Males with Polysomy Y and Females with Polysomy X
Authors
INTRODUCTION
Both polysomy Y in males (47,XYY; 48,XYYY; 48,XXYY; 49,XXYYY) and polysomy X in females (47,XXX; 48,XXXX; 49,XXXXX) may be encountered by obstetricians-gynecologists as (1) an abnormal fetal chromosome complement detected by chorionic villus sampling, amniocentesis, or percutaneous umbilical blood sampling or (2) part of an evaluation for infertility, abnormal sexual development, other congenital malformations, or mental aberrations. Polysomic sex chromosome complements may also provide valuable information concerning sex chromosome function and abnormalities of meiosis or mitosis. In this chapter epidemiologic data and clinical features of polysomy X and polysomy Y will be reviewed as well as recommendations for counseling both affected individuals and women carrying affected fetuses. The reader is referred to a previous publication for additional information.1
POLYSOMY Y IN MALES
47,XYY
FREQUENCY.
The first individual with a 47,XYY chromosomal complement (Fig. 1) was described by Sandberg and co-workers in 1961.2 For several years thereafter, relatively few cases were reported, but in 1965-1966, several investigators reported that among mentally retarded criminals the prevalence of 47,XYY was higher than expected by chance, creating a resurgence of interest in this disorder.3,4,5,6 Jacobs7 summarized the data collected from eight chromosome surveys of unselected infants.8,9,10,11,12,13,14,15 Among 39,557 males studied, 38 (0.1%) were found to have 47,XYY complements. These data correspond closely to those of two large sex chromatin surveys of newborns16,17 as well as recent data reported by Hecht and Hecht18 demonstrating the frequency of XYY in newborn males to be 1 in 975.
|
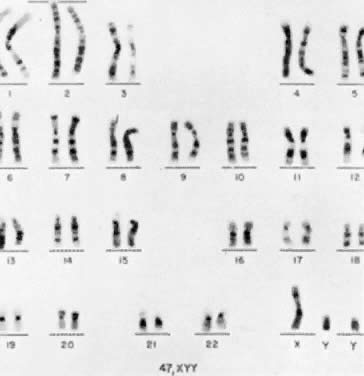
Among a total of 13,406 male infants studies, 9 (0.07%) were determined to have two Y-bodies. Subsequently, 47,XYY karyotypes were confirmed in these 9 infants. There has been some suggestion that the incidence of 47,XYY males may be lower among blacks than among whites.19
CELLULAR ETIOLOGY.
The most likely origin of 47,XYY is paternal nondisjunction at meiosis II, resulting in 24,YY spermatozoa, followed by syngamy with a normal 23,X ovum. A 24,YY spermatozoon would also be a normal product of meiosis in a male with a 47,XYY chromosomal constitution; however, this phenomenon probably accounts for relatively few 47,XYY men.20 Alternatively, postzygotic nondisjunction in a 46,XY zygote could give rise to 45,X/47,XYY mosaicism. Either exclusion of the 45,X line or selection in favor of the 47,XYY line could result in an apparently “pure” 47,XYY zygote. The occurrence of double aneuploidy, for example, 48,XYY,+21, raises the possibility that genetic factors could be important in the etiology of polysomy Y.21 Finally, parental age appears to play no important part in the genesis of 47,XYY.22
CLINICAL FINDINGS.
Psychologic and Intellectual Function.
The first psychologic studies of 47,XYY males were those of Price and co-workers,23,24 who studied inmates at a Scottish maximum security prison. Compared with 18 46,XY inmates, 9 47,XYY inmates (1) incurred their first conviction at a younger age (mean age 13.1 years for 47,XYY; 18 years for 46,XY, (2) less often had a sibling who had received a conviction (1/31 siblings of 47,XYY inmates; 13/63 sibs of 46,XY inmates), and (3) committed crimes against property more often than crimes against persons. These observations suggested that 47,XYY inmates are incarcerated for different reasons than are other inmates. Probably the most objective study was from Denmark, conducted by Witkin and co-workers,25 in which a 42% (5/12) rate of criminality was found among 47,XYY males compared with 9.3% in 4096 46,XY controls. The difference is about 4.5-fold and is significant at the 0.05 level. Hook26 suggested that despite the observation that intelligence quotients of 47,XYY males are lower than 46,XY controls in exclusively penal settings, it is not the lower intelligence per se that is exclusively responsible for the higher frequency of incarceration but rather the significant increase in risk of social maldevelopment associated with the 47,XYY genotype.
On the other hand, some investigators object to these conclusions regarding the increased frequency of social maldevelopment and predisposition to “criminality.”27 Accurate assessment of the intellectual and psychologic problems in 47,XYY individuals therefore awaits the results of larger prospective surveys of neonates or randomly ascertained adults. Robinson and associates28 summarized the findings of 43 47,XYY infants prospectively ascertained through various chromosome surveys of unselected neonates. About a third of the children showed delayed speech or language development (7 of 18), an increased frequency over their siblings and controls. IQ scores ranged from 78 to 145; however, there was a slight skew to the left in IQ distribution, with 14 of 37 children being in the 70 to 89 IQ range. Gross motor development was generally found to be normal, although there was a suggestion of fine motor problems. There currently appears to be insufficient information to make any conclusions regarding the frequency or magnitude of the increased risk for social pathology28,29 in 47,XYY individuals.
Somatic Abnormalities.
In summarizing the data concerning the 43 47,XYY neonates who were prospectively ascertained through newborn surveys, Robinson and associates28 found that birthweights, length, head circumference, and course during the first year of life were within normal ranges. There was no clear “XYY syndrome” identifiable at birth, with the majority of neonates being completely normal in appearance. There was only one major anomaly, that is, congenital hip dislocation. Eight neonates had one or more minor anomalies, including clinodactyly (2 cases) with a single crease of the fifth finger in one, inguinal hernia (2 cases), abnormal ears (2 cases), pectus carniatum, borderline large head, asymmetric head, strabismus, epicanthic folds, micrognathia, acne, philosis, simian crease, and an undefined heart murmur.
In addition to the aforementioned clinical manifestations, 47,XYY individuals are sometimes quite tall. In the Scottish prison surveys, 47,XYY inmates30 had a mean height of 71.3 inches (181.2 cm) compared with 67.2 inches (170.7 cm) for 46,XY inmates. Some investigators believe that electrocardiographic abnormalities are present31,32; however, others have not been able to confirm such abnormalities.33,34 Dermatoglyphic analyses have shown decreased total digital ridge counts consistent with the general observation of an inverse relationship between sex chromosome polysomy and total ridge count.35,36 The sizes of deciduous teeth of 47,XXY individuals have been found to be larger than controls, suggesting that the Y chromosome regulates quantitative variation of dental growth.37 Finally, abnormal electroencephalograms have been reported in 47,XYY men in psychiatric hospitals and prisons38; however, persons resident in institutions are more likely to exhibit abnormal electroencephalograms compared with normal individuals.
The evidence of fertility of 47,XYY males is limited; however, the sons of 47,XYY males usually show normal 46,XY complements.20,37,38,39,40,41 Seminiferous tubules characterized by spermatogenic arrest are detected in about 50% of 47,XYY males, and about 30% of tubules consist solely of Sertoli cells.42 Sperm counts may be low, even if testes are normal in size.42 Although somewhat controversial, the consensus is that testosterone levels are within normal ranges in most 47,XYY males. Migeon and associates43 investigated the plasma testosterone levels in 15 47,XYY males from the United States and 15 such individuals from England, whose ages ranged from 20 to 50 years. Seventeen of these 30 individuals showed a normal range of testosterone. Seven showed a level over 2 standard deviations above the mean and 6 showed a level under 2 standard deviations below the mean (normal range, 460 1 mμg/100 ml; SD, ± 125 mμg/100 ml).
GENETIC COUNSELING.
Theoretically, meiosis in 47,XYY males should lead to 50% normal gametes with a single X or Y and to 50% gametes with 24,YY or 24,XY complements; hence, 50% of the offspring should be trisomic, either 47,XYY or 47,XXY. However, offspring of 47,XYY males have usually been reported to be chromosomally normal. Some 47,XYY men have purportedly had offspring with trisomy 21,44 which raises the possibility that the abnormal sex chromosome constitution affects segregation of autosomal chromosomes as well. Further investigations are needed to understand gametogenesis in 47,XYY individuals. Until such data become available, it seems warranted to offer prenatal diagnosis in this situation.
If fetal cytogenetic studies reveal a 47,XYY complement, the couple must be fully informed regarding potential psychiatric, social, and somatic abnormalities of 47,XYY individuals. As in all cases of prenatal diagnosis, the couple must make the ultimate decision of whether to continue the pregnancy. Their decision must be supported by the physician and the entire genetic counseling team.
47,XYY and Female Phenotype
Individuals with 47,XYY cells may occasionally have female or ambiguous external genitalia and may be categorized into three groups:
- 45,X/47,XYY mosaicism. These individuals might be expected to display a wide clinical spectrum, that is, (1) almost normal male development,45 (2) ambiguous external genitalia associated either with two dysgenetic testes or with a unilateral streak gonad and a contralateral testis (mixed gonadal dysgenesis), or (3) female external genitalia and bilateral streak gonads.46 Mosaicism of the 45,X/47,XYY type has been detected in two and possibly three siblings whose parents were consanguineous47,48; therefore, in some cases this mosaicism may be influenced by genetic factors.
- Suspected 45,X/47,XYY mosaicism, 47,XYY cells may be the only demonstrable cells in individuals who have both müllerian derivatives and genital ambiguity. The presence of müllerian derivatives suggests mosaicism.
- 47,XYY individuals with female or ambiguous external genitalia but no uterus. The absence of a uterus suggests a disorder other than undetected mosaicism because a uterus is usually present in 45,X/47,XYY individuals. Some cases may represent the coincidental association of testicular feminization and 47,XYY; others are more difficult to explain and could be teratogenic in etiology.
48,XYYY
Relatively few cases of 48,XYYY have been reported.49,50,51,52,53 Presumably 48,XYYY arises from Y nondisjunction in mitosis of spermatocyte formation followed by a Y nondisjunction during meiosis I and subsequent chromatid nondisjunction of one Y during meiosis II, resulting in a sperm bearing three Y chromosomes.
Clinical features which appear similar in some 48,XYYY males include low normal intelligence quotient, behavioral problems with aggressive outbursts, repeated pulmonary infections during childhood, sparse body hair, clinodactyly of fifth fingers, acne, and hypotrophic testes.50,52 However, Hunter and Quaife51 described a patient exhibiting no stigmata other than sterility.
48,XXYY and 49,XXXYY
48,XXYY and 49,XXXYY are associated with Klinefelter's syndrome phenotype (see related chapter in this volume).
POLYSOMY X IN FEMALES
47,XXX
FREQUENCY.
Jacobs and co-workers54 first reported an individual with a 47,XXX chromosomal complement (Fig. 2) in 1959, and since that time such trisomic females have been determined to be relatively common. Jacobs7 tabulated the frequency of 47,XXX to be about 1 per 1,000 female births in reviewing seven chromosome surveys of unselected newborn infants (20 of 20,790 neonates). Hecht and Hecht18 recently confirmed this finding by reporting the frequency of XXX complement to occur in 1 of 975 female newborns. Two X-chromatin masses are detected in approximately 1 per 232 females in institutions for psychotic or neurotic individuals.55 47,XXX is rarely detected among spontaneous abortuses.7
|
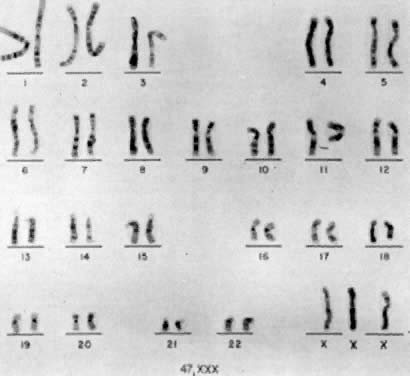
CELLULAR ETIOLOGY.
47,XXX can be the result of a nondisjunction of the two X chromosomes during meiosis I of the primary oocyte. It can also be the result of X chromatid nondisjunction during meiosis II, either of the secondary oocyte or of the secondary spermatocyte carrying an X chromosome. 48,XXXX and 49,XXXXX are due to double nondisjunction of the female germ cell. A 48,XXXX could originate from sequential nondisjunction of (1) the oogonial XX bivalents during meiosis I and (2) chromatids of one X chromosome during meiosis II, resulting in a 25,XXX ovum and, after fertilization with an X-carrying sperm, in a 48,XXXX zygote. The mechanism leading to 49,XXXXX is somewhat similar, except that chromatid nondisjunction of both X chromosomes during meiosis II has to take place to produce an ovum with four X chromosomes.
The origin of the extra X chromosome in 47,XXX patients is usually nondisjunction occurring during maternal meiosis I. Study of Xg blood group* alleles provided initial data concerning origin of the extra X chromosome56; however, Morton and associates57 found the Xg locus to be virtually uninformative about the origin of the extra X chromosome in 47,XXX women because of the long distance between the Xg locus and the centromere. This long distance makes distinction between meiosis I and meiosis II unreliable; as the distance between a gene locus and centromere increases, the chance of crossing-over between the two increases, leading to potential diagnostic discrepancies.
More recently, May and colleagues58 used X-linked DNA polymorphisms, located relatively close to the X chromosome centromere, to study parental origin of the extra X chromosome in 28 47,XXX women. Maternal nondisjunction accounted for the extra X chromosome in 26 of the 28 women; of these 26 women, the extra X chromosome was definitely shown to be an error at meiosis I in 15 women, and to an error at meiosis II in 6 women. In the remaining 5 women, the origin of the extra maternal X chromosome could not be determined.
In addition, this study revealed an association between advanced maternal age and 47,XXX offspring59 resulting from errors at maternal meiosis I but failed to reveal an association between advanced maternal age and nondisjunction occurring at maternal meiosis II. This may explain findings of Barr and colleagues,55 who found advanced maternal age to be associated with 47,XXX offspring but not as strongly as is seen in autosomal trisomies involving acrocentric chromosomes.
CLINICAL FINDINGS.
Several studies have suggested that there is an increased likelihood that 47,XXX women will exhibit developmental delay or mental illness. The magnitude of the increased risk is difficult to estimate because most 47,XXX patients have been ascertained in surveys of the mentally retarded. However, the prevalence of 47,XXX is higher among the mentally retarded than among consecutively born neonates; these observations support the apparent relationship between mental retardation and 47,XXX. Tennes and colleagues60 are prospectively studying 11 females whose 47,XXX was identified at birth. About a third have shown delayed motor and speech development. Robinson28 summarized the clinical findings from 49 children with 47,XXX complements who were studied prospectively at six different medical centers. Nineteen of 41 children studied showed delays in both receptive and expressive language development. Full-scale IQ levels were significantly lower than for siblings and controls. Five of 34 probands had IQ levels below 70. Salbenblatt and co-workers61 found 47,XXX women to exhibit both gross and fine motor dysfunction as well as abnormal sensory-motor integration. These difficulties resulted in language delays that inevitably led to poor classroom performance. In addition, 47,XXX children exhibited a delay in the age of independent walking, thereby confirming the consistency of motor dysfunction throughout development. These data must be considered as tentative because of the spectrum of ages among probands, siblings, and controls and the variety of tests used.
In addition to neurologic deficits, 47,XXX individuals exhibit dermatoglyphic abnormalities; however, it is difficult to determine whether 47,XXX individuals exhibit other somatic abnormalities more often than expected by chance. Dermatoglyphic abnormalities include decreased mean digital ridge count, increased radial loops and arches, absence of patterns in the first and second interdigital areas, wide ridges in the a-b interval, and complex patterns of the soles.62 It has been estimated that about 20% of 47,XXX women may experience delayed menarche, premature ovarian failure, and underdevelopment of secondary sexual characteristics.63,64,65 Varrela and Alvesalo66 have demonstrated taurodontism in four 47,XXX women. In addition, Lenoble and Kaplan67 described a 47,XXX woman who developed systemic lupus erythematosus (SLE), remarkable for numerous visceral localizations, multiple antinuclear antibodies, and high serum levels of IgG and IgA. However, the majority of 47,XXX individuals are normal and may be fertile.
GENETIC COUNSELING.
Theoretically, one would expect that 47,XXX women would have a 50% risk of having aneuploid offspring (i.e., 25% 46,XX; 25% 46,XY; 25% 47,XXX; 25% 47,XXY); however, most progeny are chromosomally normal. These empiric observations may reflect (1) selection against abnormal embryos, (2) preferential segregation of 24,XX into a polar body, or (3) decreased fertilizability of 24,XX oocytes. Nonetheless, various reports have suggested that 47,XXX and 46,XX/47,XXX women show some increased risk of producing chromosomally abnormal offspring. For example, in 1969, Barr and co-workers tabulated that 1 of 20 reported 47,XXX women had a 47,XXY son; 3 of 8 46,XX/47,XXX women had 47,XXY or 46,XY/47,XXY sons; and 1 of the 8 had a 46,XX/47,XXX daughter. Although controversial, there is also a suggestion that maternal trisomy X may affect the disjunction of the autosomes as well. At least two 47,XXX women have had trisomy 21 offspring.68,69 However, biases of ascertainment and reporting make the significance of the above findings uncertain. Nonetheless, it still seems prudent to offer prenatal diagnosis to 47,XXX and 46,XX/47,XXX women.
48,XXXX and 49,XXXXX
About 30 48,XXXX individuals have been reported.70,71,72,73,74,75,76 A “typical” phenotype has not yet been delineated. A few 48,XXXX have had normal appearances, whereas others have shown dysmorphic features such as simian crease, facies reminiscent of Down's syndrome, esotropia myopia, strabismus, nystagmus, and dermatoglyphic abnormalities.62,70,71,77,78,79 Alvaro-Gracia and colleagues80 described a 48,XXXX woman who developed SLE, indicating a possible association of autoimmune disorders and sex chromosome polysomy. Almost all 48,XXXX have been subnormal in intelligence; thus, 48,XXXX individuals are more likely to be retarded than are 47,XXX individuals. Hara and co-workers76 reported a pregnancy in a 48,XXXX woman which ended in the delivery of a macerated male weighing 700 gm with an omphalocele.
About a dozen cases of 49,XXXXX have been reported.81,82,83,84,85,86 Mental retardation is invariably associated with 49,XXXXX; however, major malformations are relatively uncommon, albeit higher than in 47,XXX or 48,XXXX. The anomalies most commonly present in 49,XXXXX are hypertelorism, slanting palpebral fissures, a broad nasal bridge, everted lips, esotropia, small hands and feet, abnormal teeth, clinodactyly V, a short neck, and a decreased total digital ridge count84; some of these anomalies also occur in 48,XXXX.
Although few cases of pregnancy have been reported in either 48,XXXX or 49,XXXXX women, prenatal cytogenetic diagnosis seems warranted.
Mosaicism
Mosaicism may also be associated with polysomy X. The most common mosaic complement is 46,XX/47,XXX; 47,XXX/48,XXXX and 48,XXXX/49,XXXXX have also been described.87,88
Diagnosis of Sex Chromosome Polysomy
Newborns, children, or adults suspected of having abnormal chromosome complements, including sex chromosome polysomic states, can be evaluated by cytogenetic analysis of peripheral blood lymphocytes. Women at increased risk for fetal chromosome abnormalities can now be offered several invasive prenatal diagnostic techniques for fetal cytogenetic evaluation. Chorionic villus sampling (CVS) is performed in the first-trimester (i.e., 9 to 12 weeks gestation) and can provide both rapid (2 to 4 days) results from direct analysis of uncultured cytotrophoblasts and standard culture analysis of mesenchymal core cells.89 Amniocentesis is routinely performed in the second trimester (14 to 20 weeks gestation) and provides fetal karyotypic information by analysis of cultured amniotic fluid cells. Amniocentesis can also be performed in the third trimester, and its use in the first trimester is currently being investigated.90 Percutaneous umbilical blood sampling (PUBS) can be used to obtain fetal blood samples in the second and third trimesters; analysis of fetal lymphocytes and nucleated erythrocytes can provide rapid cytogenetic results.91 However, the safety of PUBS remains to be elucidated.92
Until recently, confirmation of abnormal complements has been made by standard cytogenetic analysis (e.g., G-banded karyotyping) of cells. However, recent advancements in molecular biology techniques, specifically in situ hybridization, provides a new method for the diagnosis of polysomic states. In situ hybridization involves the hybridization of a radiolabelled probe, representing a unique sequence within the genome, to a specific chromosome in a cell during either interphase or metaphase.93 Autoradiographs show the specific area of the chromosome to which the probe was hybridized (Fig. 3). The number of chromosomes present in the cell will therefore be equal to the number of signals (discrete domains of probe hybridization) visualized. For example, a unique X-chromosome probe will exhibit three signals in a 47,XXX woman and a unique Y-chromosome probe will reveal two signals in a 47,XYY man.
|
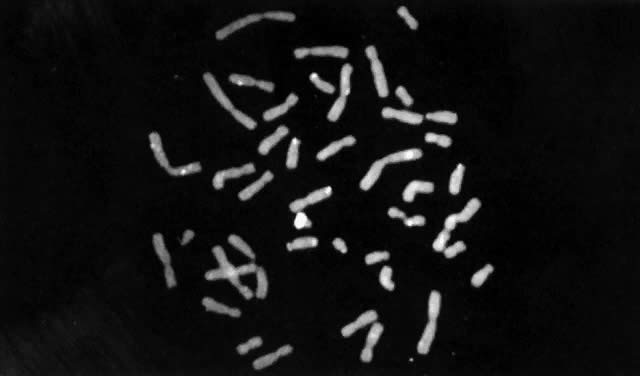
In situ hybridization has been primarily applied to analysis of cultured cells. Recently, such technology has been applied to uncultured cells, thereby allowing for rapid screening of a large number of cells for aneuploidy.94 This could prove particularly useful to evaluate suspected mosaicism. Nonetheless, for diagnostic purposes, standard cytogenetic studies should be performed whenever chromosome abnormalities, including sex chromosome polysomy, are suspected. In situ hybridization might not, for example, permit detection of structural chromosome abnormalities that can result in phenotypes similar to those resulting from numerical chromosome abnormalities.
REFERENCES
Simpson JL: Disorders of Sexual Differentiation: Etiology and Clinical Delineation. New York, Academic Press, 1976 |
|
Sandberg AA, Koeph GF, Ishihara R: An XYY human male. Lancet 2: 488, 1961 |
|
Jacobs PA, Brunton M, Melville MM et al: Aggressive behaviour, mental subnormality, and the XYY male. Nature 208: 135, 1965 |
|
Casey MD, Blank CE, Street DRK et al: YY chromosomes and anti-social behavior. Lancet 2: 859, 1966 |
|
Casey MD, Segall LJ, Street DRK et al: Sex chromosome abnormalities in two state hospitals for patients requiring special security. Nature 209: 641, 1966 |
|
Hook EB: Behavioral implications of XYY. Science 170: 139, 1973 |
|
Jacobs PA: The incidence and etiology of sex chromosome abnormalities in man. Birth Defects 15: 3, 1979 |
|
Sergovich F, Valentine GH, Chen ATL et al: Chromosome aberrations in 2159 consecutive newborn babies. N Engl J Med 280: 851, 1969 |
|
Lubs HA, Ruddle FH: Applications of quantitative karyotype to chromosome variation in 4400 consecutive newborns. In Jacobs P, Price WH, Law P (eds): Human Population Cytogenetics, pp 120–142. Edinburgh, University of Edinburgh Press, 1970 |
|
Jacobs PA, Melville M, Ratcliffe S et al: A cytogenetic survey of 11,680 newborn infants. Ann Hum Genet 37: 359, 1974 |
|
Nielsen J, Sillesen I: Incidence of chromosome aberrations among 11,148 newborn infants. Humangenetik 30: 1, 1975 |
|
Hamerton JL, Canning H, Ray M: A cytogenetic survey of 14,069 newborn infants: I. Incidence of chromosome abnormalities. Clin Genet 8: 223, 1975 |
|
Walzer S, Gerald PS: A chromosome survey of 13,751 male newborns. In Hook EB, Porter IH (eds): Population Cytogenetics, pp 45–61. New York, Academic Press, 1977 |
|
Bochkov NP, Kuleshov NP, Chebotarev AN et al: Population cytogenetic investigation of newborns in Moscow. Humangenetik 22: 139, 1974 |
|
Lin CC, Gedeon MM, Griffith P et al: Chromosome analysis of 930 consecutive newborn children using quinacrine fluorescent banding technique. Hum Genet 31: 315, 1976 |
|
Robinson A, Goad WB, Puck TT et al: Studies on chromosomal nondisjunction in man. III. Am J Hum Genet 21: 466, 1969 |
|
Bell AG, Corey PN: A sex chromatin and Y body survey of Toronto newborns. Can J Genet Cytol 16: 239, 1974 |
|
Hecht F, Hecht BK: Aneuploidy in Humans: Dimensions, demography and dangers of abnormal numbers of chromosomes. In Vig BK, Sandberg AA (eds): Aneuploidy. Part A: Incidence and Etiology, pp 9–49. New York, Alan R. Liss, 1987 |
|
Hook EB, Porter IH: Human population cytogenetics: Comments on racial differences in frequency of chromosome abnormalities, putative clustering of Down's syndrome and radiation studies. In Hook EB, Porter IH (eds): Population Cytogenetics, pp 353–365. New York, Academic Press, 1977 |
|
Thompson H, Melnyk J, Hecht F: Reproduction and meiosis in XYY. Lancet 2: 831, 1967 |
|
Laxova R, McKeown MA, Saldaña P et al: A case of XYY Down's syndrome confirmed by autoradiography. J Med Genet 8: 215, 1971 |
|
Court-Brown WM: Males with an XYY sex chromosome complement. J Med Genet 5: 341, 1968 |
|
Price WH, Strong JA, Whatmore PB et al: Criminal patients with XYY sex-chromosome complement. Lancet 1: 565, 1966 |
|
Price WH, Whatmore PB: Behaviour disorders and pattern of crime among XYY males identified at a maximum security hospital. Br Med J 1: 533, 1967 |
|
Witkin HA, Mednick SA, Schulsinger F et al: Criminality in XYY and XXY men. Science 193: 547, 1976 |
|
Hook EB: Extra sex chromosomes and human behavior: The nature of the evidence regarding XYY, XXY, XXYY, and XX genotypes. In Vallet HL, Porter IH (eds): Genetic Aspects of Sexual Differentiation. New York, Academic Press, 1978 |
|
Kessler S, Moos RH: Behavioral aspects of chromosomal disorders. Annu Rev Med 24: 89, 1973 |
|
Robinson A, Lubs HA, Nielsen J et al: Summary of clinical findings: Profiles of children with 47,XXY, 47,XXX and 47,XYY karyotypes. Birth Defects 15: 261, 1979 |
|
Walzer S, Gerard PS, Shah SA: The XYY genotype. Annu Rev Med 29: 563, 1978 |
|
Jacobs PA, Price WH, Court-Brown WM et al: Chromosome studies on men in a maximum security hospital. Ann Human Genet 31: 339, 1968 |
|
Price WH: The electrocardiogram in males with extra Y chromosomes. Lancet 1: 1106, 1968 |
|
Price WH, Lauder IJ, Wilson J: The electrocardiogram and sex chromosome aneuploidy. Clin Genet 6: 1, 1974 |
|
Valentine GH, McClelland MA, Sergovich FR: The growth and development of four XYY infants. Pediatrics 48: 583, 1971 |
|
Vianna AM, Frota-Pressoa O, Lion MF et al: Searching for XYY males through electrocardiograms. J Med Genet 9: 155, 1972 |
|
Holt SB: Dermatoglyphics and sex chromosomes. In Rashad MN, Morton WRM (eds): Genital Anomalies, p 375. Springfield, Il, Charles C. Thomas, 1969 |
|
Borgoankar DS, Mules E: Comments on patients with sex chromosome aneuploidy: Dermatoglyphes, parental ages, Xg blood group. J Med Genet 7: 345, 1970 |
|
Alvesalo L, Kari M: Sizes of deciduous teeth in 47,XYY males. Am J Hum Genet 29: 485, 1977 |
|
Volavka J, Mednick SA, Sergeant J et al: Electroencephalograms of XYY and XXX men. Br J Psychiatry 130: 43, 1977 |
|
Melnyk J, Thompson H, Rucci AJ et al: Failure of transmission of the extra chromosome in subjects with 47,XYY karyotype. Lancet 2: 797, 1969 |
|
Owen DR: The 47,XYY male. Psychol Bull 78: 209, 1972 |
|
Baghdassarian A, Bayard F, Borgoankar DS: Testicular function in XYY men. Johns Hopkins Med J 136: 15, 1975 |
|
Skakkebaek NE, Hulten M, Jacobsen P et al: Quantification of human seminiferous epithelium: II. Histological studies in eight 47,XYY men. J Reprod Fertil 32: 391, 1973 |
|
Migeon CR, quoted by Jones HR Jr, Scott WM: Hermaphroditism, Genital Anomalies, and Related Endocrine Disorders, 2nd ed. Baltimore, Williams & Wilkins, 1971 |
|
Jones HR Jr, Scott WM: Hermaphroditism, Genital Anomalies, and Related Endocrine Disorders, 2nd ed. Baltimore, Williams & Wilkins, 1971 |
|
Court-Brown WM, Price WH, Jacobs PA: Further information on the identity of 47,XYY males. Br Med J 2: 235, 1968 |
|
Suñe MV, Centeno JV, Salzano FM: Gonadoblastoma in a phenotypic female with 45,X/47,XYY mosaicism. J Med Genet 7: 410, 1970 |
|
Goldstein A, Hausknecht R, Hsu LY et al: Sex chromosome mosaicism in 3 sibs. Am J Obstet Gynecol 107: 108, 1970 |
|
Hsu LYF, Hirschhorn K, Goldstein A et al: Familial chromosomal mosaicism, genetic aspects. Ann Hum Genet 33: 343, 1970 |
|
Townes PC, Ziegler NA, Lenhard LW: A patient with 48 chromosomes (XYYY). Lancet 1: 1041, 1965 |
|
Schoepflin GS, Centerwall WR: 48,XYYY: A new syndrome? J Med Genet 9: 356, 1972 |
|
Hunter H, Quaife R: A 48,XYYY male: A somatic and psychiatric description. J Med Genet 10: 80, 1973 |
|
Ridler MAC, Lax R, Mitchell MJ et al: An adult male with XYYY sex chromosomes. Clin Genet 4: 69, 1973 |
|
Sele B, Bachelot Y, Richard J et al: Les hommes 48,XYYY. Pediatrie 30: 601, 1975 |
|
Jacobs PA, Blake AG, Court-Brown WM et al: Evidence on the existence of the human “super female.” Lancet 2: 423, 1959 |
|
Barr ML, Sergovich RF, Carr DH et al: The triplo-X female: An appraisal based on a study of 12 cases and a review of the literature. Can Med Assoc J 101: 247, 1969 |
|
Sanger R, Tippett P, Gavin J: Xg groups and sex abnormalities in people of Northern European ancestry. J Med Genet 8: 417, 1971 |
|
Morton NE, Wu D, Jacobs PA: Origin of sex chromosome aneuploidy. Ann Hum Genet 52: 85, 1988 |
|
May KM, Jacobs PA, Lee M et al: The parental origin of the extra X chromosome in 47,XXX females. Am J Hum Genet 46: 754, 1990 |
|
Court-Brown WM, Law P, Smith PG: Sex chromosome aneuploidy and parental age. Ann Hum Genet 33: 1, 1969 |
|
Tennes K, Puck M, Bruant K et al: A developmental study of girls with trisomy X. Am J Hum Genet 27: 71, 1975 |
|
Salbenblatt JA, Meyers DC, Bender BG et al: Gross and fine motor development in 45,X and 47,XXX girls. Pediatrics 84: 678, 1989 |
|
Schaumann B, Alter M: Dermatoglyphics in Medical Disorders. New York, Springer-Verlag, 1976 |
|
Johnston AW, Ferguson-Smith MA, Handmacker SD et al: The triple X syndrome: Clinical, pathological and chromosomal studies in three mentally retarded cases. Br Med J 2: 1046, 1961 |
|
Ferrier P: Cytogénétique de la différentiation sexuelle chez l'homme. J Génét Hum 14: 1, 1965 |
|
Smith HC, Seale JP, Posen S: Premature ovarian failure in a triple X female. J Obstet Gynaecol Br Commonw 81: 405, 1974 |
|
Varrela J, Alvesalo L: Taurodontism in females with extra X chromosomes. J Craniofac Genet Dev Biol 9: 129, 1989 |
|
Lenoble L, Kaplan G: Lupus erythemateux dissemine chez une femme atteinte d'un hypogonadisme primaire 47XXX. Rev Med Interne 8: 430, 1987 |
|
Kadotoni T, Ohama K, Makino S: A case of 21-trisomic Down's syndrome from the triplo-X mother. Proc Jpn Acad Med 46: 709, 1970 |
|
Singer J, Sachdeva S, Smith GF et al: Triple X female with a Down's syndrome offspring. J Med Genet 9: 238, 1972 |
|
Carr DH, Barr ML, Plunkett ER: An XXXX sex chromosome complex in two mentally defective families. Can Med Assoc J 84: 131, 1961 |
|
Park IJ, Tyson JE, Jones HW Jr: A 48,XXXX female with mental retardation. Obstet Gynecol 35: 248, 1970 |
|
Blackston RA, Chen ATL: A case of 48,XXXX female with normal intelligence. J Med Genet 9: 230, 1972 |
|
Waldbaum R, Vandervelde-Staquet MF, Lefebvre C: Syndrome 48,XXXX chez un nourrisson. J Génét Hum 21: 43, 1973 |
|
Gardner RJM, Veale AMO, Sands VE et al: XXXX syndrome: Case report and a note on genetic counseling and fertility. Humangenetik 17: 323, 1973 |
|
Peña SDJ, Ray M, Douglas G et al: A 48,XXXX female. J Med Genet 11: 211, 1974 |
|
Hara S, Haywood BD, Davis KK et al: A black female with the 48,XXXX chromosome constitution. Ann J Ment Defic 79: 464, 1975 |
|
DeGrouchy J, Brissaud HE, Richardet JM et al: Syndrome 48,XXXX chez une enfant de six ans: Transmission anormale du group Xg. Ann Genet 11: 120, 1968 |
|
Telfer MA, Richardson CE et al: Divergent phenotypes among 48,XXXX and 47,XXX females. Am J Hum Genet 22: 326, 1970 |
|
Berkeley MF, Faed MW: A female with the 48,XXXX karyotype. J Hum Genet 7: 83, 1970 |
|
Alvaro-Gracia JM, Humbria A, Garcia-Vicuna R et al: Systemic lupus erythematosus and tetrasomy-X. J Rheumatol 16: 1486, 1989 |
|
Kesaree N, Woller PV, Samson M: A phenotypic female with 49 chromosomes, presumably XXXXX. J Pediatr 63: 1099, 1963 |
|
Brody J, Fitzgerald MG, Spiers ASD: A female child with 5 X chromosomes. J Pediatr 70: 105, 1967 |
|
Zajaczkowsa K, Korniszeqski L, Wolff-Plsdowska A: A case of quintuple-X syndrome (49,XXXXX). J Ment Def Res 14: 305, 1970 |
|
Sergovich FR, Uilenberg C, Pozsonyi J: The 49,XXXXX chromosome constitution: Similarities to the 49,XXXXY condition. J Pediatr 78: 285, 1971 |
|
Yamada Y, Neriishi S: Penta X (49,XXXXX) chromosome constitution: A case report. Jpn J Hum Genet 16: 15, 1971 |
|
Larget-Piet L, Rivron J, Baillif P et al: Syndrome 49,XXXXX chez une fille de 5 ans. Ann Genet 15: 115, 1972 |
|
Cooke P, Black JA, Curtis DJ: Comparative clinical studies and X chromosome behaviour in a case of XXXX/XXXXX mosaicism. J Med Genet 9: 235, 1972 |
|
Bergemann E: Manifestations familiale du karyotype triplo-X: Communication du karyotype triplo-X communication preliminale. J Génét Hum 10: 370, 1962 |
|
Shulman LP, Elias S: Chorionic villus sampling. Pediatr Ann 18: 714, 1989 |
|
Penso CA, Frigoletto FD Jr: Early amniocentesis. Sem Perinatol 14: 465, 1990 |
|
Tipton RE, Tharapel AT, Chang HT et al: Rapid chromosome analysis using spontaneously dividing cells derived from umbilical cord blood (fetal and neonatal). Am J Obstet Gynecol 161: 1546, 1989 |
|
Shulman LP, Elias S: Percutaneous umbilical blood sampling, fetal skin sampling, and fetal liver biopsy. Sem Perinatol 14: 456, 1990 |
|
Pinkel D, Straume T, Gray JW: Cytogenetic analysis using quantitative, high-sensitivity, fluorescence hybridization. Proc Natl Acad Sci USA 83: 2934, 1986 |
|
Cremer T, Popp S, Emmerich P et al: Rapid metaphase and interphase detection of radiation-induced chromosome aberrations in human lymphocytes by chromosomal suppression in situ hybridization. Cytometry 11: 110, 1990 |