocrinol Metab. 2007;3(3):249-59. |
|
Haddow JE, Palomaki GE, Allan WC, Williams JR, Knight GJ, Gagnon J, et al. Maternal thyroid deficiency during pregnancy and subsequent neuropsychological development of the child. N Engl J Med. 1999;341(8):549. |
|
Ghassabian A, El Marroun H, Peeters RP, Jaddoe VW, Hofman A, Verhulst FC, et al. Downstream effects of maternal hypothyroxinemia in early pregnancy: nonverbal IQ and brain morphology in school-age children. J Clin Endocrinol Metab. 2014;99(7):2383-90. |
|
Henrichs J, Schenk JJ, Roza SJ, van den Berg MP, Schmidt HG, Steegers EA, et al. Maternal psychological distress and fetal growth trajectories: the Generation R Study. Psychological medicine. 2010;40(4):633. |
|
Päkkilä F, Männistö T, Pouta A, Hartikainen AL, Ruokonen A, Surcel HM, et al. The impact of gestational thyroid hormone concentrations on ADHD symptoms of the child. J Clin Endocrinol Metab. 2014;99(1):E1-8. |
|
Pearce EN, Oken E, Gillman MW, Lee SL, Magnani B, Platek D, et al. Association of first-trimester thyroid function test values with thyroperoxidase antibody status, smoking, and multivitamin use. Endocrine practice : official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists. 2008;14(1):33. |
|
Yassa L, Marqusee E, Fawcett R, Alexander EK. Thyroid hormone early adjustment in pregnancy (the THERAPY) trial. The Journal of clinical endocrinology and metabolism. 2010;95(7):3234. |
|
Alexander EK, Marqusee E, Lawrence J, Jarolim P, Fischer GA, Larsen PR. Timing and magnitude of increases in levothyroxine requirements during pregnancy in women with hypothyroidism. The New England journal of medicine. 2004;351(3):241. |
|
Cooper DS, Doherty GM, Haugen BR, Kloos RT, Lee SL, Mandel SJ, et al. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid. 2009;19(11):1167. |
|
Garber JR, Cobin RH, Gharib H, Hennessey JV, Klein I, Mechanick JI, et al. Clinical practice guidelines for hypothyroidism in adults: cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid Association. Thyroid : official journal of the American Thyroid Association. 2012;22(12):1200. |
|
Obican SG, Jahnke GD, Soldin OP, Scialli AR. Teratology public affairs committee position paper: Iodine deficiency in pregnancy. Birth defects research.Part A, Clinical and molecular teratology. 2012. |
|
Network ICftCoIDDG. FAQs about iodine nutrition [December 16, 2014]. Available from: http://www.ign.org/p142000355.html#p4. |
|
Leung AM, Avram AM, Brenner AV, Duntas LH, Ehrenkranz J, Hennessey JV, et al. Potential Risks of Excess Iodine Ingestion and Exposure: Statement by the American Thyroid Association Public Health Committee. Thyroid. 2014. |
|
Mazzaferri EL. Approach to the pregnant patient with thyroid cancer. J Clin Endocrinol Metab. 2011;96(2):265-72. |
|
Moosa M, Mazzaferri EL. Outcome of differentiated thyroid cancer diagnosed in pregnant women. J Clin Endocrinol Metab. 1997;82(9):2862-6. |
|
Messuti I, Corvisieri S, Bardesono F, Rapa I, Giorcelli J, Pellerito R, et al. Impact of pregnancy on prognosis of differentiated thyroid cancer: clinical and molecular features. Eur J Endocrinol. 2014;170(5):659-66. |
|
Stagnaro-Green A. Approach to the patient with postpartum thyroiditis. J Clin Endocrinol Metab. 2012;97(2):334-42. |
|
Stuckey BG, Kent GN, Ward LC, Brown SJ, Walsh JP. Postpartum thyroid dysfunction and the long-term risk of hypothyroidism: results from a 12-year follow-up study of women with and without postpartum thyroid dysfunction. Clin Endocrinol (Oxf). 2010;73(3):389-95. |
|
Lazarus JH, Ammari F, Oretti R, Parkes AB, Richards CJ, Harris B. Clinical aspects of recurrent postpartum thyroiditis. Br J Gen Pract. 1997;47(418):305-8. |