
Alloimmune Hemolytic Disease of the Fetus and Newborn (Erythroblastosis Fetalis): Diagnosis, Management, and Prevention
Authors
INTRODUCTION
Although Hippocrates may have described fetal hemolytic disease (erythroblastosis fetalis, hemolytic disease of the newborn), the first recorded report is that of a French midwife in 1609, who reported the birth of twins: the first was grossly edematous (hydrops fetalis) and died promptly; the second, in good condition at birth, became deeply jaundiced (icterus gravis) and died a few days later.
Diamond and co-workers, in 1932, were the first to show that hydrops fetalis, icterus gravis, and severe anemia of the newborn were the same disease albeit of differing severity, characterized by hemolysis of the red blood cells (RBCs) of the fetus and newborn and an outpouring of immature nucleated RBCs (erythroblasts). Darrow theorized that the hemolysis was due to the transplacental passage of a maternal antibody, antifetal hemoglobin, into the fetal circulation. Her theory was correct, but her postulated antigen (fetal hemoglobin) and antibody were untenable.
The epoch-making experiments of Landsteiner and Wiener in 1940 revealed the true state of affairs. They produced a rhesus monkey RBC antiserum in rabbits and guinea pigs. The rhesus (Rh) antisera produced agglutination of the RBCs of 85% of a group of white subjects (Rh positive) but not of the remaining 15% (Rh negative). This experiment laid the foundation for safe blood transfusion, the determination of the cause of Rh hemolytic disease, and the development of the science of human anthropology. Although the rhesus monkey antigen, now named LW, is not exactly the same as the human Rh antigen (Rh-negative people usually are weakly LW positive), this in no way diminishes the importance of Landsteiner and Wiener's work.
In the same year, Wiener and Peters demonstrated that most transfusion reactions were due to transfusion of Rh-negative patients with Rh-positive blood. In the following year, Levine and associates showed that the presence of Rh antibodies in Rh-negative pregnant women was the cause of erythroblastosis.
Since this work in the 1940s, the complexities of the Rh blood group system have been unraveled. There are more than 40 antigens in the system. Other blood group systems, some of clinical importance, have been discovered. Sensitive methods of screening for blood group antibodies and of measuring their strength are available, and the pathogenesis of maternal blood group immunization and of erythroblastosis fetalis is now known. The severity of erythroblastosis can be assessed accurately in utero. Methods of treatment of both fetus and newborn are now available, and since 1967, there has been a method of preventing Rh immunization, which has reduced its incidence greatly in the past 30 years.
THE Rh SYSTEM
The Rh blood group system is still the most important factor in erythroblastosis fetalis. It is made up of a family of inherited antigens. Although Wiener's theory of a single gene locus occupied by a pair of complex agglutinogens is the most accurate,1 the nomenclature and theories of inheritance of Fisher and Race2 are simpler and more practical. They theorize that there are three pairs of Rh antigens, commonly Dd, Cc, and Ee. The presence of D indicates an Rh(D)-positive person. The absence of D, not the presence of d, which has never been proved to exist, denotes an Rh(D)-negative person. The production of anti-D in Rh(D)-negative women causes erythroblastosis fetalis in Rh(D)-positive fetuses. The antigens are inherited in two sets of three, one set from each parent. CDe(R1), c(d)e(r), and cDE(R2) are the most common sets. CDE(Rz) is uncommon in whites but not rare in Native Americans. C(d)E(ry) is exceedingly rare.
Slightly fewer than half of Rh-positive people are homozygous for D (i.e., they have inherited a set of antigens containing D from both sets of parents). The remainder are heterozygous for D (i.e., they have inherited a D-containing set from only one parent). The D zygosity of an Rh-positive mate of an Rh-negative woman is important. If he is homozygous, all of his children will be Rh-positive; if he is heterozygous, there is an equal chance that the fetus will be Rh- negative or Rh-positive. Only Rh-positive fetuses can cause Rh immunization, and only Rh-positive fetuses are affected by the Rh antibody produced.
Because anti-d has never been determined, a man's zygosity for D can only be determined if he fathers two infants who have received different sets of antigens from him. Because certain sets of antigens are more common than others (Table 1), determination of the presence or absence of the other Rh antigens, C, E, c, and e, indicates the likely, but not certain, zygosity of the father for D (Table 2).
TABLE 1. Rh Gene Frequencies in a White Canadian Population of 2000 Unrelated Adults
Gene Complex | Frequency (%) |
CDe (R1) | 41 |
cde (r) | 39 |
cDE (R2) | 16 |
cDe (R0) | 2.2 |
Cde (r') | 1.1 |
cdE (r”) | 0.6 |
CDE (R2) | 0.08 |
CdE (ry) | 0.00 |
(Lewis M, Kaita H, Chown B: The inheritance of the Rh blood groups: Frequencies in 1000 unrelated Caucasian families consisting of 2000 parents and 2806 children. Vox Sang 20:502, 1971)
TABLE 2. Zygosity for Rh(D) of D-Positive Husband (Mother D Negative)*
Antigens | A | B | C |
Present in | (Most Likely | (Less Likely | (Least Likely |
Father | Rh Genotype) | Rh Genotype) | Rh Genotype) |
1. CDe | CDe/CDe (R1R1) | CDe/Cde (R1r') |
|
| Homozygous | Heterozygous |
|
2. CDce | CDe/cde (R1r) | CDe/cDe (R1R”) | Cde/cDe (r'R0) |
| Heterozygous | Homozygous | Heterozygous |
3. CDEce | CDe/cDE (R1R2) | Cde/cDE (r'R2) | CDE/cDe (R2R0) |
| Homozygous | CDe/cdE (R1r”) | Homozygous |
|
| CDE/cde (R2r) |
|
|
| Heterozygous |
|
4. DEc | cDE/cDE (R2R2) | cDE/cdE (R2r”) |
|
| Homozygous | Heterozygous |
|
5. DEce | cDE/cde (R2r) | cDE/cDe (R2R0) | cdE/cDe (r”R0) |
| Heterozygous | Homozygous | Heterozygous |
6. Dce | cDe/cde (R0r) | cDe/cDe (R0R0) |
|
| Heterozygous | Homozygous |
|
* Genotypes 1A and 4A can never be proved because the infant will be only one paternal genotype (CDe in 1A and cDE in 4A). The remainder of the father's possible genotypes can be proved only if he produces children of two different genotypes.
(Bowman JM, Friesen RF: Rh-isoimmunization. In Goodwin JW, Godden JO, Chance G (eds): Perinatal Medicine, p 93. Baltimore, Williams & Wilkins, 1976)
There are many more antigens in the Rh system than the five described. Forty-three antibodies have been discovered that delineate Rh antigens that are alleles at one or other of the five loci. Cw, an allele of C, and Du, an allele of D, are not uncommon. Rarely, a Du mother carrying a D-positive fetus may produce anti-D, which on one occasion has been reported to cause hydrops fetalis.3 Even more uncommonly, an Rh-negative mother carrying a Du fetus may become Rh immunized. The presence of D variants is more common in blacks. Tippett and Sanger4 have described six degrees of D variants in these partially Rh-positive people.
Rh-ANTIGEN STRUCTURE AND DISTRIBUTION
The Rh(D) antigen (approximate molecular weight, 30,000 kd) is associated with the membrane skeleton of the RBC.5 It appears to be a proteolipid.6 Unlike the ABO antigens, which are ubiquitous, the Rh antigens appear to be confined to the RBC membrane. They are an essential component of the membrane. The rare people who lack Rh antigens (Rh null) have defective RBC membranes and mild to moderate hemolytic anemia. There is some, albeit disputed, evidence that the Rh(D) may be present in human trophoblast.7
In 1992, the Rh(D) and Rh(non-D) polypeptides were cloned. Cartron and his group8 have determined that the Rh blood group locus is the product of two homologous structural genes, one of which encodes the Cc Ee polypeptide; the other (missing in Rh(D)-negative people) encodes the D antigen polypeptide. The predicted translation of the Rh(D) mRNA is a 417-aminoacid product with a molecular weight of 45,000. The D and Cc/Ee polypeptides differ by 36 aminoacid substitutions (8.4% divergence). The similarity between the two genes supports the belief that they evolved by duplication of a common ancestral gene.
Rh negativity is a trait of white people. In most whites, the incidence is 15% to 16%. In Finland, it is only 11% to 12%. In the Basques, the incidence is about 35%. Races other than white, millennia ago, were probably all Rh(D) positive. They owe their present incidence of Rh negativity to intermingling of white genes (Native Americans and Inuits, about 1%; American blacks, 7% to 8%; Indo-Eurasians, about about 2% to 4%; Asiatic Chinese and Japanese, almost zero).
PATHOGENESIS OF Rh IMMUNIZATION
Transfusion
Before the discovery of the Rh blood group system, blood transfusion was a common cause of Rh immunization. It is still the most common cause of non-D blood group immunization because transfused blood is compatible only for ABO and D. After blood transfusion, 1% to 2% of people develop blood group antibodies; most are of little consequence. Anti-c, Kell, and occasionally E, C, and Fya may cause erythroblastosis as severe as that caused by anti-D.
Fetomaternal Transplacental Hemorrhage
Despite prohibition of transfusions of D-positive blood into D-negative people, a significant incidence of Rh immunization persists. Wiener9 theorized that Rh immunization could only be due to the transplacental passage of fetal D-positive RBCs into the circulation of Rh-negative women during pregnancy or at the time of delivery. This theory was proved by Chown.10 After delivery of an anemic D-positive infant, an Rh-negative primipara was shown to have 5% circulating Rh-positive fetal RBCs. Within 20 days after delivery, she developed a strong Rh antibody.
Determination of the incidence and size of fetomaternal transplacental hemorrhage (TPH), using the acid elution test of Kleihauer and colleagues,11 is now possible (Fig. 1). The Kleihauer test has allowed the determination of the incidence and size of fetal TPH at varying gestations, at delivery, and after obstetric procedures such as amniocentesis, external version, and manual removal of the placenta.
|
Studies show that 75% of women have a fetomaternal TPH during pregnancy or at the time of delivery. In about half of these women, the TPH does not exceed 0.1 mL of fetal blood. Less than 1% have more than 5 mL, and less than 0.25% have more than 30 mL of fetal blood in their circulations. Certain obstetric situations increase the risk of TPH: antepartum bleeding, toxemia of pregnancy, external version, cesarean section, and manual removal of the placenta.
As pregnancy progresses, the incidence and size of fetomaternal TPH increase from less than 0.1 mL in about 10% of women in the first trimester to 45% in the third trimester.
Amniocentesis, particularly if not carried out under ultrasound guidance, is a hazard (11.2% incidence of TPH after amniocentesis before the advent of ultrasound in one series).12
A significant number of abortions, both spontaneous (5%) and therapeutic (20% to 25%), are associated with fetomaternal TPH. The D antigen is well developed by 30 to 45 days' gestation. After therapeutic abortion, about 4% of women may have TPH in excess of 0.2 mL of fetal blood.
Rh IMMUNE RESPONSE
Primary Immune Response
The primary immune response develops slowly. In experimental Rh immunization of male volunteers, 8 to 9 weeks elapsed before the response was apparent; indeed, it may not be detectable for 6 months.
The primary response usually is weak and predominantly immunoglobulin M (IgM). IgM anti-D does not cross the placenta and does not cause fetal RBC hemolysis. Most Rh-negative women convert to IgG anti-D production. IgG anti-D traverses the placenta and does produce fetal RBC hemolysis.
Secondary Immune Response
Once the primary response has occurred, a second exposure to Rh-positive RBCs produces a rapid (within days) increase in anti-D, which is for the most part IgG. Subsequent exposure may produce even higher levels. If the intervals between antigen exposure are long, the subsequent exposure commonly is associated with a marked increase in Rh-antibody titer and increased avidity (binding ability) of the antibody for Rh antigen. The greater the avidity of the Rh antibody for Rh antigen (binding constant), the greater is the severity of Rh hemolytic disease.
DETECTION AND MEASUREMENT OF Rh ANTIBODIES
Saline
Only IgM anti-D agglutinates Rh antibodies suspended in isotonic saline. The IgG anti-D molecule is too small to bridge the distance between RBCs suspended in saline. Although it coats the Rh-positive RBCs, it cannot agglutinate them. If a maternal serum contains only IgG anti-D (most do), no saline RBC agglutination occurs. In the early Rh era, when saline-suspended RBC antibody screening tests were the only tests available, great confusion ensued. Many Rh-negative women giving birth to severely affected erythroblastotic infants had no apparent Rh antibodies in their circulations.
Colloid
Wiener13 noted that Rh antibodies (IgG) that did not agglutinate Rh-positive RBCs when they were suspended in saline promptly did so when the same RBCs were suspended in a colloid medium such as gum acacia or albumin. The colloid media have a higher dielectric constant, which reduces the membrane negative electrical potential, bringing the RBCs closer together. IgG anti-D then can bridge the gap and agglutinate the RBCs. The most commonly used colloid medium is bovine serum albumin.14
IgM anti-D also agglutinates RBCs suspended in a colloid medium. When the saline and colloid (albumin) titers are at the same level, the amount of IgG anti-D present is unknown. Mixing such sera with dithiothreitol disrupts IgM sulfhydryl bonds, leaving IgG intact. Subsequent retitration in colloid allows determination of the true IgG anti-D level.
Indirect Antihuman Globulin
Antihuman globulin is produced when human serum (or specific human IgG) is injected into other animals (rabbits, guinea pigs, goats). The animals recognize the human material as foreign and produce an antihuman globulin (Coombs' serum).
Rh-positive RBCs after incubation with serum being screened for anti-D or after incubation with doubling dilutions of serum known to contain anti-D are then washed four times in isotonic saline. The washing removes nonadherent serum proteins but not any anti-D because it is adherent to the D antigenic sites on the RBC membrane. The washed RBCs are then suspended in the animal antihuman globulin (Coombs') serum. If the RBCs are coated with anti-D, agglutination occurs, and the test result is positive. The reciprocal of the greatest serum dilution in which agglutination occurs is the indirect antiglobulin titer.15 This titer is a more sensitive antibody screening and titration method than are colloid techniques. Indirect antiglobulin titers usually are one to three dilutions greater than are colloid (albumin) titers.
Enzyme
Incubation of RBCs with enzymes, such as trypsin, bromelin, ficin, or papain, reduces the membrane electrical potential. The RBCs suspended in saline lie in closer apposition. They are agglutinable by IgG anti-D. Enzyme techniques are the most sensitive manual Rh-antibody screening methods.16
Autoanalyzer
Autoanalyzer techniques are available to detect and measure Rh antibodies (the bromelin method of Rosenfield and Haber17; the low ionic polybrene method of Lalezari18). Autoanalyzer methods are the most sensitive techniques for the determination of Rh antibodies; however, an Rh antibody demonstrable only by Autoanalyzer and not by any manual method may not represent true Rh immunization. The bromelin autoanalyzer technique has been modified by Moore to allow quantitation of serum anti-D.19
DOSE OF D ANTIGEN NECESSARY TO PRODUCE IMMUNIZATION
Rh Immunization Experiments
Amounts of Rh-positive blood required to produce Rh immunization may be small. In one study, half of volunteers were immunized by 10 mL of blood. In other experiments, two thirds were immunized by five injections of 3.5 mL, 80% by one injection of 0.5 mL of D-positive RBCs,20 and 30% by repeated injections of 0.1 mL of RBCs.21 The incidence of Rh immunization depends on the dose of Rh-positive RBCs: 15% after 1 mL, 33% after 40 mL, and 65% to 70% after 250 mL. Secondary immune responses occur after exposure to much smaller amounts (as little as 0.1 mL of Rh-positive RBCs).
Rh Immunization Clinical Studies
Kleihauer fetal cell studies during pregnancy and immediately after delivery allow the determination of the risk of Rh immunization in relation to the presence and size of fetomaternal TPH. If the TPH always is less than 0.1 mL of RBCs, the incidence of Rh immunization detectable up to 6 months after delivery is 3%21; when volumes exceed 0.4 mL, the incidence is 22%.20 Because in 75% to 80% of pregnancies, TPH (if any) are always less than 0.1 mL, most women are Rh immunized as a result of small or nondetectable TPH.
FREQUENCY OF Rh IMMUNIZATION
The incidence of Rh immunization demonstrable within 6 months after delivery of the first Rh-positive, ABO compatible infant is 8% to 9%. Nevanlinna,22 by mounting a secondary Rh immune response in the next Rh-positive pregnancy, observed that about the same number who had no detectable Rh antibodies after delivery demonstrated that they also were Rh immunized by the previous Rhpositive pregnancy, a phenomenon he called sensibilization. Therefore, the true incidence of Rh immunization as a result of the first Rh-positive, ABO-compatible pregnancy is about 16%.
A woman not immunized by the first such pregnancy is at about the same risk in a second Rh-positive, ABO-compatible pregnancy. As parity increases and the number of women capable of an Rh immune response diminishes because they have become immunized, the number who mount an immune response will decrease because of a greater residual number of nonresponders. By the time an Rh-negative woman has completed five ABO-compatible, Rh-positive pregnancies, there is more than a 50% probability that she will be Rh immunized. Before the Rh prevention era, 0.8% to 1.0% of pregnant women were Rh immunized.
ABO incompatibility confers partial protection for a mother against Rh immunization. The incidence of Rh immunization 6 months after delivery of an ABO-incompatible Rh-positive infant is 1.5% to 2.0%.20 Partial protection probably is due to rapid intravascular hemolysis of the ABO-incompatible Rh-positive RBCs, with sequestration of Rh-positive stroma in the liver, an organ with poorer antibody-forming potential than the spleen, which is the site of stroma sequestration when extravascular RBC destruction occurs. ABO incompatibility confers substantial protection against the primary Rh immune response; it confers no protection against the secondary Rh immune response.
Rh immunization during pregnancy, once thought to be a rare phenomenon, is not uncommon. In one series, 1.8% (62 of 3533) of Rh-negative women without evidence of Rh immunization in early pregnancy were Rh immunized during pregnancy or within 3 days after delivery23 (Table 3). Rh immunization during pregnancy accounts for 14% of the 13% of Rh-negative women who become Rh immunized by an Rh-positive pregnancy (16% in the 80% with ABO-compatible, 1.5% in the 20% with ABO-incompatible fetuses; 13% overall). Rh immunization during pregnancy is a major factor in Rh immunization prevention and is discussed further later in this chapter.
TABLE 3. Rh Immunization During Pregnancy or Within 3 Days After Delivery (Manitoba, March 1, 1967, to December 15, 1974)
| No. of |
|
| Rh-Negative | No. (and %) |
Blood Group of Infant | Pregnant Women | Rh Immunized |
Rh positive, ABO compatible | 2859 | 58 (2.0) |
Rh positive, ABO incompatible | 674 | 4 (0.6) |
Total of all women* | 3533 | 62 (1.8) |
*Primigravidas and multigravidas given Rh immune globulin after every preceding Rh-positive pregnancy and abortion.
(Bowman JM, Chown B, Lewis M et al: Rh-immunization during pregnancy: Antenatal prophylaxis. Can Med Assoc J 118:623, 1978)
As already noted, fetomaternal TPH occurs after abortion. The risk of Rh immunization occurring after spontaneous abortion is about 1.5% to 2%, increasing the later in gestation that abortion occurs. It is considerably higher after therapeutic abortion (about 4%). Women who are Rh immunized after the relatively small TPHs that occur at the time of abortion are good responders. They commonly have severely affected infants in subsequent pregnancies. Although the risk of Rh immunization after abortion at 6 to 8 weeks' gestation is small, it becomes significant by 10 to 12 weeks.
PATHOGENESIS OF Rh ERYTHROBLASTOSIS FETALIS
Erythropoiesis is present in the yolk sac of the human embryo by the third week of gestation. Rh antigen has been found in the RBC membrane by the sixth week. By 8 to 10 weeks' gestation, RBC production has begun in the liver and spleen. Under normal conditions, erythropoiesis has shifted to, and is confined to the bone marrow by the sixth month. In the presence of fetal anemia, due to either hemolysis or blood loss, erythropoiesis may persist in the liver and spleen and may be extreme.
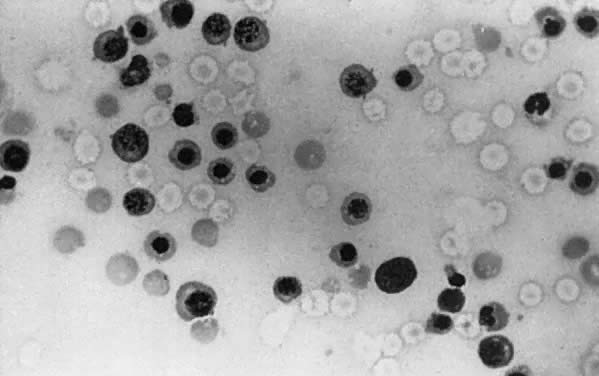
The fundamental cause of erythroblastosis fetalis is hemolysis of Rh-positive fetal RBCs by maternal anti-D IgG antibody. Hemolysis causes fetal anemia, which stimulates the production of erythropoietin. Erythropoiesis increases. Fetal marrow RBC production cannot keep up with the RBC destruction, and extramedullary erythropoiesis (spleen, liver, kidneys, adrenals) recurs. Hepatosplenomegaly (Fig. 2) is a hallmark of erythroblastosis fetalis.
|
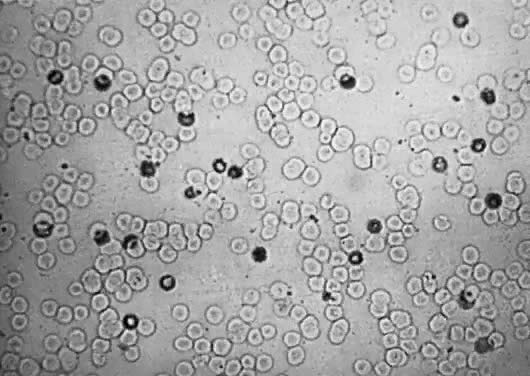
In the presence of extramedullary erythropoiesis, RBC maturation is poorly controlled. Immature nucleated RBCs, from normoblasts to early erythroblasts (Fig. 3), are poured into the circulation.
|
Mechanism of Red Blood Cell Hemolysis
COMPLEMENT MEDIATED.
When antibody fixes complement, such as anti-A and anti-B, severe RBC damage occurs. Large defects are produced in the RBC membrane. Intravascular hemolysis with hemoglobinemia and hemoglobinuria occurs. RBC debris is picked up for the most part in the liver, where it is phagocytized by the reticuloendothelial cells in the microcirculation.
NON–COMPLEMENT MEDIATED.
When antibodies do not fix complement, such as anti-D, either IgG or IgM, the mechanism of hemolysis is different. It is more subtle but in the end as destructive as that of anti-A or anti-B. When anti-D attaches to the D antigen in the RBC membrane, the attraction of macrophages for the coated RBCs (chemotaxis) is increased. The coated RBCs adhere to the macrophages, forming rosettes, particularly in the spleen, where the circulation slows and the hematocrit increases, bringing RBCs and macrophages into close apposition. Electron microscopy reveals macrophage pseudopods attaching to the RBC membrane, puckering and invaginating it.24 A portion of the membrane breaks off, and the defect seals. Loss of membrane substance causes sphering of the RBC, with greater rigidity and loss of deformability. Even if the RBC escapes the macrophage, it is damaged, with greater osmotic fragility and likelihood of lysis. Rh-antibody-mediated RBC destruction follows the chain of events outlined: rosette formation around macrophages in the spleen, loss of RBC membrane with sphering and rigidity, and osmotic fragility with lysis and phagocytosis of RBC fragments.
DEGREE OF Rh DISEASE
Degree of severity of erythroblastosis is determined by the amount of maternal anti-D IgG present, its avidity for the D antigen (binding constant), and the ability of the erythroblastotic fetus to replace the hemolyzed RBCs without developing hepatic damage, portal obstruction, and hydrops fetalis (Table 4).
TABLE 4. Classification of Severity of Erythroblastosis
Degree of Severity | Incidence (%) |
Mild: No treatment is required. Indirect bilirubin does not exceed 16–20 mg/dl. Anemia is minimal. | 45–50 |
Moderate: Fetal hydrops will not develop. Anemia is moderate. There is severe jaundice and risk of kernicterus unless infant is treated after birth. | 25–30 |
Severe: Fetal hydrops will develop in utero. | 20–25 |
Before 34 weeks gestation | 10–12 |
After 34 weeks gestation | 10–12 |
Mild
About half of the affected infants are mildly or not anemic at birth (cord hemoglobin exceeds 12 g/dL). They are not dangerously hyperbilirubinemic (cord bilirubin levels are less than 3.5 mg/dL). Their RBCs, however, are coated with anti-D, making them direct antiglobulin (Coombs' test) positive, the hallmark of all types of alloimmune hemolytic disease other than ABO.
In these infants, significant anemia (less than 10 g/dL) does not develop, nor does significant hyperbilirubinemia (more than 20 mg/dL in mature infants, 15 mg to 18 mg/dL in premature infants). They require no treatment and survive intact, as they did 60 years ago before the Rh blood group system was known and before any treatment was available.
Moderate
In about 25% of affected infants, the disease is more severe. Erythropoiesis is adequate to prevent severe anemia but not so great that hepatic hypertrophy and circulatory obstruction occur. The fetus is delivered in good condition at or near term. Before birth, the products of fetal RBC hemolysis are transported across the placenta and metabolized by the mother. After delivery, infants must rely on their own resources to metabolize and excrete the products of RBC destruction.
When an RBC is destroyed, globin is split from hemoglobin, leaving the prosthetic pigment heme. Heme is rapidly converted to indirect bilirubin, which is neurotoxic. With increased RBC destruction, there is increased production of indirect bilirubin. At birth, an infant's ability to metabolize indirect bilirubin is severely limited. Infants' livers are deficient in the transport protein Y and the microsomal enzyme uridine diphosphoglucuronyl transferase, which are responsible for the transfer of indirect bilirubin across the liver cell membrane into the cytoplasm and then for the conjugation into water-soluble, nontoxic bilirubin diglucuronide (direct bilirubin), which is then excreted by way of the biliary canaliculi down the bile ducts into the small intestine.
Indirect bilirubin is water insoluble and lipid soluble and can circulate only if it is bound to the plasma protein carrier albumin. When the bilirubin-binding sites on albumin are saturated, unbound free indirect bilirubin appears. It cannot remain in a watery medium such as plasma and diffuses into high lipid content tissues. Free indirect bilirubin diffuses across the lipid-containing neuron membrane and interferes with vital metabolic processes, causing ballooning of the neural mitochondria. The neuron dies. Bilirubin accumulates within the dead neurons, producing the characteristic yellow color found at autopsy (kernicterus).
Infants who develop bilirubin encephalopathy (kernicterus) are deeply jaundiced. Signs of brain dysfunction appear on the third to the fifth day. They become lethargic and hypertonic, lying in a position of opisthotonos (Fig. 4). Their necks are extended, and their wrists, elbows, and knees are flexed. They are rigid and suck poorly. Their grasp and Moro reflexes disappear, and they may convulse. Apnea develops, and death occurs in 90% of such infants. In the remaining 10% of infants, jaundice lessens, and hypertonicity diminishes. Initially, the neonates appear well. As they grow older, signs of severe neurologic damage inevitably appear. Most are profoundly deaf. Most have cerebral palsy of the spastic choreoathetoid type. Intellectual retardation is variable and may be severe. Even when retardation is mild, learning and functioning are impeded by the severe deafness and abnormal movements produced by the spastic choreoathetosis.
|

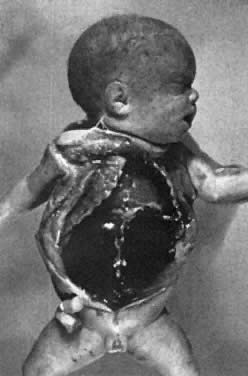
Severe
The remaining 20% to 25% of erythroblastotic fetuses are so severely affected that they develop ascites and generalized edema (hydrops fetalis; Fig. 5). Hydrops develops in half of these fetuses between 18 and 34 weeks' gestation and in the other half between 34 and 40 weeks' gestation.
|

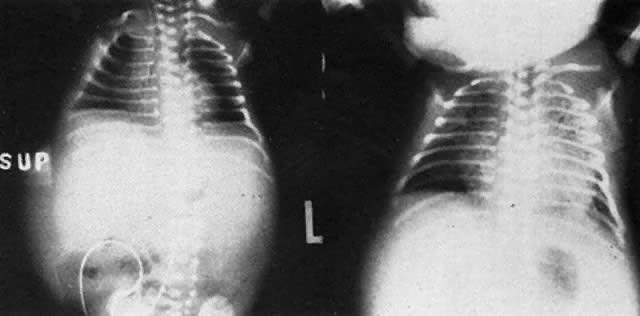
The earlier belief that hydrops was due to hypervolemic heart failure may not be correct. Although treatment measures carried out to keep hydropic neonates alive inevitably precipitate heart failure (Fig. 6), at birth, most hydropic infants are not hypervolemic, nor are they in heart failure.25 Hepatic hypertrophy with portal hypertension and hepatocellular damage is a more likely cause of hydrops fetalis.26
|
With severe RBC destruction, hepatic erythropoiesis and hepatic enlargement become extreme. Hepatic cords and hepatic circulation are distorted by erythropoietic tissue. Portal and umbilical venous hypertension develops. The placenta becomes thick and edematous; cytotrophoblast persists. Placental perfusion is reduced, and ascites appears. As progressively greater distortion of hepatic cords by islets of erythropoiesis occurs, hepatic circulation and hepatocyte function are reduced. Hypoalbuminemia develops and produces generalized anasarca. Pleural and pericardial effusions appear. Death in utero usually occurs. If a hydropic fetus is born alive, ventilation usually is impossible because of pulmonary edema and compression hypoplasia of the lungs.
Although the cause of hydrops fetalis just presented is a theory, it fits the observed facts. It explains the variable degree of fetal anemia noted with Rh hydrops fetalis because the degree of hepatic hypertrophy, portal hypertension, and hepatocellular damage—not anemia—are its basic causes. Although anemia usually is severe, hydrops may occur with hemoglobin levels well above 7 g/dL; conversely, an affected fetus may not be hydropic with hemoglobin levels well below 5 g/dL (in one instance, 2.5 g/dL).
SCREENING WOMEN AND FETUSES AT RISK
Antenatal Screening
Unless physicians send a blood sample from each patient at her first prenatal visit for blood grouping and antibody screening, they will not know which of their patients are Rh negative and at risk of Rh immunization and which are already immunized and have fetuses at risk of erythroblastosis fetalis. For this reason, a blood sample should be sent for Rh grouping and antibody screening from every woman at her first prenatal examination. This policy should be mandatory for all patients no matter what their parity or what reports have been received from prior pregnancies. Rh mistyping may have occurred in a previous pregnancy. Atypical blood group immunization may occur in an Rh-positive woman, particularly if she has been transfused. Atypical blood group antibodies, such as anti-c and anti-Kell, can produce erythroblastosis as severe as that produced by anti-D.
Screening Women Without Blood Group Antibodies
An Rh-positive woman without atypical antibodies early in pregnancy is unlikely to develop lethal atypical blood group antibodies as her pregnancy progresses. For this reason, and because there is no method of preventing atypical blood group immunization, frequent antibody screening in an Rhpositive pregnant woman is not indicated. Because atypical isoimmunization occasionally develops that is sufficient to make a fetus at significant risk, however, it is a wise precaution to rescreen an Rh-positive unimmunized woman at 28 weeks' gestation.
An Rh-negative pregnant woman should have her ABO status and the Rh and ABO status of the father determined. If the father is Rh negative, the infant should be Rh negative and the mother at no risk of immunization. Because extramarital pregnancies do occur, she should be rescreened at 32 to 36 weeks' gestation, and at the time of delivery, the Rh status of her baby should be determined from a cord blood sample.
The father, if Rh positive, should have his ABO group and Rh phenotype determined. From his phenotype, the likelihood of his D zygosity can be estimated (see Table 2). If he appears to be heterozygous, the risk of Rh immunization is halved. If he is A, B, or AB and the mother is group O, there is a 65% chance that the fetus is ABO incompatible, reducing the risk of Rh immunization to about 1.5% if the conceptus is Rh positive. If the ABO and Rh genotype of the father are known, the risk of Rh immunization can be estimated (Table 5).
TABLE 5. Approximate Risk of Rh Immunization
Factor | Risk (%) |
1. Father Rh negative, infant Rh negative | 0 |
2. Father Rh positive homozygous, ABO compatible | 16 |
3. Father Rh positive homozygous, ABO incompatible, ABO of infant unknown | 7 |
4. Infant Rh positive, ABO incompatible | 1.5–2 |
5. Father Rh positive heterozygous, ABO compatible | 8 |
6. Father Rh positive heterozygous, ABO incompatible, ABO and Rh of infant unknown | 3.5 |
Regular retesting of an Rh-negative pregnant woman who has an Rh-positive mate must be carried out. Because an initial immune response rarely occurs before 20 weeks' gestation, further testing should be carried out at 18 to 20 weeks and every 4 weeks thereafter.
Rh-negative unimmunized women in labor must be managed carefully. Both cesarean section and manual removal of the placenta increase the risk of TPH and the hazard of Rh immunization if the fetus is Rh positive. Amniocentesis carried out on an unimmunized woman for any reason (fetal genetics or pulmonary maturity) puts an Rh-negative mother at risk of fetal TPH and Rh immunization. Although carrying out the amniocentesis under ultrasound guidance reduces the risk of TPH, it does not eliminate it.
After the infant is born, cord and maternal blood must be tested. ABO, Rh, and direct antiglobulin (Coombs') tests are carried out on the cord blood sample; Rh-antibody screening and Kleihauer testing (if available) for TPH are carried out on the maternal sample. Most cases of Rh immunization are found after small or undemonstrable fetomaternal TPH. Kleihauer screening detects the 1 woman in 400 who has a TPH greater than 30 mL of fetal blood, who may not be protected against Rh immunization by the standard postdelivery dose of Rh immune globulin.
Screening Women With Blood Group Antibodies
Those responsible for the antenatal care of the immunized Rh-negative woman must determine the degree of severity of erythroblastosis (if any) in the fetus. If the fetus is severely affected, the physician must be aware of the gestation at which hydrops is likely to occur. Treatment measures that increase the likelihood of survival of the fetus who would otherwise become hydropic are not without risk to the fetus and must be reserved for fetuses at such risk. Only the fetus who will be hydropic before 34 weeks' gestation should undergo fetal blood transfusions, and only these fetuses and those who will otherwise become hydropic between 34 weeks' gestation and term should be exposed to the hazards of induced early delivery. Because fetal transfusions and early delivery pose risks to the fetus, the latest period for initial transfusion and for delivery that is compatible with the prevention of hydrops and with intact survival should be selected. The risk of death due to trauma at fetal transfusion (intraperitoneal) is 10% to 15% at 22 to 23 weeks' gestation and falls to 3% to 5% after 28 weeks. Although modern tertiary-level intensive care nursery facilities have increased the chances of premature infant survival, risk of death in the premature, severely affected, erythroblastotic infant is still significant at 32 weeks' gestation.
ASSESSMENT OF THE DEGREE OF ERYTHROBLASTOSIS FETALIS
Six parameters can help to assess the degree of severity of hemolytic disease: (1) history of preceding disease, (2) maternal antibody titers, (3) cell-mediated maternal antibody functional assays, (4) amniotic fluid spectrophotometric measurements, (5) ultrasonographic assessment, and (6) direct fetal blood sampling.
History of Preceding Disease
Two common patterns of severity of erythroblastosis fetalis are seen. The degree of severity of disease may remain unchanged in subsequent pregnancies or become progressively worse. On rare occasions, it may become less severe. Once a pattern of mild or moderate disease is established, it tends to remain. Hydrops, however, may occur after several mildly or moderately affected infants result from preceding pregnancies. The pattern of subsequent Rh disease may depend greatly on the size and frequency of further Rh-positive fetomaternal RBC TPH in the succeeding pregnancy. Significant fetomaternal TPH occurs in 75% of pregnant women. After one hydropic birth, there is a 90% likelihood that the next affected fetus will also become hydropic.
History is not helpful in the first affected pregnancy, wherein 8% to 10% of fetuses become hydropic before term. It is of little value when there is a history of hydrops but the father is heterozygous for D. Also, prior history of hydrops does not indicate when in the next pregnancy the fetus will become hydropic, although as a general rule, hydrops develops at the same time or earlier in gestation. Methods of treating the fetus destined to become hydropic before 32 to 34 weeks' gestation (fetal transfusions) are different from the methods used in treating fetuses destined to become hydropic after 34 weeks' gestation (early delivery).
Maternal Antibody Titration
Despite some beliefs to the contrary, Rh-antibody measurements, carried out by the same experienced technologists using the same methods and the same test cells, are of help in predicting the severity of erythroblastosis. Because other factors vary, such as the Rh-antibody–binding constant, the amount of D antigen on the RBC membrane, and the ability of the fetus to maintain a reasonable circulating RBC hemoglobin without compromising hepatocellular function and umbilical portal venous circulation, Rh-antibody titrations predict only the fetus at risk. They do not predict the severity of erythroblastosis with such accuracy that treatment measures may be undertaken on the basis of antibody titrations alone.
Methods of titration vary from laboratory to laboratory. The Rh-antibody titer that puts a fetus at risk of developing hydrops must be determined for each laboratory (in the Winnipeg Rh Laboratory, the significant titer is 1:16 in albumin, which puts the fetus at a 10% risk of becoming hydropic before term).
Indirect antiglobulin titer is used by many laboratories. It is more sensitive (usually positive one to three dilutions higher) than albumin titers. The indirect antiglobulin titer that puts the fetus at risk may be 1:32 to 1:64 but must be determined individually for each laboratory.
Antibody titers are of little help if there is a history of severe erythroblastosis but the father is heterozygous for D. Only rarely does the antibody titer rise, indicating an affected fetus. More commonly, the titer does not change, and the Rh status (Rh-positive and severely affected or Rh-negative and unaffected) of the fetus is not known. This problem has now been solved, as is outlined later.
Because the antibody titer is the basis for selecting the mother and her fetus who are at risk and require further investigation, regular antibody titrations must be carried out during pregnancy. The first titration should be made on a blood sample obtained at a patient's first prenatal visit. Subsequent titrations should be made at 18 weeks, at 20 to 22 weeks, and every 2 weeks thereafter.
Antibody measurement and history of severity of prior Rh disease together are insufficient to allow proper management of an Rh-immunized woman and her affected fetus. At the Winnipeg General Hospital, in an 8-year period from 1954 to 1961 in which 426 Rh-immunized pregnancies were managed, there were 67 perinatal deaths from erythroblastosis and 54 infants who survived only because they were delivered early (as early as 32 weeks' gestation).27 In just 62% of the 121 most severely affected fetuses was severity of erythroblastosis predicted accurately on the basis of history and antibody titer. If more accurate prediction of severity had been possible, half of the 67 deaths might have been prevented by treatment measures available at that time.
Cell-Mediated Maternal Antibody Functional Assays
Because of the relatively poor correlation between antibody titrations and severity of hemolytic disease, various assays have been developed that reflect the binding constant or avidity of the antibody for the antigen on the RBC membrane and therefore its ability to produce severe hemolysis. These assays include the monocyte monolayer assay28,29 and the antibody-dependent cellular cytotoxicity (ADCC) test using lymphocytes,30 monocytes,31 and monocyte chemiluminescence.32
Each one of these assays has its advocates. Three recent papers have compared the functional assays. Hadley and colleagues33 compared monocyte chemiluminescence, K-cell lymphocytes ADCC, monocyte-macrophage ADCC, and a rosette assay using U937 cells. They determined that monocyte-based (i.e., monocyte chemiluminescence and monocyte ADCC) functional assays predicted severity of disease better than lymphocyte-based assays (U937 cells and K-lymphocyte ADCC). Zupanska and co-workers28 reported that a monocyte-based assay correlated better with clinical severity of hemolytic disease than did rosette assays using lymphocytes. Mollison34 reported on a survey of nine European laboratories using functional assays to test sera from mothers delivering babies with varying degrees of hemolytic disease. Correct test results were as follows: ADCC (monocytes), 60%; ADCC (lymphocytes), 57%; chemiluminescence, 51%; rosetting and phagocytosis with peripheral monocytes, 41%; rosetting and phagocytosis with U937 cells or cultured macrophages, 32%. The assays appeared to be more accurate in predicting mild or minimal disease than in predicting severe disease. Because all of these assays measure the potential lethality of the maternal antibody, they are incapable of differentiating the antigen-negative fetus from the antigen-positive fetus.
A 1991 report cast doubt on the ability of the monocyte-macrophage assay to predict severity of hemolytic disease of the newborn.35 In sera from 41 pregnant women with potentially dangerous blood group antibodies who delivered affected babies, there was no correlation between the hematocrit of a fetal blood sample obtained at cordocentesis and the monocyte-macrophage assay.35 Therefore, although the functional tests listed may be of value in helping to determine the fetus at risk and, in some pregnancies, of removing the need for invasive measures such as amniocentesis and fetal blood sampling, they do not replace these invaluable perinatal management parameters in differentiating the fetus who requires treatment in utero from the fetus who does not.
AMNIOTIC FLUID DOD 450 MEASUREMENT.
For many years, obstetricians have observed that the amniotic fluid surrounding a fetus with severe Rh disease is yellow stained. The yellow material is bilirubin. Bevis36 carried out the first spectrophotometric measurements of bilirubin in amniotic fluid in 1956. Bilirubin, which absorbs visible light at wavelengths between 420 and 460 nm, can be measured most accurately by spectrophotometry.
The primary source of amniotic fluid is the fetus, who swallows it and voids into it. The probable sources of bilirubin in amniotic fluid are tracheal and pulmonary secretions, which are yellow in fetuses with severe Rh disease.
In 1961, Liley37 reported on a technique of amniotic fluid spectrophotometry that allows accurate comparison of measurements from one laboratory to another. Amniotic fluid must be protected from light, which destroys bilirubin. It is centrifuged and filtered, and optical density measurements are made in a good-quality spectrophotometer and recorded over the visible 700- to 350-nm range. The measurements are plotted on semilogarithmic paper (Fig. 7). Wavelength is the linear horizontal coordinate; optical density is the logarithmic vertical coordinate. The readings are connected. The deflection from linearity at 450 nm (ΔOD 450) is related to the severity of Rh disease. In fluid not contaminated with blood, a second rise at 405 nm is caused by heme pigment, which denotes severe hemolysis. To calculate the deflection from linearity at 450 nm, a tangent is drawn connecting the reading at 550 nm with the reading at 365 nm. The measurement from the intersection of the tangent with the 450-nm wavelength line to the actual amniotic fluid optical density at 450 nm gives the optical density rise at 450 nm (ΔOD 450; see Fig. 7).
|
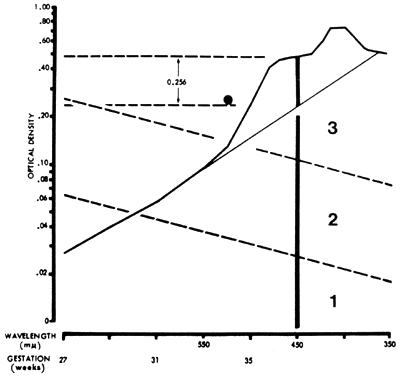
The ΔOD 450 is then replotted using gestation as the linear coordinate (Fig. 8; see Fig. 7). This replotting is necessary because the normal fetus produces bilirubin in the amniotic fluid early in gestation, which gradually diminishes with maturation of the fetus. Liley37 reported his experience with amniotic fluids taken from 101 Rh-immunized women after 28 weeks' gestation. From his assessment of severity of Rh disease in the 101 infants after delivery, he divided his graph into three zones (see Fig. 7 and Fig. 8). Fluids that fell into zone 1 indicated mild or no hemolysis but a 10% possibility that exchange transfusion would be required after birth. Fluids that fell into zone 3 indicated severe hemolysis with the great likelihood of hydrops and fetal death within 7 to 10 days. Fluids that fell into zone 2 indicated intermediate disease, becoming greater in severity as the ΔOD 450 rose toward the zone 3 boundary.
|
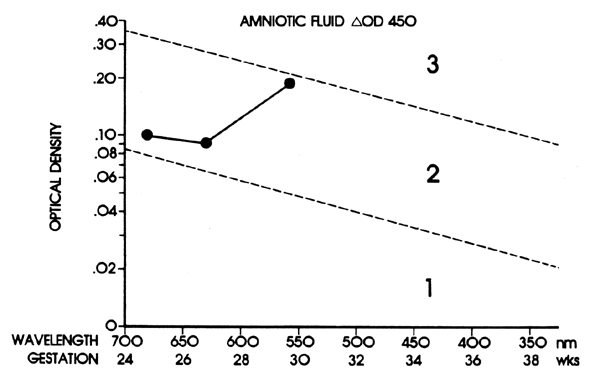
The zone boundaries slope downward (see Fig. 7 and Fig. 8), demonstrating that bilirubin present in normal pregnancies early in gestation decreases as gestation advances. One amniotic fluid measurement after 30 weeks' gestation often is of value in predicting severity of Rh disease. Early in gestation, one reading, unless it is very high (more than 0.350), may be of little value. Also, the degree of disease may change as pregnancy progresses. Serial amniotic fluid measurements, connecting each ΔOD 450 reading and measuring the slope of the reading, provide a more accurate prediction of the severity of Rh disease (see Fig. 8).
Because the affected fetus may become hydropic by 18 to 20 weeks' gestation, and because fetal salvage is possible with fetal transfusions begun as early as 19 to 20 weeks' gestation,38 the degree of severity of Rh disease must be known as early as 19 to 20 weeks' gestation. To accomplish this, the initial amniocentesis and ΔOD 450 measurements should be undertaken at 18 to 19 weeks' gestation. In early gestation, the true zone boundaries are unknown. They are probably parabolic and reach their highest levels at 22 to 24 weeks' gestation.
Nicolaides and associates39 noted little, if any, relation between amniotic fluid ΔOD 450 readings and fetal circulating hemoglobin concentrations between 18 and 25 weeks' gestation. I have observed, as have Nicolaides and associates,39 that ΔOD 450 readings in unaffected pregnancies peak at 23 to 25 weeks' gestation, rendering the true zone boundaries parabolic. Thus, I have modified the Liley zone boundaries, declining them downward before 24 weeks' gestation at the same angle as the angle of inclination after 24 weeks' gestation (Fig. 9). Because fetal blood sampling (described later), followed if necessary by intravascular fetal transfusions, is the most accurate means of determining the presence and severity of hemolytic disease and is the preferred treatment in utero, I recommend fetal blood sampling if a single or final ΔOD 450 reading is at the 65% level of zone 2 modified before 24 weeks' gestation.
|
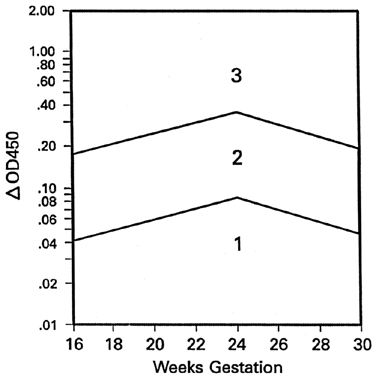
Because of the high risk of fetomaternal TPH that accompanies fetal blood sampling, I do not recommend abandonment of amniocentesis, which carries a 2% to 2.5% risk of fetal TPH. Serial amniotic fluid ΔOD 450 measurements still have a place in the management of alloimmune hemolytic disease. Nevertheless, recognizing that serial ΔOD 450 measurements before 24 weeks' gestation may not be completely accurate, I also recommend proceeding to fetal blood sampling if the antibody titers are high, if there is a prior history of severe hemolytic disease, and if amniotic fluid ΔOD 450 readings are either equivocal (middle of modified zone 2) or unobtainable because of an anterior placenta.
On the basis of this information and experience at the Rh Laboratory in Winnipeg based on 3351 ΔOD 450 measurements carried out on 1170 immunized pregnant women in the 22-year period ending January 31, 1983, I make the following observations (Table 6):
TABLE 6. Amniotic Fluid Spectrophotometry: Liley Method*
Zone of Last Fluid | No. of | Prediction | Life-Threatening |
Examination | Women | Inaccurate (%) | Inaccuracy (%) |
1 | 280 | 2.2 | 1.1 |
2 | 560 | 8.4 | 3.4 |
3 | 330 | 1.5 | 0.6 |
Total | 1170 | 5.0 | 2.1 |
*Data from Rh Laboratory, Winnipeg, December 15, 1961, through January 31, 1983: 3351 amniotic fluids, 1170 immunized women.
- A ΔOD 450 reading greater than 0.400 at any time in pregnancy is associated with hydrops 65% of the time.
- Hydrops may be present with ΔOD 450 readings as low as 0.200 to 0.250 at 28 weeks' gestation.
- Conversely, rarely, an Rh-negative unaffected fetus may have ΔOD 450 readings of 0.200 to 0.250 at 22 to 24 weeks' gestation.
- 4. When ΔOD 450 readings are in the 80% to 85% upper zone 2 area, delay in fetal transfusion may be associated with hydrops fetalis 7 to 14 days later.
- Disease may be fulminant with midzone readings of 0.160 at 23 to 0.240 at 27 weeks' gestation, followed by zone 3 readings of 0.385 at 25 weeks and 0.370 at 29 weeks, with hydrops present at the second amniocenteses.
OTHER AMNIOTIC FLUID MEASUREMENTS.
Many other methods of measurement of amniotic fluid have been reported, with each author extolling his or her method as an advance in improving accuracy of prediction of Rh disease. As pointed out by Bartsch in 1970,41 none is an improvement over the Liley method. Experience and judgment of the person reviewing the amniotic fluid measurements and predicting the severity of the disease are more important than the method of measurement used.
AMNIOCENTESIS.
Technique.
Because the risk of fetomaternal TPH, with consequent increasing antibody titers and increasing severity of Rh disease, is always present, amniocentesis should be carried out only after careful ultrasound placental localization. If the placenta is on the anterior uterine wall, amniocentesis should be performed under direct real-time ultrasound guidance. The use of ultrasound is of great value in determining the position of a pocket of amniotic fluid and the site and depth of insertion of the needle required to obtain a blood-free sample of amniotic fluid without traversing the placenta.
If amniocentesis must be carried out without the use of ultrasound, a suprapubic approach may be less likely to encounter the placenta. Nevertheless, amniocentesis carried out without ultrasound guidance is associated with a 10% to 11% risk of fetomaternal TPH and increased severity of Rh disease.12
Amniocentesis always should be carried out using careful aseptic technique and local anesthesia. A clotted blood sample should be taken before and 5 minutes after amniocentesis. The needle insertion site is determined by palpation and ultrasound examination. The site is prepared with a suitable antiseptic, draped, and infiltrated with a local anesthetic. The operator introduces a lumbar puncture needle (20 or 22 gauge) through the abdominal wall into the uterus to the depth at which ultrasound has indicated a pocket of amniotic fluid. The stylet is removed, and 10 to 15 mL of fluid are aspirated. If fluid cannot be removed, the needle is withdrawn or inserted slightly. Rotation of the needle may aid the flow of fluid. Normally, amniotic fluid is slightly turbid, and turbidity increases as term is approached. Various degrees of yellow coloration are present. The fluid is protected from light and sent for ΔOD 450 measurement. After 31 weeks' gestation, it is also examined for the present of pulmonary maturity (lecithin/sphingomyelin [L:S] ratio of more than 2.0:1, phosphatidylglycerol [PG] of more than 2%). The maternal blood samples are sent for Kleihauer fetal cell screening and for antibody titration.
Risks.
Maternal risks from amniocentesis are negligible. If the procedure is carried out properly, there should be no risk of infection. Rarely, abruptio placentae or precipitation of labor occurs after amniocentesis.
Fetal hazards, although not great, are not inconsequential. Direct trauma from the needle has been reported but is rare.42 The chief hazards—placental trauma, fetomaternal TPH, rising antibody titers, and increased severity of Rh disease—were described previously. Fetal exsanguination, a theoretical hazard, must be rare after amniocentesis.
Sources of Error in Amniotic Fluid Diagnosis.
Other fetal conditions or contaminants can distort the ΔOD 450 measurement and produce serious errors. Maternal or fetal blood (oxyhemoglobin) causes sharp 580-, 540-, and 415-nm peaks (Fig. 10), which obscure any 450-nm peak and render the fluid valueless. Although smaller amounts of blood, if removed quickly, do not obscure the 450-nm peak, a small amount of plasma (particularly fetal plasma) can cause a spurious increase in the ΔOD 450 measurement, with resultant misinterpretation of severity of Rh disease. Methemalbumin, usually an indicator of severe disease, produces a 405-nm peak that diminishes the ΔOD 450 measurement. Meconium contamination of amniotic fluid markedly distorts the ΔOD 450 measurement, shifting the peak toward 420 nm. Exposure of the fluid to daylight or fluorescent light destroys bilirubin, producing a falsely low ΔOD 450 measurement.
|
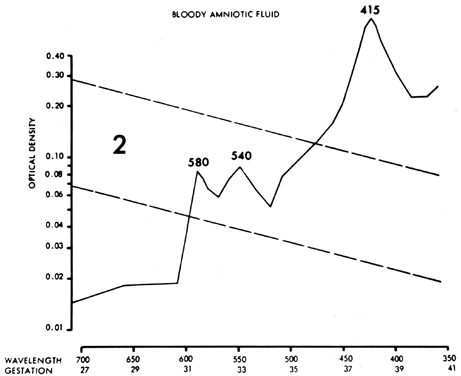
Maternal urine or fetal ascitic fluid occasionally is aspirated instead of amniotic fluid. Urine produces no 450-nm peak. Ascitic fluid is clear, bright yellow, and more viscous than amniotic fluid. It appears different from the more turbid, less viscous amniotic fluid. The absorption peak of ascitic fluid is much higher, often requiring dilution before it can be measured. The absorption peak is deviated toward 460 nm.
Congenital abnormalities of the fetus, such as anencephaly, open spina bifida, and obstructive lesion of the gastrointestinal tract such as tracheoesophageal fistula and duodenal atresia, produce hydramnios and marked rises in the ΔOD 450 measurement, which may be misleading if the mother is Rh immunized.
Despite these potential errors, I believe that amniotic fluid spectrophotometry is an accurate biologic screening test. Life-threatening inaccuracies occurred in only 2.1% of the 1170 women who underwent 3351 amniotic fluid examinations.
Criteria and Timing.
Because amniocentesis is not without some hazard to the fetus, it should be undertaken only when the history or antibody titer indicates that the fetus is at risk of developing hydrops and dying. Using the history and antibody criteria already outlined as guidelines, only half of Rh-immunized pregnant women require amniocentesis. Amniocentesis should be undertaken at 18 to 19 weeks' gestation, irrespective of the titer, if there is a history of previous stillbirth or an infant who required exchange transfusion. If there is no such history, amniocentesis should be carried out only if there is an antibody titer indicating a risk of hydrops (more than 1:16 in albumin in my institution). If the critical titer is present before 18 weeks, the initial procedure is carried out at 18 to 19 weeks' gestation. If the critical titer is reached after 18 weeks' gestation, amniocentesis should be carried out within 5 to 7 days. Amniocentesis should be performed at 5- to 21-day intervals, depending on the ΔOD 450 measurement of the preceding amniotic fluid and the direction of the slope of the serial ΔOD 450 measurements already taken. Amniotic fluid ΔOD 450 readings may increase rapidly. It may be necessary to carry out amniocentesis at weekly intervals for 6 to 8 weeks before the ΔOD 450 slope is such that the interval may be increased or definitive treatment such as fetal transfusion or early delivery may be carried out.
ULTRASOUND IN THE MANAGEMENT OF THE Rh-IMMUNIZED MOTHER AND AFFECTED FETUS.
Ultrasound imaging techniques, introduced in the late 1970s, constitute a major advance in the management of maternal blood group alloimmunization.43 Ultrasound allows an estimation of placental and hepatic size and the presence or absence of edema, ascites, and other effusions (i.e., the presence or absence of hydrops fetalis; Fig. 11 and Fig. 12). Ultrasound is of great help in assessing fetal well-being (measuring the fetal biophysical profile), has increased the accuracy of placental localization, and has sharply reduced the incidence of placental trauma at amniocentesis.
|
|

ULTRASOUND AND FETAL TRANSFUSION.
Real-time ultrasound is essential in directing the transfusion needle with the least possible hazard during both intraperitoneal and intravascular transfusions. After intraperitoneal transfusion, ultrasound examination confirms the presence of blood in the fetal peritoneal cavity, and serial examinations monitor its absorption. At the time of direct fetal intravascular transfusion, ultrasound observation of characteristic turbulence within the fetal umbilical blood vessel, as the blood is injected, confirms that it has been transfused into the fetal circulation. Although ultrasound establishes the presence of hydrops with great accuracy, it may not make the diagnosis of impending hydrops until hydrops has developed. After fetal transfusions, however, ultrasound biophysical profile scoring provides an accurate assessment of fetal condition and of whether improvement or deterioration is occurring.
PERCUTANEOUS UMBILICAL BLOOD SAMPLING.
With the development of sophisticated ultrasound equipment and the availability of perinatologists skilled in its use, percutaneous fetal umbilical blood sampling became feasible in the mid-1980s.44 This procedure allows the direct measurement of all blood parameters that can be measured after birth (hemoglobin, hematocrit, blood groups, direct antiglobulin testing, serum bilirubin levels, platelet and leukocyte counts, serum protein levels, and fetal blood gases). Fetal blood sampling is by far the most accurate means of determining the degree of severity of fetal hemolytic disease (in the absence of hydrops) and the need for fetal treatment measures.
Fetal blood sampling is a relatively benign procedure, carrying with it a traumatic fetal mortality rate of a fraction of 1%.44 Because it carries a high risk of fetomaternal TPH, its use is recommended only when serial amniotic fluid ΔOD 450 readings rise into the upper 65% to 75% level of zone 2 or when an anterior placenta cannot be avoided at amniocentesis and maternal pregnancy history or maternal alloantibody titers place the fetus at risk. Fetal blood sampling may be possible as early as 18 weeks' gestation; it usually is feasible by 20 to 21 weeks' gestation. The preferred sampling site is from the umbilical vessel (preferably the vein) at its insertion into the placenta. For this reason, the procedure is technically easier if the placenta is implanted on the anterior uterine wall.
DETERMINATION OF FETAL D ANTIGEN STATUS BY POLYMERASE CHAIN REACTION.
As referred to earlier, the Rh gene locus (which is on chromosome 1p34–p36), consists of two homologous genes designating the antigens CcEe and D. The sequences of the two genes are 96% identical, suggesting that they arose through the duplication of a single ancestral gene. The gene CcEe encodes the Cc and Ee proteins, probably by alternate splicing of a primary transcript.8 The gene D encodes the D protein, which is absent on both chromosomes of D-negative people. The presence or absence of the D gene determines whether the fetus is Rh(D) positive or Rh(D) negative.
The cloning of the CcEe and D complementary DNA now provides a means of determining the fetal Rh(D) type in DNA obtained by chorionic villus biopsy or amniocentesis. Bennett and colleagues45 were able to determine the Rh(D) type with 100% accuracy by polymerase chain reaction (PCR) assays on 30 samples of fetal blood cells, obtained either from chorionic villus sampling or amniocentesis. It is now possible, therefore, to determine the D status of a fetus whose mother is Rh alloimmunized and whose father appears to be heterozygous for D from fetal cells in amniotic fluid or chorionic villus samples, making the diagnosis of a D-positive, affected fetus, in whom further invasive diagnostic tests are required, or a D-negative, unaffected fetus, in whom no further invasive tests are required.
Rh variants at the Rh gene and Rh RNA transcript levels reflect a certain degree of polymorphism; rare gene deletions also may cause a D-positive variant person to group by PCR as D negative, depending on the exon probe used and the exon deletion present. Whether these rare variants are of clinical significance in respect to fetal D typing in Rh-alloimmunized women is uncertain (they probably are not). Because of such variants, an absolute diagnosis of fetal D status may require multiplex assays targeting more than one exon along the D gene.46 Using a single exon probe only, however, my laboratory has had 100% concordance in determining fetal D status to date. Fetal Kell status also can be determined in the same manner.47
Because fetal nucleated erythroid precursors are present in the maternal circulation early in pregnancy, these cells also may be used for D antigen status determination by PCR methods. Thus, the ability to determine D antigen fetal status by a maternal blood sample PCR assay is now possible.48 The method has not reached the level of accuracy needed to be applicable in clinical situations, but this should be possible in the future. Once this accuracy is reached, a total noninvasive method will be available to determine fetal Rh(D) antigen status, when the mother is Rh(D) alloimmunized.
TREATMENT OF THE MOTHER AND AFFECTED FETUS
When Hydrops Is Not a Factor
In pregnancies in which amniocentesis has not been necessary and in those in which final ΔOD 450 measurements are below the middle of zone 2, the mother may be allowed to deliver spontaneously. If she is obstetrically suitable and her dates are firm, labor might be induced and the infant delivered at 38.5 to 39 weeks' gestation. If there is a history of severe Rh disease or an Rh-antibody titer exceeding 16 in albumin, fetal PCR Rh(D)-negative fetal typing or a final zone 1 or low zone 2 fluid indicates that the fetus will be Rh negative, and the mother may be allowed to deliver spontaneously. If Rh(D) PCR typing is positive or if the final ΔOD 450 reading has risen into the upper 50% to 70% area of zone 2 by 35 to 37 weeks' gestation, delivery should be undertaken at 36.5 to 38.5 weeks' gestation provided that the mother's dates are firm and she is obstetrically suitable; she should not be allowed to remain undelivered past 39.5 to 40 weeks' gestation. Although the fetus will not be hydropic, it will probably require prompt treatment after delivery. Such infants are better delivered 2 to 3 weeks before term. If gestation is in doubt, delivery should not be carried out until amniotic fluid tests (L:S ratio and PG) have been done and indicate pulmonary maturity.
When Risk of Hydrops is a Factor
About half of the 20% to 25% of fetuses doomed to become hydropic become so after 34 weeks' gestation (see Table 4). If the final amniotic fluid reading falls into the upper 75% to 80% of zone 2 or if an initial reading is in zone 3 after 34 weeks' gestation, prompt delivery should be undertaken, provided that there is evidence that the fetal lungs are mature. If the placenta is situated anteriorly, a fetal blood sampling procedure should be considered before delivery.
INTRAUTERINE TRANSFUSIONS FOR FETAL HEMOLYTIC DISEASE
Intraperitoneal Fetal Transfusion
In 1961, induced early delivery could not be carried out earlier than 31 to 32 weeks' gestation without encountering prohibitive mortality from prematurity and severe Rh disease.
Eight percent of fetuses become hydropic before 32 weeks' gestation. The introduction by Liley49 in 1963 of fetal intraperitoneal transfusion (IPT) completely altered the outlook for these severely affected fetuses.
Physiology
It has been known since the turn of the century that RBCs placed in the peritoneal cavity are absorbed and function normally. At one time, IPT was a favorite method of transfusing children with thalassemia. It was abandoned in favor of vascular transfusion because of the severe discomfort that it caused. Absorption is through the subdiaphragmatic lymphatic lacunae, up the right lymphatic duct, into the venous circulation. Diaphragmatic movements are necessary for absorption to occur.50 In the absence of hydrops, 10% to 12% of infused RBCs are absorbed daily. The presence of ascites does not prevent absorption,51 although the rate of absorption is more variable. When the hydropic fetus is moribund and not breathing, no donor RBCs are absorbed.50
Peritoneal capacity limits the volume of RBCs infused. If the volume is such that intraperitoneal pressure exceeds umbilical venous pressure, placental blood flow to the fetus stops, and the fetus dies.52 In nearly every case, RBC volumes infused safely can be calculated by the following formula: gestation in weeks minus 20 multiplied by 10 mL (i.e., 40 mL at 24 weeks, 80 mL at 28 weeks).
Accurate calculation of donor hemoglobin concentration in the fetus at any time after transfusion allows appropriate spacing of IPT and selection of the proper gestation (after 33 to 34 weeks) for delivery. After IPT, 85% of the transfused RBCs are in the fetoplacental circulation. Residual donor hemoglobin levels in the fetus may be estimated within 1.5 g/dL 95% of the time using the following parameters: 0.85 times the fraction of donor RBCs in the fetoplacental circulation; fetal weight at the gestation donor hemoglobin levels; 125 mL/kg as fetoplacental blood volume; and 1/120 of the donor hemoglobin infused as the daily attrition rate of donor RBCs. For example, the donor RBC hemoglobin concentration at 28 weeks' gestation (fetal weight estimated to be 1150 g) 10 days after intrauterine transfusion (IUT) of 65 mL of donor RBCs with a hemoglobin concentration of 30 g/dL can be determined as follows:

After subsequent IPT, estimations of residual circulating donor hemoglobin concentrations for each fetal transfusion at any period after the last transfusion can be made and added together to give the estimated donor hemoglobin level at that gestation.
These calculations can be used to determine the time for the next fetal transfusion (or for delivery), with the requirement that the donor hemoglobin level be kept above 10 to 11 g/dL at all times. Because one transfusion rarely raises donor hemoglobin levels above 10 g/dL, a second transfusion is carried out 9 to 12 days after the first. Subsequent transfusion intervals are 3.5- to 4-week intervals, with the last transfusion rarely being given after 32 weeks' gestation. Delivery is carried out 3.5 to 4 weeks after the last transfusion, usually between 34 and 36 weeks' gestation.
Blood for Intrauterine Transfusion
Group O, Rh-negative, Kell-negative RBCs are used for IUT if the alloantibody is anti-D. If the alloantibody is non-D in the Rh system, group O, Kell-negative, maternal serum compatible RBCs are used. If the alloantibody is outside the Rh system, group O, Rh-negative, Kell-negative RBCs are used that are missing the offending antigen to which the mother is alloimmunized. The blood should have been drawn from the donor within 48 to 96 hours of the transfusion. The donor RBCs are carefully cross-matched against maternal serum; these women are prolific antibody producers, some having as many as four to six alloantibodies other than their primary antibody. In such circumstances, finding a compatible donor may be difficult. Before the transfusion, the unit is tightly packed, and all plasma and buffy coat are removed. Immediately before the IUT, 10 to 15 mL of sterile, isotonic saline is added so that the hemoglobin and hematocrit of the packed RBCs are 27 to 30 g/dL and 85% to 90%, respectively.
The donor unit must be human immunodeficiency virus negative, hepatitis C virus negative, and HBsAg negative and should be irradiated, although the risk of graft-versus-host disease occurring if nonirradiated blood is used is small.
The Intrauterine Transfusion Team
The number of candidates for IUT is decreasing as the number of immunized Rh-negative women declines because of the success of Rh prophylaxis. IUT techniques, either intraperitoneal or direct intravascular, appear simple, but they are not. Complete management of the immunized mother and baby, before and after birth, requires not only an experienced obstetrician but also neonatal, ultrasound, and laboratory personnel and services of the highest order. Procedures should be carried out only in a tertiary-level perinatal center. A team approach is essential. Each center in which fetal transfusions are performed should treat at least five or six fetuses annually, on whom 15 to 20 transfusions are carried out. To reach this volume of patients, the IUT team must have all transfusion candidates referred from a population base of 6 million (75,000 to 90,000 deliveries per year). Only under such circumstances can the team's expertise in IUT and overall management of severely affected fetuses and infants with hemolytic disease be maintained.
Fetal Transfusions of the Hydropic Fetus
Although the goals are to predict impending hydrops and to prevent it by beginning fetal transfusions before hydrops has developed, in 28% to 33% of cases, hydrops is present at first transfusion because of late referral, or it develops between the first and second IPTs because one IPT does not raise donor hemoglobin levels high enough to shut off hepatic erythropoiesis and interrupt the hepatic chain of events that leads to the development of ascites and hydrops fetalis.
Although direct intravascular transfusion (IVT) is the procedure of choice when hydrops is encountered, significant salvage of hydropic fetuses has been produced with IPT in the past. In the ultrasound era in Winnipeg, 60% of hydropic fetuses (18 of 30) were salvaged with IPT; however, none of eight moribund, nonbreathing hydropic fetuses subjected to IPT survived.
Hydrops fetalis may be reversed by fetal transfusions. Reversal is more common when hydrops is found at the second IPT for the first time. Reversal is common when hydrops is encountered at the first direct IVT. As donor hemoglobin levels rise after transfusion, erythropoietin levels fall. If the fetus remains alive, hepatic erythropoiesis diminishes, intrahepatic circulation improves, portal and umbilical venous pressures fall, hepatocellular function improves, serum albumin levels rise, and ascites and fetal anasarca disappear.
Direct Intravascular Fetal Transfusion
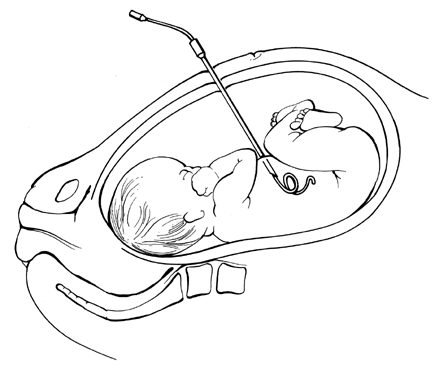
Pioneering attempts at direct IVT into either a fetal or placental vessel approached through a hysterotomy incision were attempted in the mid-1960s.53,54,55 The results were abysmal because the women almost invariably went into labor. In 1981, Rodeck and associates56 introduced direct transfusion through a fetoscope. Few others have achieved his skill with the fetoscope. Blood, meconium, or turbidity in the amniotic fluid makes fetoscopic visualization of the fetal blood vessels impossible. With the advent of fetal blood sampling, by the early to mid-1980s44 it became possible to follow the sampling procedure with direct IVT.57,58,59,60,61 Under ultrasound guidance, the tip of a 22- or 20-gauge spinal needle is introduced into a umbilical blood vessel, preferably the vein but occasionally the artery, at its insertion into the placenta and rarely at its insertion into the fetal abdomen.
Advantages of Fetal Blood Sampling and Intravascular Fetal Transfusion
As stated earlier, in the absence of hydrops, direct measurement of fetal blood parameters is the most accurate method of determining the severity of hemolytic disease, far more accurate than amniotic fluid ΔOD 450 measurements and ultrasonographic assessments. The latter two parameters, however, combined with past history and maternal antibody titers, can identify the fetus at risk who requires a fetal blood sampling procedure.
Direct IVT, as a method of transfusing the severely affected fetus, does not rely on diaphragmatic movement to increase hemoglobin levels. It is, therefore, capable of salvaging the moribund nonbreathing fetus, provided that the fetus still has umbilical blood flow. Also, direct IVT raises circulating hemoglobin levels in the fetus immediately, rather than taking the 8 to 10 days required for IPT.
Selection of Patients
Fetal transfusion is hazardous for the fetus. Only fetuses proven to be at high risk of hydrops or hydropic before 33 to 34 weeks' gestation should undergo fetal transfusion. Patients are selected for IPT on the basis of amniotic fluid findings. Women undergoing amniocentesis are selected on the basis of history and antibody criteria already outlined. If the indications are present, amniocentesis should be carried out initially as early as 18 to 19 weeks' gestation. IPT is indicated when serial ΔOD 450 readings reach the upper 80% to 85% area of zone 2 before 30 weeks' gestation (see Fig. 8) or reach zone 3 between 30 and 33 weeks' gestation. Fetal transfusions may be indicated at somewhat lower readings if serial ΔOD 450 readings show a sharp slope rising toward the zone 3 boundary, indicating rapidly progressive disease.
IVT, the preferred transfusion method, is indicated if, at fetal blood sampling, the circulating hemoglobin concentration in the fetus is less than 10 to 11 g/dL. Fetal blood sampling is carried out when serial ΔOD 450 readings reach the 65% to 75% level of zone 2 at any gestation or when the placenta is anterior and maternal history or antibody titer place the fetus at risk.
Technique of Intraperitoneal Transfusion
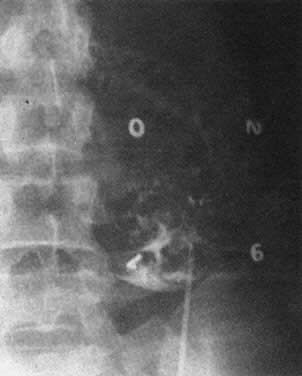
Real-time ultrasonography is used to determine the site for the needle insertion on the maternal abdomen and the depth that the needle must be inserted to enter the fetal peritoneal cavity. After aseptic and local anesthetic preparation, the operator directs the needle (an 18-cm, 16-gauge Tuohy needle) through a 5-mm maternal skin incision into the fetal peritoneal cavity under direct ultrasound guidance, having selected the site and depth of needle insertion on the basis of real-time ultrasound assessment. Often, but not invariably, the real-time ultrasound image shows the needle tip in the fetal abdomen. As the needle enters the fetal abdominal wall, the operator may note a feeling of resistance that disappears as the tip enters the peritoneal cavity. The stylet is removed, and an epidural catheter with its tip and side holes removed is passed down the needle. If the catheter passes through the end of the needle, it is lying free in a cavity, which should be the fetal peritoneal cavity (Fig. 13). Twenty-five to 30 cm of catheter is passed down the needle. The needle is pulled back over the catheter to lie on the maternal abdomen, and 1 to 2 mL of radiopaque contrast medium is injected through the catheter. An anteroposterior radiograph is taken. If the catheter lies free in the peritoneal cavity, the contrast-filled catheter is seen within the peritoneal cavity, and contrast appears within the cavity, outlining the negative shadow of liver and semilunes of small bowel (Fig. 14).
|
|
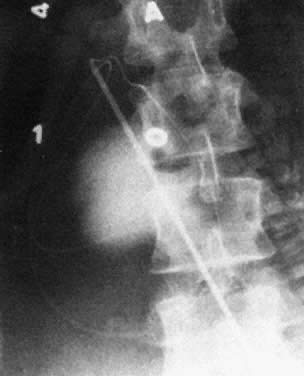
Diagnosis of fetal ascites should be made by ultrasound before the fetal transfusion. Ascitic fluid obtained at first transfusion is characteristically bright yellow, clear, and slightly viscous because of its protein content. It looks different from the dull yellow, slightly turbid, less viscous amniotic fluid. If ascites is present at second and subsequent fetal transfusions, it is mixed with residual donor blood to various degrees. When ascites is present, a radiograph taken after injection of contrast medium reveals characteristic diffusion of dye into the ascitic fluid, with no visible small bowel semilunes or other landmarks (Fig. 15).
|
When the contrast injection radiograph demonstrates the catheter lying in the fetal peritoneal cavity (see Fig. 14), the transfusion is started. The infusion is carried out in 10-mL aliquots, with each 10-mL injection taking 3 to 6 minutes. The syringe is rinsed after each injection. The fetal heart rate is monitored by Doppler ultrasound at the end of each 10-mL injection and continuously for the last 10 to 15 mL of the infusion. The volume infused is calculated according to the following formula: gestation (in weeks) minus 20 multiplied by 10 mL. If the fetus is in good condition, fetal heart rate increases to 160 to 190 beats/min during the procedure. Fetal bradycardia early in the transfusion, a rare event, is ominous, indicating the probability of transfusion trauma and fetal death. Fetal bradycardia toward the end of the transfusion is uncommon and is an indication for prompt termination of the procedure because intraperitoneal pressure may be approaching umbilical venous pressure.
After completion of the procedure, the catheter is removed slowly with continuous heart rate monitoring. Marked bradycardia of vagal origin may occur. Further catheter removal should be delayed until the fetal heart rate improves. The needle site is swabbed with iodine, and a small dressing is applied. Fetal heart rate is monitored periodically after the transfusion. Fetal movement may be markedly reduced for the first 24 hours after the procedure. The mother should be reassured that this is not an ominous sign. She usually can be discharged 24 to 36 hours after the procedure.
If gross ascites is noted (see Fig. 15), every effort should be made to aspirate a significant quantity of fluid through the needle before the catheter is passed. After passage of the catheter and injection of contrast, if the postinjection radiograph reveals a considerable amount of residual ascitic fluid, a further volume of fluid (as much as possible) should be aspirated through the catheter.
If a greater volume of ascitic fluid is aspirated than the planned RBC volume to be injected, the total volume to be transfused may be increased but should remain 15 to 20 mL less than the volume of ascitic fluid removed. IPT is no longer recommended for treatment of the hydropic fetus; direct IVT is the preferred method of transfusion. Indeed, direct IVT is now the preferred route for all fetal transfusions.
Problems With Intraperitoneal Transfusion
Although IPT represented a major advance in the management of severe erythroblastosis fetalis, it was associated with some serious problems. The procedure is of no value for the nonbreathing moribund hydropic fetus. In this situation, RBCs are not absorbed, and the fetus dies. Also, when the placenta is implanted on the anterior uterine wall and must be transfixed by the Tuohy needle to enter the fetal peritoneal cavity, the traumatic death rate per procedure is 7%. Moreover, after IPT, there is a 30% spontaneous labor rate per patient, with delivery earlier than otherwise planned; however, most of such deliveries occur after 30 weeks' gestation. Seventy percent of fetuses born after spontaneous labor survive. Finally, although serial amniotic fluid ΔOD 450 measurements have increased the accuracy of prediction of severity of hemolytic disease, inaccuracies do occur—the occasional only moderately affected fetus despite a zone 3 reading and, less commonly, hydrops despite only a moderate (60%) zone 2 reading.
Technique of Direct Intravascular Fetal Transfusion
Because of the previously outlined problems with IPT, direct IVT has become the fetal transfusion procedure of choice. When it has been decided from amniotic fluid and ultrasound assessment that fetal blood sampling and direct IVT are required, the mother is heavily premedicated and transferred to the fetal assessment and therapy unit. A highly skilled, experienced perinatal ultrasonographer is an essential component of the IVT group. Great care and considerable time are spent in identifying the target blood vessel (Fig. 16). Packed RBCs used for IVT are of the same hemoglobin and maternal cross-match compatibility as those used for IPT. Immediately before the planned procedure, with great care, 8 to 10 mL of sterile saline is added to the packed RBC unit. A blood transfusion set is inserted into one of the outlet ports of the packed RBC unit. The side arm of a metal stopcock is inserted into the other end of the transfusion tubing. A length of sterile connector tubing is attached to the male end of the stopcock, and a 10-mL glass syringe with finger-ring assembly is attached to the other end of the stopcock. Packed RBCs are run down the transfusion tubing through the stopcock into the extension tubing. The stopcock lever is then set at an angle so that blood will not run down the transfusion tubing in any direction. With a 45-degree turn of the stopcock in one direction, the syringe may be filled with the packed RBC unit; changing the lever 90 degrees then allows blood to be transfused down the extension tubing. Packed RBCs treated in this manner can be readily infused through a 22-gauge spinal needle without hemolysis.
|
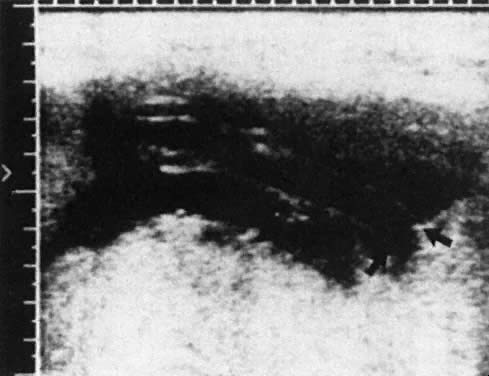
After careful aseptic preparation of the maternal abdomen, the obstetric surgeon drapes the maternal abdomen, infiltrates the skin with local anesthetic, and inserts a 20- or 22-gauge spinal needle tip through the maternal skin immediately over the target site selected previously by the obstetric ultrasonographer. The ultrasound transducer, enclosed in sterile plastic, is applied to the maternal abdomen immediately adjacent to the needle insertion site. The ultrasonographer and venipuncturist select a plane and position the transducer to allow simultaneous ultrasonographic identification of the blood vessel target site and the needle tip. The needle is guided toward the blood vessel, with appropriate ultrasound directed corrections to keep the tip on target.
In most instances, the needle tip can be identified as it penetrates the target blood vessel. Because the needle tip is being advanced in a three-dimensional plane using ultrasound, which is in two dimensions, the operator cannot always be certain that the needle tip has been correctly inserted into the lumen of the vein. When the tip is at the proper depth, the needle stylet is withdrawn, and aspiration is attempted using a lightly heparinized 1-mL tuberculin syringe. If nothing is obtained or if amniotic fluid is aspirated, withdrawal, but not removal, of the needle tip and reinsertion under ultrasound guidance are carried out. If a free-flowing blood sample is aspirated, the tip of the needle probably is positioned properly in the fetal blood vessel. At initial fetal blood sampling, this may be confirmed by determining that the blood is fetal by alkaline denaturation screening. After the first IVT, the amount of donor blood present in the fetal circulation obscures any alkaline denaturation test to confirm its fetal origin.
Confirmation that the needle tip is placed properly is done easily and accurately by injecting 0.5 mL of sterile saline. If the needle tip is in the vein, streaming ultrasound turbulence is seen as the saline passes down the vein. Conversely, if the needle tip is in the artery, the turbulence of the saline may be seen moving in the opposite direction onto the surface of the placenta. If turbulence can be seen in the amniotic fluid, the needle tip is in the amniotic cavity. If no turbulence can be seen, the needle tip may be dislodged and embedded in the umbilical cord. In this situation, injection of blood should not be carried out because life-threatening umbilical vein compression may occur. Only if streaming of saline in the vessel can be seen should blood infusion be begun.
An immediate hemoglobin estimation is carried out on the fetal blood sample. Subsequently, complete investigation of the fetal blood sample is carried out in the Rh Laboratory.
If the immediate hemoglobin reading is below 10 to 11 g/dL, the IVT proceeds. If fetal movements are likely to disturb the needle insertion (posterior cord insertion), the fetus is paralyzed by the intravenous injection of pancuronium. The obstetric venipuncturist inserts the filled blood transfusion tubing connector into the needle hub and holds it firmly. The obstetric ultrasonographer continuously watches the blood flow turbulence in the fetal blood vessel. The transfusionist (the third member of the transfusion team) transfuses the packed RBCs in 10-mL aliquots. Because there is only 2 or 3 mm of needle tip within the fetal blood vessel, which may be as small as 2 to 3 mm in diameter, the tip is in danger of early dislodgment. For this reason, infusion of packed RBCs is carried out as rapidly as possible. Each aliquot is transfused in 1 to 2 minutes until the desired blood transfusion volume is attained. The usual transfusion volume is 50 mL/kg of estimated nonhydropic fetal weight. Because of the expansile placental vascular bed, large RBC volumes transfused rapidly are tolerated. The ultrasonographer monitors fetal heart rate and cardiac ventricular size. If there is evidence of significant bradycardia or marked ventricular dilation (a rare event), the transfusion is discontinued before the full volume is administered. Because the aim is to shut off erythropoiesis so that hepatosplenomegaly diminishes and hydrops is prevented or reversed, a second procedure is carried out as soon as it is estimated that the total circulating hemoglobin concentration has dropped into the range of 9 to 10 g/dL. The average donor hemoglobin attrition rate is 0.4 g/dL/d. Subsequent IVTs usually are required every 3 to 4 weeks after the estimated residual circulating hemoglobin concentration has dropped into the range of 9 to 10 g/dL.
Intravascular Transfusion of a Hydropic Fetus
In the presence of hydrops fetalis, IVT is preferred over IPT. IVT has the potential for salvaging all hydropic fetuses, even those who are moribund and not breathing, with the exception of those that are dying and have no blood flow in their umbilical vessels. The procedure is not different from IVT of a nonhydropic fetus, other than that the volume administered in the first transfusion frequently is confined to 30 to 40 mL/kg of estimated nonhydropic fetal body weight.
When the transfusion has been completed, the needle is cleared with 0.5 mL of normal saline. A 1-mL posttransfusion blood sample is withdrawn, again into a heparinized tuberculin syringe. All blood samples, before and after transfusion, are tested for hemoglobin, hematocrit, bilirubin, Kleihauer fetal/adult RBC ratios, platelet counts, plasma protein, and blood gas estimations. The initial pretransfusion blood sample is blood grouped and direct Coombs' tested.
Intrauterine Transfusion Survival Rates
The Winnipeg overall salvage rate with IPT from January 1964 to October 1986 (the last date that a fetus was given IPT alone) was 63% (222 of 353 fetuses transfused 862 times).
Survival rates with IPT and IVT in the modern ultrasound era are outlined in Table 7, which shows IVT survival rates that are superior to IPT survival rates in every category. More IVTs than IPTs are required per patient (4.3 versus 2.7), for three reasons. First, IVTs are being carried out earlier in gestation. Second, perinatal mortality per procedure is less, allowing multiple procedures in more patients. Third, because of somewhat smaller IVT volumes, the intervals between IVTs are modestly shorter and the gestation of delivery moderately longer (37 to 38 weeks).
TABLE 7. Intrauterine Transfusions (IUT): Winnipeg Ultrasound Era
| 204 IPT | 696 IVT |
| (July 1980-October 1986) | (May 1986-May 1997) |
Fetuses | 75 | 161* |
Survivors | 57 (76%) | 140 (88%) |
Nonhydrops | 45 | 111* |
Survivors | 39 (86.7%) | 104 (95%) |
Hydrops | 30 | 50 |
Incidence | 40% | 31% |
Survivors | 18 (60%) | 36 (72%) |
Nonmoribund hydrops | 22 | 31 |
Survivors | 18 (82%) | 24 (77%) |
Moribund hydrops | 8 | 19 |
Survivors | 0 (0%) | 12 (63%) |
IUT per fetus | 2.7 | 4.3 |
Risk per IUT | 3.5% | 0.8% |
*Two in utero are expected to survive.
IPT, intraperitoneal transfusion; IVT, intravenous transfusion.
Pros and Cons of Intravascular Transfusion and Intraperitoneal Transfusion
If feasible, IVT is the procedure of choice. Only through IVT can the moribund, nonbreathing, hydropic fetus be salvaged; 12 of 19 (63%) such fetuses in the Winnipeg series survived. What is gratifying is the much lower overall risk with IVT than with IPT (0.8% versus 3.5% per procedure).
Although the risk of IVT is materially less, this low risk can only be achieved when the obstetric ultrasonographer and the obstetric venipuncturist have great skill and experience with the procedure. Otherwise, there are hazards that increase the risk of IVT: overtransfusion, with cardiac failure (not observed in the Winnipeg series); exsanguination (a problem, particularly if the fetus is hydropic, thrombocytopenic, and the cord insertion posterior62); and inadvertent injection of blood into the cord around the vein, producing a cord hematoma, compressing and interfering with umbilical blood flow. This third hazard has caused fetal death and can be prevented only by an alert, experienced obstetric ultrasonographer.
Despite the great advantages of IVT, there are two situations in which IPT may still be required; therefore, the skill in carrying out IPT must be maintained. One is the rare situation, before 20 to 21 weeks' gestation, when the cord vessels may be too small for successful venipuncture. The second is the more common situation late in pregnancy (after 30 weeks' gestation) when, after several successful IVTs, increasing fetal size totally obscures a posterior cord vessel insertion, making venipuncture impossible.
Development in Fetal Transfusion Survivors
Physical and intellectual development in most fetal transfusion survivors is normal. Because prematurity may be marked (as early as 27 weeks' gestation) and residual severity of disease great, some problems may be anticipated. In the Winnipeg Rh Laboratory, of 101 children tested at 18 months or later, 87 appeared completely normal, 10 had minor developmental delay (probably transient and due to prematurity), and 4 were definitely abnormal: 1 had a normal IQ but spastic hemiparesis; 2 had IQs of 75 and 85; and the 4th, in whom fetal hydrops had been reversed, had cerebral agenesis that was probably unrelated to the hydrops.
SUPPRESSION OF Rh ALLOIMMUNIZATION
Since the mid-1940s, attempts have been made to suppress the strength of already developed maternal RBC immunization. Rh hapten63 has no value. The benefit of Rh-positive RBC stroma touted by Biermé and associates64 has been refuted by Gold and colleagues.65 The value of administration of promethazine hydrochloride put forward by Gusdon and co-workers66 has not been confirmed by others. Similarly, administration of Rh immune globulin (RhIG), of great value in Rh prevention, has been shown to be ineffective in suppressing Rh immunization, no matter how weak, once Rh immunization has begun.67,68
The two suppressive modalities of some benefit in reducing maternal antibody levels are intensive plasma exchange69,70 and administration of intravenous immune serum globulin (IVIG).71,72 With intensive plasma exchange (10 to 20 L weekly), maternal alloantibody can be reduced as much as 75%. After 6 to 8 weeks, however, even with continued plasma exchange, antibody levels tend to rebound. Venous access often becomes a problem, with the need for placement of arteriovenous shunts. The plasma removed must be replaced, at least partially, with blood fractions (albumin and IVIG) to reduce antibody feedback rebound and to keep maternal serum albumin and IgG at adequate levels (more than 3 g/dL and 300 mg/dL, respectively). Plasma exchange is tedious, costly, and uncomfortable. It is not without some risk to the mother, including bacterial sepsis, particularly if arteriovenous shunts have been implanted. The only expectation with the use of intensive plasma exchange is that fetal treatment measures be delayed until the fetus is greater than 22 to 24 weeks' gestation.
The institution of plasma exchange should never preclude or delay the use of definitive investigative procedures, such as amniocentesis or fetal blood sampling. Plasma exchange should be reserved for the mother of a fetus whose father is homozygous for the antigen to which she is immunized and who has a prior history of hydrops, at or before 24 to 26 weeks' gestation. In this situation, intensive plasma exchange should be begun at 10 to 12 weeks' gestation when transfer of maternal IgG is beginning, with initial amniocentesis at 18 weeks' gestation, fetal blood sampling at 19 to 22 weeks' gestation, or both.
Reports have been made of the benefits of high-dose IVIG administration in severely alloimmunized pregnant women.71,72 With doses of 1 g/kg of maternal body weight weekly, circulating maternal alloantibody levels can be reduced by as much as half, primarily as a result of the negative feedback produced by total circulating maternal IgG levels of 2.5 to 3 g/dL, readily achieved by a weekly dose of 1 g/kg. Further benefits of IGIV therapy may be due to interference with transfer of maternal antibody across the placenta by trophoblastic FC-receptor saturation and, similarly, by reduction of IgG-coated fetal RBC hemolysis by fetal reticuloendothelial FC-receptor saturation with the injected IGIV.
If IGIV therapy is considered, it should be used only in the same situation as intensive plasma exchange, beginning at 8 to 10 weeks' gestation. The recommended dose is 1 g/kg of maternal body weight at weekly intervals. Again, amniocentesis, fetal blood sampling, or both should not be delayed.
DELIVERY OF THE FETUS WITH ERYTHROBLASTOSIS
If history, antibody levels, and amniotic fluid measurements indicate that the fetus is mildly affected, delivery should be allowed to take place spontaneously. The infant may be delivered in a regional perinatal center or a community hospital, provided that the necessary personnel and resources are present to monitor cord and infant blood parameters and to undertake exchange transfusion if it should prove necessary. Fetal heart rate monitoring should be available if induction of labor is being carried out.
On the other hand, the severely affected fetus who may or may not have undergone fetal transfusions and requires early delivery needs perinatal management facilities of the highest quality. Delivery should be carried out in this instance only in a tertiary-level center where personnel and facilities are available to manage a sick premature infant who may be hydropic.
If amniotic fluid measurements indicate the need for interference at 34 weeks' gestation, the amniotic fluid should be examined for pulmonary maturity (L:S ratio greater than 2:1, PG present). If the lungs are mature, delivery should be undertaken. If they are not, administration of dexamethasone with delivery in 48 hours or fetal blood sampling and IVT repeated in 10 days with delivery at 37 to 37.5 weeks' gestation should be considered. If delivery has to be undertaken before 32 weeks' gestation or if the fetus is in a breech position, delivery should be by cesarean section, preferably with the use of epidural anesthesia. Otherwise, induction of labor with vaginal delivery should be attempted. Fetal heart rate monitoring during labor is essential. If delivery has not taken place within 24 hours after rupture of the membranes, cesarean section should be carried out.
If fetal heart rate monitoring indicates the likelihood of fetal distress, a fetal scalp blood sample for pH determination should be obtained. If it indicates fetal jeopardy (pH less than 7.2) and if immediate vaginal delivery is not possible, cesarean section should be promptly undertaken. Every effort should be directed to ensuring that adequate oxygenation of the fetus takes place during labor and at delivery. After delivery, the cord should be clamped and cut within 10 to 15 seconds; 10 to 15 mL of blood should be expressed from the placental side of the cord into a heparinized tube. Blood grouping, direct Coombs' testing, and hemoglobin and serum bilirubin estimations are carried out on this sample. The infant's nasopharynx and oropharynx are suctioned gently and quickly, and the neonate is transferred immediately to a neonatologist experienced in the care of erythroblastotic infants. Treatment of these very ill, anemic, premature infants requires the resources of a highly developed tertiary-level neonatal intensive care unit.
PREVENTION OF Rh IMMUNIZATION
Whereas our ability to suppress Rh immunization is doubtful, our ability to prevent Rh immunization is unquestionable, representing a major advance in the management of Rh-negative pregnant women. In 1900, the same year that Landsteiner described the ABO blood group system, Von Dungern, from ox RBC rabbit injection studies, proved the axiom that active immunization to an antigen (ox RBCs) is prevented by the presence of passive antibodies to the antigen (rabbit ox RBC antibodies).73 It is a comment on the rapidity of dissemination of scientific knowledge that this piece of basic research was not applied to the clinical problem of Rh prevention until 25 years after the discovery of the Rh blood group system and the elucidation of the pathogenesis of Rh immunization.
In the mid-1960s, almost simultaneously in New York74 and Liverpool, England75, and shortly thereafter in Winnipeg,76 Rh prevention experiments were carried out. Rh-negative male volunteers were given Rh-positive RBCs followed by Rh antibody in the form of high-titer plasma75 or anti-D IgG.74,76 In all these experiments, Rh immunization was prevented by the administration of Rh antibody.
These centers then carried out clinical trials in which Rh-negative, unimmunized women were given anti-D IgG after they were delivered of Rh-positive infants. The clinical trials were successful in preventing Rh immunization when anti-D IgG was given within 72 hours after delivery of an Rh-positive infant77 (Table 8).
TABLE 8. Western Canada Rh Prevention Trial (March 1, 1967, to January 31, 1968*)
| Given RhIG | Not Given RhIG |
Rh-Negative Women | After Delivery | After Delivery |
Total women in trial | 1216 | 500 |
Rh immunized 6 to 9 months after delivery | 0 | 36 (7.2%) |
* Only Rh-negative women delivered to ABO-compatible Rh-positive infants entered the trial.
(Chown B, Duff AM, James J et al: Prevention of primary Rh-immunization: First report of the Western Canadian Trial. Can Med Assoc J 100;1021, 1969)
Anti-D IgG has been licensed in North America and Europe since 1968. One prophylactic intramuscular dose in North America is about 300 μg. In Europe and Australia, the standard doses are 100 μg and 125 μg, which have been shown to be just about as effective.
Anti-D IgG prevents Rh immunization from developing with two provisions: it must be given in sufficient dose, and it must be given before Rh immunization has begun. The mechanism by which Rh antibody prevents Rh immunization is unknown. It may exert its effect by a negative feedback phenomenon or by blocking Rh-antigenic determinants on the RBC membrane from coming into contact with the surface receptors on the immunocyte. IgM anti-D, once thought to have an enhancing effect on Rh immunization, is now known to have no effect on enhancement or prevention of Rh immunization.
Standard Rh Prevention Procedure
Because all successful clinical trials of Rh prevention were based on injection of anti-D IgG within 72 hours of birth, the period required in some instances to obtain cord blood findings, the recommendation following licensure of anti-D IgG was that all Rh-negative unimmunized women be given one prophylactic dose within 72 hours after delivery of an Rh-positive infant. This recommendation applies to Rh-negative mothers of all Rh-positive infants, including the 20% whose infants are ABO incompatible, because the protection against Rh immunization conferred by ABO incompatibility is only partial.
Because rare but undoubted instances of Rh immunization appear within 72 hours after delivery,23 anti-D IgG should be given as soon after delivery as the infant is determined to be Rh-positive and the mother at risk. If the Rh status of the infant is not available 72 hours after delivery, the mother should be given anti-D IgG with the understanding that there is a one third chance that her infant is Rh negative and that she therefore is not at risk. It is preferable to give a mother anti-D IgG unnecessarily than to withhold treatment of a woman who then becomes immunized. Conversely, if for any reason a woman at risk has not been given anti-D IgG within 72 hours, she should be treated as soon as she is determined to be at risk. It has been shown experimentally that at least partial protection is afforded by giving anti-D IgG up to 13 days after exposure to Rh-positive RBCs.78 Rh prophylaxis therefore is recommended up to 28 days after delivery, with the understanding, however, that the longer prophylaxis after delivery is delayed, the less likely it is to be effective.
Residual Rh Prophylaxis Problems
Although Rh immunization has been reduced by at least 80% in most areas since the introduction of Rh prophylaxis, some problems remain.
FAILURE OF Rh PROPHYLAXIS AFTER DELIVERY.
There is still an occasional unimmunized Rh-negative woman who is not treated after delivery of an Rh-positive infant. This may be due to failure of a mother to seek prenatal care, failure of the physician to carry out prenatal blood testing, or failure of the hospital delivery unit to carry out postdelivery cord and maternal blood testing.
Physicians rendering prenatal and obstetric care must know who their Rh-negative patients are. A blood sample from every pregnant woman should be screened at her first prenatal visit. If she is Rh negative, she should be rechecked regularly. At delivery, cord and maternal blood must be tested. If the infant is Rh positive and the mother is not Rh immunized, she should be given Rh prophylaxis. ABO incompatibility and the presence of a weakly positive cord RBC direct antiglobulin (Coombs') test are not a contraindication to Rh prophylaxis. The infant has ABO erythroblastosis; the mother should be given anti-D IgG. It is the physician's responsibility to ensure that all Rh-negative, unimmunized patients receive RhIG if they deliver Rh-positive infants.
AFTER ABORTION.
Rh-negative women who have an abortion are at risk of Rh immunization (1.5% to 2% if spontaneous, 4% to 5% if therapeutic). Women who are Rh immunized because of abortion are good responders. They often have severely affected fetuses in subsequent pregnancies. Because the hospital stay may be brief, particularly at the time of spontaneous abortion, the Rh status of the mother may not be known before discharge. The physician caring for the woman who aborts must know her Rh status. If she is Rh negative, the physician must ensure that she is given anti-D IgG. Because fetal TPHs after abortions in the first trimester usually are small, a dose of anti-D IgG as low as 50 μg, if available, is protective.
AFTER AMNIOCENTESIS.
If the placenta is implanted on the anterior uterine wall, there is a risk that it will be traumatized at amniocentesis, with subsequent fetal TPH.12 An Rh-negative unimmunized woman undergoing genetic amniocentesis or amniocentesis for determination of pulmonary maturity who has a TPH is at risk of Rh immunization. Ultrasound placental localization reduces but does not remove the risk of placental trauma.79 Similarly, a negative Kleihauer test after amniocentesis does not exclude a small TPH. For these reasons, all Rh-negative unimmunized women undergoing amniocentesis should be given anti-D IgG. After amniocentesis, 300 mmg of anti-D should be given because a protective effect must be exerted over several weeks. A second dose should be given 12 weeks after the first if the woman is undelivered. Despite the fears of some investigators, anti-D IgG given after amniocentesis does not harm the conceptus. This has been shown by the thousands of normal infants born after antenatal Rh prophylaxis.
AFTER MASSIVE TRANSPLACENTAL HEMORRHAGE.
The protective effect of anti-D IgG is dose dependent. It has been shown experimentally that one prophylactic dose of about 300 mmg prevents Rh immunization up to an exposure of about 30 mL of Rh-positive blood (12 to 15 mL of RBCs).80 If the exposure is greater, protection is partial. The incidence of Rh immunization after injection of 30 to 450 mL of Rh-positive blood into Rh-negative male volunteers and administration of one dose of anti-D IgG (300 μg) is about 26%.81 In Winnipeg, of 9000 pregnancies studied, only 0.23% of women had fetal TPH in excess of 30 mL of blood; thus, the Rh immunization rate because of failure to diagnose massive TPH and administration of only one dose of anti-D IgG is low (0.23% of 26%, or 0.06% of all Rh-negative women treated in Winnipeg). Although routine fetal Kleihauer cell screening at delivery is recommended, failure to diagnose and treat massive fetal TPH is a rare cause of failure of Rh prophylaxis.
Massive TPH usually occurs at the time of delivery. If TPH is diagnosed after delivery of an Rh-positive infant, multiple vials of anti-D IgG (300 μg) should be given intramuscularly as follows: two vials if the TPH is between 25 and 50 mL of blood; three vials if the TPH is between 50 and 75 mL, and so on. It is recommended that the contents of four vials (1200 μg) be given intramuscularly every 12 hours until the total dose has been administered. After administration of the total dose of anti-D IgG, the result of the Kleihauer test should be negative, and passive anti-D should be present in the maternal circulation.
In the rare event that massive TPH is diagnosed during pregnancy (nearly always in the third trimester), there are three considerations: the risk of fetal exsanguination, the risk of Rh immunization, and the risk, if large doses of anti-D IgG are given, that the Rh-positive RBCs in the fetal circulation will be hemolyzed.
As much as 600 μg (two vials) of anti-D IgG may be given intramuscularly to a mother without risk of dangerous RBC hemolysis in the fetus. Larger doses may be safe, but evidence is not available that they are. If fetal cells clear quickly after a dose of two vials and if passive anti-D is present, neither Rh immunization of the mother nor exsanguination of the fetus is likely. If fetal cells persist or if the original hemorrhage was greater than 100 mL, the hazards of maternal Rh immunization and fetal exsanguination are great. Prompt delivery is indicated if there is evidence of fetal maturity. The infant should be examined and transfused immediately if there is evidence of shock or a low central venous pressure. If the infant is Rh positive, the mother should be given anti-D IgG according to the formula outlined.
If the father appears to be heterozygous for D, there is a dilemma. The fetus may be Rh negative, with only a risk of fetal exsanguination, or Rh positive, with the three risks described. When TPH of greater than 25 mL is present, two vials of anti-D IgG should be administered. If Kleihauer testing reveals no change in the number of fetal RBCs in the maternal circulation, and if passive antibody is present, the fetus is probably Rh negative. Fetal Rh status can be proved by PCR testing of amniotic fluid. The fetus should be assessed regularly by ultrasonography and the pregnancy allowed to continue to term, provided that the fetal condition is good and the TPH is less than 100 mL of blood and is not increasing.
If an Rh-negative woman is inadvertently transfused with large volumes of Rh-positive blood, direct attempts at prevention may produce severe anemia. If the Rh-positive blood transfusion exceeds two units of blood (900 mL), Rh prevention should not be attempted unless she is a child or in her childbearing years. If prevention is necessary, a preliminary 1.5 mL blood volume exchange transfusion should be carried out using cross-matched ABO-compatible Rh-negative blood. The exchange will remove 75% of the previously transfused Rh-positive RBCs. The dose of anti-D IgG may then be calculated on the basis of 35% of the volume of Rh-positive blood transfused. Differential agglutination studies 48 to 72 hours after the administration of the calculated dose of anti-D should reveal no circulating Rh-positive RBCs, and passive anti-D should be readily demonstrable.
Rh IMMUNIZATION DURING PREGNANCY: FAILURE OF POSTDELIVERY Rh PROPHYLAXIS.
In 1.8% of Rh-negative women beginning an Rh-positive pregnancy without evidence of Rh immunization, there is evidence of Rh immunization during the pregnancy or within 3 days after delivery.23 Postdelivery Rh prophylaxis does not prevent Rh immunization in this group of women. In Manitoba, 62 of 3533 such women were Rh immunized during pregnancy (see Table 3), 5 before 28 weeks' gestation. Similar findings have been reported from Hamilton, Ontario82 and from Sweden,83,84 and a somewhat lower incidence was reported from Finland.85 The incidence of Rh immunization during pregnancy in the United States is unknown, but it does occur,86 and there is no reason to believe that it is any less than in Canada and Sweden.
Rh immunization during pregnancy, therefore, accounts for about 14% of all cases of Rh immunization (if no Rh prophylaxis is given) and is the most important cause of residual Rh immunization. In Manitoba, it accounted for 69 (41%) of 167 Rh-immunized pregnancies encountered in a 3-year period ending October 31, 1976. Severity of subsequent Rh disease in fetuses of women Rh immunized during pregnancy is just as great as in fetuses of women Rh immunized after delivery.
A clinical trial of Rh prophylaxis (300 μg of anti-D IgG given at 28 and 34 weeks' gestation) reduced the incidence of Rh immunization during pregnancy from 1.8% to 0.1%.23 A service program of administration of one dose (300 μg of intramuscular anti-D87 or 300 μg of intravenous anti-D88) was equally successful (Table 9).
TABLE 9. Antenatal Rh Prophylaxis from (December 1, 1968, through December 31, 1982)
Rh-Negative Women | No. of Women |
Treated antepartum and | 12,215 |
delivered of Rh-positive infants | 8,422 |
Rh immunization expected at delivery | 137 |
Rh immunization observed | 8* |
Antenatal protection rate | 94% |
* Four were multigravidas not treated antenatally in a past pregnancy.
Administration of anti-D IgG to the mother during pregnancy does not pose a risk to the fetus. If one dose is given at 28 weeks' gestation, the cord RBCs are uniformly direct Coombs' test negative. When a dose is given at 28 weeks and 34 weeks, 35% of Rh-positive infants have cord RBCs that are weakly direct Coombs' test positive; however, none will show evidence of anemia or hyperbilirubinemia.
The desirability of an antenatal Rh prophylaxis program depends on the incidence of Rh immunization during pregnancy and the cost of anti-D IgG. Half again as much anti-D IgG must be given during pregnancy as after delivery because one third of infants born of Rh-negative mothers and Rh-positive fathers are Rh negative. Thus, institution of antenatal Rh prophylaxis increases anti-D use by a factor of 2.5. Because antenatal Rh prophylaxis prevents about one seventh as much Rh immunization as does postpartum prophylaxis, the anti-D cost of preventing one case of Rh immunization during pregnancy is 11 times the cost of preventing one case of Rh immunization after delivery. Nevertheless, antenatal Rh prophylaxis is cost-effective and is highly recommended (one dose at 28 weeks' gestation).
Rh IMMUNIZATION IN INFANCY: THE GRANDMOTHER THEORY.
Rh immunization in infancy as a result of maternofetal TPH from an Rh-positive mother to an Rh-negative infant at delivery has been postulated.89 Sixty percent of mothers of Rh-negative infants are Rh positive. Careful studies have refuted earlier evidence that maternofetal TPH is a common occurrence.90 Some reports of a high incidence of Rh immunization during infancy91,92 have not been substantiated86,93 and are untenable based on the low incidence of Rh immunization if antenatal, postnatal, and postabortion prophylaxis is carried out. Rh immunization of the Rh-negative neonate occurs in less than 0.2% of Rh-negative infants born of Rh-positive mothers. Administration of anti-D IgG to Rh-negative infants born of Rh-positive mothers is not recommended.
THE PRESENCE OF WEAK ANTI-D: ATTEMPTS AT SUPPRESSION.
An occasional woman has a weak Rh antibody, demonstrable only by an autoanalyzer method or by an Auto Analyzer plus a manual enzyme-treated RBC technique; these women may not be Rh immunized. In one study, 206 of 236 such women showed no progression of Rh immunization despite delivery of Rh-positive infants. Women who have Rh antibodies demonstrable only by autoanalyzer should be given Rh prophylaxis if they deliver Rh-positive infants.
In cases in which the antibody is demonstrable by an enzyme technique but not by an indirect antiglobulin (Coombs') test, administration of anti-D IgG has been unsuccessful. Twenty-one of 36 women with such antibodies developed full-blown Rh immunization despite administration of anti-D IgG at 6-week intervals during pregnancy.67 Anti-D IgG administration to these women is not recommended. If the tests are equivocal and if there is any doubt about the specificity of the antibody found, Rh prophylaxis is recommended with the understanding that it may be ineffective.
REACTIONS TO ANTI-D IMMUNOGLOBULIN G.
Anti-D IgG is prepared by the cold ethanol method originally described by Cohn in 1940. Anti-D IgG made by this process is effective and has a low incidence of untoward reactions. It does contain small amounts of IgA and IgM and traces of other plasma proteins. Some women, after a second or third injection, develop transient rashes, pain, and swelling at the injection site. There was one report of severe anaphylaxis after the use of Cohn-prepared anti-D IgG.94 Cohn-prepared anti-D IgG is anticomplementary and must not be given intravenously. The yield of anti-D in the IgG prepared by the Cohn method is at best only 40% to 50% and may be as low as 35% of that in the starting plasma.
Anti-D IgG preparations suitable for intravenous use have been prepared by various methods. Acid-hydrolyzed anti-D IgG has been produced and used successfully in France. Because the source of the acid hydrolyzed anti-D is a Cohn preparation, its efficiency of yield and minor risk of anaphylaxis are no different from any other Cohn-prepared anti-D preparation.95
Hoppe and associates96 have prepared anti-D IgG by an ion-exchange column method using DEAE Sephadex. This method has been adapted for use in North America, and an anti-D IgG prepared by ion exchange is now licensed in Canada and the United States.88 Anti-D prepared by ion exchange is pure and has a low total protein content. There is no demonstrable IgA or IgM. Its anticomplementary activity is low, and it may be safely given intravenously.
The efficiency of yield of anti-D in the ion-exchange preparation is 85% to 90%. When given intravenously, anti-D, microgram for microgram, is twice as effective. Therefore, half the dose (120 μg) given after delivery is as effective as 250 μg to 300 μg of the Cohn product given intramuscularly. Because its half-life is no greater when given intravenously, however, the antenatal prophylaxis dose must be about 300 μg to provide protection for the 12-week period between the time of injection and the time of delivery. Clinical trials and service programs in which thousands of doses of ion-exchange anti-D have been given intravenously show that it is at least as successful in preventing Rh immunization as cold ethanol-produced anti-D IgG.
The advantages of ion-exchange anti-D IgG are as follows: greater purity, and therefore less likelihood of production of a reaction; less cost because the efficiency of yield is greater and ion-exchange preparation is cheaper; less dose because it is given by the intravenous route; and less discomfort because of the use of the intravenous route.
Current Rh Prophylaxis Recommendation
- An Rh-negative unimmunized woman delivered of an Rh- positive infant should be given one prophylactic dose of anti-D IgG irrespective of the ABO status of her child as soon after delivery as the neonate is known to be Rh positive. In no event should prophylaxis be delayed past 72 hours after delivery.
- An Rh-negative unimmunized woman who aborts should be given Rh prophylaxis unless the father of the conceptus is known to be Rh negative.
- An Rh-negative unimmunized woman undergoing amniocentesis should be given one standard dose of anti-D IgG (300 μg) unless the father of the conceptus is know to be Rh negative. This dose should be repeated in 12 weeks if she remains undelivered.
- One prophylactic dose of anti-D IgG (300 μg) should be administered to an Rh-negative unimmunized woman at 28 weeks' gestation unless the father of the conceptus is known to be Rh negative.
- An Rh-negative unimmunized woman who has a massive fetal TPH of Rh-positive blood should be given one prophylactic dose of 300 μg of anti-D for every 25 mL of Rh-positive blood in her circulation. If an intravenous preparation is used, 120 μg should be given for every 15 mL of Rh-positive fetal blood in her circulation.
- An Rh-negative woman who has an Rh antibody demonstrated only by an autoanalyzer method should be given prophylaxis if she is delivered of an Rh-positive baby. An Rh-negative woman who has an Rh antibody demonstrable by both an autoanalyzer and an enzyme method but not by an indirect antiglobulin (Coombs') text will not be protected by administration of anti-D after delivery of an Rh-positive infant. She should be treated, however, if there is any doubt regarding the validity of the enzyme reaction.
ATYPICAL BLOOD GROUP IMMUNIZATION
Because of the progressive reduction in Rh immunization due to Rh prophylaxis, atypical immunization is assuming greater importance. Most cases of atypical immunization are caused by blood transfusion because so-called compatible transfusions are compatible only for ABO and for D in the Rh system. After blood transfusion, the recipient is exposed to many blood group antigens that she does not possess. After transfusion, 1% to 2% of patients develop atypical antibodies.
Few of such antibodies are of clinical importance, although they may make the subsequent procurement of compatible blood difficult and may produce transfusion reactions. Most, such as anti-Lea, Leb, M, N, and P, almost never produce erythroblastosis. Rarely, anti-Lua may cause erythroblastosis, as may antibodies to low-incidence antigens such as Wra.
Anti-c, anti-Kell, and less commonly anti-E, anti-C, anti-Fya and anti-Jka may produce erythroblastosis as severe as that produced by anti-D. Anti-Kell, although a potent antibody, rarely causes problems because it nearly always is produced by blood transfusion and because 90% of fathers of the fetuses of Kell-immunized women are Kell negative. Anti-c is another matter because two thirds of fathers are c positive, of which about one half are homozygous for c. Hydrops from anti-c occurs. Anti-Fya, anti-JKa, anti-E, and anti-C, which usually produce mild disease, on rare occasion can cause hydrops.
Of 300 fetuses referred to the Rh Laboratory in Winnipeg for fetal transfusions, the responsible antibody was anti-D in 261; anti-Kell in 22; anti-c in 12; and anti-E, anti-C, anti-k, anti-Fya, and anti-Jka in one each.
When a pregnant woman is found to be immunized against any blood group antigen set out in the first two columns of Table 10 or against an antigen for which the effect on the fetus of such immunization is unknown, she should be monitored in exactly the same way as if she were Rh negative and Rh immunized. Antibody titrations should be carried out at the same intervals, and amniocentesis should be performed for the same indications. Early delivery, fetal transfusions, or both are carried out for the same amniotic fluid ΔOD 450 indications.
TABLE 10. Atypical Maternal Blood Group Antibodies: Association with Clinical Hemolytic Disease of Fetus and Newborn.
Common | Uncommon | Very Rare | Never |
Anti- | Anti- | Anti- | Anti- |
c | e | S | Lea |
K | C | U | Leb |
E | Ce | M | P |
| Kpa | Fyb |
|
| Kpb | N |
|
| cE | Doa |
|
| k | Coa |
|
| s | Dia |
|
| Wra | Dib |
|
| Fya | Lua |
|
| jKd | Yta |
|
|
| Jkb |
|
(Bowman Jm: Maternal blood group immunization. In Creasy RK, Resnik R (eds): Maternal-Fetal Medicine: Principles and Practice. Philadelphia, WB Saunders, 1984).
Anti-Kell alloimmunization is a special case. There are several reports of severe Kell alloimmune disease with misleading low ΔOD 450 values.97,98 Anti-Kell has been postulated to cause destruction of poorly hemoglobinized marrow erythroid precursors,99 which may explain the reports of more severe Kell hemolytic disease of the newborn (HDN) than was predicted from the ΔOD 450 readings.97,98 Therefore, if a significant anti-Kell antibody is present, fetal blood sampling should be carried out at lower ΔOD 450 readings. Indeed, if there is a past history of Kell hydrops, an early amniocentesis should be carried out to determine the Kell status of the fetus by PCR testing, and fetal blood sampling should be carried out at 20 to 22 weeks' gestation.
Whereas anti-c and anti-Kell erythroblastosis differ little in severity from Rh erythroblastosis, ABO erythroblastosis is different and should be managed differently. Although anti-A and anti-B bind complement and produce severe, life-threatening intravascular hemolysis if ABO-incompatible blood is transfused, ABO erythroblastosis is mild. Hyperbilirubinemia may develop, which may cause kernicterus if left untreated, but hydrops almost never occurs, and anemia is never more than moderate.
The reasons for the mildness of ABO erythroblastosis are that the fetal RBC membrane has fewer A and B antigenic sites; most anti-A and anti-B is IgM and does not cross into the fetal circulation; the small amount of anti-A or anti-B that is IgG and does cross into the fetal circulation has many antigenic sites in tissue and secretions other than on the RBCs to which it can bind. Because only a small amount of antibody is fixed to each RBC membrane, the direct antiglobulin test is only weakly positive when cord RBCs are tested and may be negative when capillary blood is tested at 1 or 2 days of age.
ABO erythroblastosis, which may occur in the first pregnancy, is not an obstetric problem because hydrops and severe anemia at birth rarely occur. Amniocentesis and early delivery are not indicated. Anti-A and anti-B titers are of no value in predicting the probability of clinical ABO erythroblastosis, although ABO erythroblastosis only occurs if some of the maternal anti-A or anti-B is IgG and hemolytic.
Infants with ABO erythroblastosis usually are group A or B and have group O mothers. Twenty-five to 30% of such infants have weakly direct antiglobulin (Coombs' test) positive cord RBCs. Only a small number of these infants develop early and severe jaundice. In one report, in a 12-year period from 1954 to 1965, 2500 (28%) of 9000 ABO-incompatible infants had cord RBCs that were weakly direct antiglobulin positive.100 Only 41 (1.6%) of the 2500 infants required exchange transfusion. ABO erythroblastosis is entirely a pediatric problem and is concerned with the management of hyperbilirubinemia and the prevention of kernicterus.
REFERENCES
Wiener AS, Wexler IB: Heredity of the Blood Groups. New York, Grune & Stratton, 1958 |
|
Race RR. The Rh genotype and Fisher's theory. Blood 3(Special Issue No 2):27, 1948 |
|
Lacey PA, Caskey CR, Werner DJ et al: Fatal hemolytic disease of the newborn due to anti-D in an Rh positive Du variant mother. Transfusion 23: 91, 1983 |
|
Tippett P, Sanger R: Observations on subdivisions of the Rh antigen D. Vox Sang 7: 9, 1962 |
|
Gahmberg CG, Karhi KK: Association of Rho (D) polypeptides with the membrane skeleton in Rho (D)-positive human red cells. J Immunol 133: 334, 1984 |
|
Brown PJ, Evans JP, Sinor LT et al: The Rhesus D antigen: A dicyclohexylcarbodiimide-binding proteolipid. Am J Pathol 110: 127, 1983 |
|
Goto S, Nishi H, Tomoda A: Blood group Rh-D factor in human trophoblast determined by immunofluorescent method. Am J Obstet Gynecol 137: 707, 1980 |
|
Le Van Kim C, Mouro I, Chrif-Zahar B et al: Molecular cloning and primary structure of the human blood group RhD polypeptide. Proc Natl Acad Sci USA 89: 10925, 1992 |
|
Wiener AS: Diagnosis and treatment of anemia of the newborn caused by occult placental hemorrhage. Am J Obstet Gynecol 56: 717, 1948 |
|
Chown B: Anemia from bleeding of the fetus into the mother's circulation. Lancet 1: 1213, 1954 |
|
Kleihauer E, Braun H, Betke K: Demonstration von fetalem haemoglobin in den erythrozyten eines blutausstriches. Klin Wochenschr 35: 637, 1957 |
|
Peddle LJ: Increase of antibody titer following amniocentesis. Am J Obstet Gynecol 100: 567, 1968 |
|
Wiener AS: Conglutination test for Rh sensitization. J Lab Clin Med 30: 662, 1945 |
|
Lewis M, Chown B: A short albumin method for the determination of isohemagglutinins, particularly incomplete Rh antibodies. J Lab Clin Med 50: 494, 1957 |
|
Coombs RRA, Mourant AE, Race RR: A new test for the detection of weak and “incomplete” Rh agglutinins. Br J Exp Pathol 26: 255, 1945 |
|
Lewis M, Kaita H, Chown B: Kell typing in the capillary tube. J Lab Clin Med 52: 163, 1958 |
|
Rosenfield RE, Haber GV: Detection and measurement of homologous human hemagglutinins: Automation in analytical chemistry. Technicon Symposia 503, 1966 |
|
Lalezari P: A polybrene method for the detection of red cell antibodies. Fed Proc 26: 756, 1967 |
|
Moore BPL: Automation in the blood transfusion laboratory. I. Antibody detection and quantitation in the Technicon AutoAnalyzer. Can Med Assoc J 100: 381, 1969 |
|
Woodrow JC: Rh immunization and its prevention. In Jensen KG, Killmann SA (eds): Series Hematologica III, Vol 3, p 33. Copenhagen, Munksgaard, 1970 |
|
Zipursky A, Israels LG: The pathogenesis and prevention of Rh immunization. Can Med Assoc J 97: 1245, 1967 |
|
Nevanlinna HR: Factors affecting maternal Rh immunization. Ann Med Exp Biol 31 (Fenn suppl 2): 1, 1953 |
|
Bowman JM, Chown B, Lewis M et al: Rh-immunization during pregnancy: Antenatal prophylaxis. Can Med Assoc J 118: 623, 1978 |
|
Lobuglio AF, Cotran RS, Jandl JH: Red cells coated with immunoglobulin G: Binding and sphering by mononuclear cells in man. Science 158: 1582, 1967 |
|
Phibbs RH, Johnson P, Tooley WH: Cardio-respiratory status of erythroblastotic infants. II. Blood volume, hematocrit and serum albumin concentration in relation to hydrops fetalis. Pediatrics 53: 13, 1974 |
|
James LS: Shock in the newborn in relation to hydrops. In Robertson JG, Dambrosio F (eds): International Symposium on the Management of the Rh problem. Annali Obstet Ginec Special Number:193, 1970 |
|
Bowman JM, Pollock JM: Amniotic fluid spectrophotometry and early delivery in the management of erythroblastosis fetalis. Pediatrics 35: 815, 1965 |
|
Zupanska B, Brojer E, Richards Y et al: Serological and immunological characteristics of maternal anti-Rh(D) antibodies in predicting the severity of haemolytic disease of the newborn. Vox Sang 56: 247, 1989 |
|
Nance SJ, Nelson JM, Horenstein J et al: Monocyte monolayer assay: An efficient noninvasive technique for predicting the severity of hemolytic disease of the newborn. Am J Clin Pathol 92: 89, 1989 |
|
Urbaniak SJ, Greiss MA, Crawford RJ, Fergusson MJ: Prediction of the outcome of Rhesus haemolytic disease of the newborn: Additional information using an ADCC assay. Vox Sang 46: 323, 1984 |
|
Engelfriet CP, Brouwers HAA, Huiskes E et al: Prognostic value of the ADCC with monocytes and maternal antibodies for haemolytic disease of the newborn, p 162 [Abstract]. Sydney, Australia, Book of Abstracts, 21st Congr. ISH and 29th Congr ISBT, 1986 |
|
Hadley AG, Kumpel BM, Merry AH: The chemiluminescent response of human monocytes to red cells sensitized with monoclonal anti-Rh(D) antibodies. Clin Lab Haematol 10: 377, 1988 |
|
Hadley AG, Kumpel BM, Leader KA et al: Correlation of serological, quantitative and cell-mediated functional assays of maternal alloantibodies with the severity of haemolytic disease of the newborn. Br J Haematol 77: 221, 1991 |
|
Mollison P: Results of tests with different cellular bioassays in relation to severity of RhD haemolytic disease: Report from nine collaborating laboratories. Vox Sang 60: 225, 1991 |
|
Brown SJ, Perkins JT, Sosler SD et al: The monocyte- monolayer assay does not predict severity of hemolytic disease of the newborn. Transfusion 31 (suppl 193): 53S, 1991 |
|
Bevis DCA: Blood pigments in haemolytic disease of the newborn. J Obstet Gynaecol Br Emp 63: 68, 1956 |
|
Liley AW: Liquor amnii analysis in management of pregnancy complicated by rhesus immunization. Am J Obstet Gynecol 82: 1359, 1961 |
|
Bowman JM: Management of Rh-isoimmunization. Obstet Gynecol 52: 1, 1978 |
|
Nicolaides KH, Rodeck CH, Mibashan MD, Kemp JR: Have Liley charts outlived their usefulness? Am J Obstet Gynecol 155: 90, 1986 |
|
Bowman JM, Pollock JM, Manning FA et al: Maternal Kell blood group alloimmunization. Obstet Gynecol 79: 239, 1992 |
|
Bartsch FK: Bilirubin in the amniotic fluid: A review. In Robertson JG, Dambrosio F (eds): International Symposium on the Management of the Rh problem. Annali Obstet Ginec Special Number:73, 1970 |
|
Rehder H, Weitzel H: Intrauterine amputation after amniocentesis. Lancet 1: 832, 1978 |
|
Chitkara U, Wilkins I, Lynch L et al: The role of sonography in assessing severity of fetal anemia in Rh- and Kell-isoimmunized pregnancies. Obstet Gynecol 71: 393, 1988 |
|
Daffos F, Capella-Pavlovsky M, Forestier F: Fetal blood sampling during pregnancy with use of a needle guided by ultrasound: A study of 606 consecutive cases. Am J Obstet Gynecol 153: 655, 1985 |
|
Bennett PR, Le Van Kim C, Colin Y et al: Prenatal determination of fetal RhD type by DNA amplification. N Engl J Med 329: 607, 1993 |
|
Hyland CA, Wolter LC, Saul A: Identification and analysis of Rh genes: Application of PCR and RFLP typing tests. Transfus Med Rev 1X: 289, 1995 |
|
Avent ND, Martin PG: Kell typing by allele-specific PCR (ASP) Br J Haematol 93:728, 1996 |
|
Lo Y-MD, Bowel PJ, Selinger M et al: Prenatal determination of fetal RhD status by analysis of peripheral blood of rhesus negative mothers [Letter]. Lancet 341: 1147, 1993 |
|
Liley AW: Intrauterine transfusion of fetus in haemolytic disease. Br Med J 2: 1107, 1963 |
|
Menticoglou SM, Harman CR, Manning FA et al: Intraperitoneal fetal transfusion: Paralysis inhibits red cell absorption. Fetal Ther 2: 154, 1987 |
|
Lewis M, Bowman JM, Pollock JM et al: Absorption of red cells from the peritoneal cavity of an hydropic twin. Transfusion 13: 37, 1973 |
|
Crosby WM, Brobmann GF, Chang ACK: Intrauterine transfusion and fetal death: Relationship of intraperitoneal pressure to umbilical vein blood flow. Am J Obstet Gynecol 108: 135, 1970 |
|
Adamsons K Jr, Freda VJ, James LS et al: Prenatal treatment of erythroblastosis fetalis following hysterotomy. Pediatrics 35: 848, 1965 |
|
Asensio SH, Figueroa-Longo JG, Pelegrina A. Intrauterine exchange transfusion. Am J Obstet Gynecol 95:1129, 1966 |
|
Seelen J, Van Kessel H, Eskes T et al: A new method of exchange transfusion in utero: Cannulation of vessels on the fetal side of the human placenta. Am J Obstet Gynecol 95: 872, 1966 |
|
Rodeck CH, Holman CA, Karnicki J et al: Direct intravascular fetal blood transfusion by fetoscopy in severe rhesus isoimmunisation. Lancet 1: 652, 1981 |
|
De Crespigny LC, Robinson HP, Quinn M et al: Ultrasound-guided blood transfusion for severe rhesus isoimmunization. Obstet Gynecol 66: 529, 1985 |
|
Berkowitz RL, Chitkara U, Goldberg JD et al: Intrauterine intravascular transfusions for severe red blood cell isoimmunization: Ultrasound guided percutaneous approach. Am J Obstet Gynecol 155: 574, 1986 |
|
Nicolaides KH, Soothill PW, Clewell W et al: Rh disease: Intravascular fetal blood transfusion by cordocentesis. Fetal Ther 1: 185, 1986 |
|
Seeds JW, Bowes WA: Ultrasound-guided intravascular transfusion in severe rhesus immunization. Am J Obstet Gynecol 154: 1105, 1986 |
|
Grannum PAT, Copel JA, Moya FR et al: The reversal of hydrops fetalis by intravascular intrauterine transfusion in severe isoimmune fetal anemia. Am J Obstet Gynecol 158: 914, 1988 |
|
Harman CR, Bowman JM, Menticoglou SM et al: Profound fetal thrombocytopenia in Rhesus disease: Serious hazard at intravascular transfusion. Lancet 2: 741, 1988 |
|
Carter BB: Preliminary report on a substance which inhibits anti-Rh serum. Am J Clin Pathol 17: 646, 1947 |
|
Biermé SJ, Blanc M, Abbal M et al: Oral Rh treatment for severely immunized mothers. Lancet 1: 604, 1979 |
|
Gold WR Jr, Queenan JT, Woody J et al: Oral desensitization in Rh disease. Am J Obstet Gynecol 146: 980, 1983 |
|
Gusdon JP Jr, Caudle MR, Herbst GA et al: Phagocytosis and erythroblastosis: 1. Modification of the neonatal response by promethazine hydrochloride. Am J Obstet Gynecol 125: 224, 1976 |
|
Bowman JM, Pollock JM: Reversal of Rh alloimmunization: Fact or fancy? Vox Sang 47: 209, 1984 |
|
De Silva M, Contreras M, Mollison PL: Failure of passively administered anti-Rh to prevent secondary Rh immune responses. Vox Sang 48: 178, 1985 |
|
Graham-Pole J, Barr W, Willoughby MLN: Continuous flow plasmapheresis in management of severe Rhesus disease. Br Med J 1: 1185, 1977 |
|
Robinson EAE, Tovey LAD: Intensive plasma exchange in the management of severe Rh disease. Br J Haematol 45: 621, 1980 |
|
Berlin G, Selbing A, Ryden G: Rhesus haemolytic disease treated with high-dose intravenous immunoglobulin [Letter]. Lancet 1: 1153, 1985 |
|
de la Camara C, Arrieta R, Gonzalez A et al: High-dose intravenous immunoglobulin as the sole prenatal treatment for severe Rh immunization. N Engl J Med 318: 519, 1988 |
|
Von Dungern F: Beitrage zur Immunitatslehr. Munch Med Wochenschr 47: 677, 1900 |
|
Freda VJ, Gorman JG, Pollack W: Successful prevention of experimental Rh sensitization in man with an anti-Rh gamma 2-globulin antibody preparation: A preliminary report. Transfusion 4: 26, 1964 |
|
Clarke CA, Donohoe WTA, McConnell RB et al: Further experimental studies in the prevention of Rh-haemolytic disease. Br Med J 1: 979, 1963 |
|
Zipursky A, Pollock J, Yeow R et al: The pathogenesis and prevention of Rh-immunization in pregnancy. Proceedings of the II Congress of the International Society of Blood Transfusion. Sydney, 1966. Bibl Haematol 29 (I): 280, 1968 |
|
Chown B, Duff AM, James J et al: Prevention of primary Rh immunization: First report of the Western Canadian Trial. Can Med Assoc J 100: 1021, 1969 |
|
Samson D, Mollison PL: Effect on primary Rh-immunization of delayed administration of anti-Rh. Immunology 28: 349, 1975 |
|
Bowman JM, Pollock JM: Transplacental fetal hemorrhage after amniocentesis. Obstet Gynecol 66: 749, 1985 |
|
Pollack W, Ascari WQ, Crispin JF et al: Studies on Rh prophylaxis. II. Rh immune prophylaxis after transfusions with Rh-positive blood. Transfusion 11: 340, 1971 |
|
Pollack W, Ascari WQ, Kockesky RJ et al: Studies on Rh prophylaxis. I. Relationship between doses of anti-Rh and size of antigenic stimulus. Transfusion 11: 333, 1971 |
|
Zipursky A, Blajchman M: The Hamilton Rh prevention studies. In Proceedings of the McMaster Conference on Prevention of Rh Immunization. September 28–30, 1977. Vox Sang 36: 50, 1979 |
|
Hermann M, Kjellman H: Rh prophylaxis with immune globulin anti-D administered during pregnancy and after delivery. In Proceedings of the McMaster Conference on Prevention of Rh Immunization. September 28–30, 1977. Vox Sang 36: 50, 1979 |
|
Bartsch F, Sandberg L: Incidence of anti-D at delivery in previously non-immunized Rh-negative mothers with Rh-positive babies. In Proceedings of the McMaster Conference on Prevention of Rh Immunization. September 28–30, 1977. Vox Sang 36: 50, 1979 |
|
Eklund J, Nevalinna HR: Rh antibody appearance during pregnancy in Finland. In Proceedings of the McMaster Conference on Prevention of Rh Immunization. September 28–30, 1977. Vox Sang 36: 50, 1979 |
|
Scott JR, Beer AE, Guy LR et al: Pathogenesis of Rh immunization in primigravidas: Fetomaternal versus maternofetal bleeding. Obstet Gynecol 49: 9, 1977 |
|
Bowman JM, Pollock JM: Antenatal Rh prophylaxis: 28 weeks' gestation service program. Can Med Assoc J 118: 627, 1978 |
|
Bowman JM, Friesen AD, Pollock JM et al: WinRho: Rh immune globulin prepared by ion exchange for intravenous use. Can Med Assoc J 123: 1121, 1980 |
|
Taylor JF: Sensitization of Rh-negative daughters by their Rh-positive mothers. N Engl J Med 276: 547, 1967 |
|
Cohen F, Zuelzer WW: The transplacental passage of maternal erythrocytes into the fetus. Am J Obstet Gynecol 93: 566, 1965 |
|
Bowen FW, Renfield M: The detection of anti-D in Rho (D)-negative infants born of Rho (D)-positive mothers. Pediatr Res 10: 213, 1976 |
|
Carapella-de Luca E, Casadei AM, Pascone R et al: Maternofetal transfusion during delivery and sensitization of the newborn against Rhesus D-antigen. Vox Sang 34: 241, 1978 |
|
Bernard B, Presley M, Caudillo G et al: Maternal fetal hemorrhage: Incidence and sensitization [Abstract]. Pediatr Res 11: 467, 1977 |
|
Rivat L, Parent M, Rivat C: Accident survenu apres injection de gamma-globulines anti-Rh du a la presence D/ anti-corps anti-yA. Presse Med 78: 2072, 1970 |
|
Jouvenceaux AC, Adenot N, Berthoux F et al: Gammaglobuline anti-D lyophilisee intra-veineuse pour la prevention de l'immunization anti-Rh. Rev Fr Transfus 12 (suppl): 341, 1969 |
|
Hoppe HH, Mester T, Hennig W et al: Prevention of Rh-immunization: Modified production of IgG anti-Rh for intravenous application by ion exchange chromatography (IEC). Vox Sang 25: 308, 1973 |
|
Caine ME, Mueller-Heubach E: Kell sensitization in pregnancy. Am J Obstet Gynecol 154: 85, 1986 |
|
Hadi HA, Robertson A: Kell sensitization, hydrops, and low delta OD 450. J Maternal-Fetal Med 1: 293, 1992 |
|
Vaughan JI, Warwick R, Letsky E et al: Erythropoietic suppression in fetal anemia because of Kell alloimmunization. Am J Obstet Gynecol 171: 247, 1994 |
|
Bowman JM: Neonatal management. In Queenan JT (ed): Modern Management of the Rh Problem, 2nd ed, p 233. Hagerstown, MD, Harper & Row, 1977 |