
Male Pseudohermaphroditism Due to Androgen Insensitivity or 5α-Reductase Deficiency
Authors
INTRODUCTION
Male pseudohermaphrodites are persons with a Y-chromosome whose external genitalia fail to develop as expected for normal males. Causes of male pseudohermaphroditism include cytogenetic abnormalities, teratogenic causes, defects in testosterone biosynthesis, and defects in androgen action. This chapter focuses on perturbations involving the androgen action. Disorders in this category include androgen insensitivity (caused by defects in the androgen receptor gene) and 5α-reductase deficiency (caused by defects in the gene steroid reductase 5) (Table 1). Diagnosis and management are now standard and well established.1, 2 The term disorders of sexual development is now widely used to describe this group of disorders, as applied elsewhere.1 In that nomenclature the disorders discussed in this chapter would be classified under 46,XY DSD (Disorders of sexual development, disorders in androgen synthesis or action).
Table 1. 5α-reductase deficiency and androgen insensitivity syndromes (AIS)
| 5α-Reductase deficiency | Complete AIS (CAIS) | Partial AIS (PAIS) |
Karyotype | 46,XY | 46,XY | 46,XY |
Inheritance | Autosomal recessive | X-linked recessive | X-linked recessive |
Chromosome location | 2 (p23) | X (q11–12) | X (q11–12) |
Hormone levels | |||
Testosterone | Normal male | Normal male | Normal male |
Dihydrotestosterone | Decreased | Normal or increased | Normal or increased |
Luteinizing hormone | Increased | Increased | Increased |
Estrogens | Normal male | Slightly increased | Slightly increased |
Anatomic development | |||
External genitalia |
| ||
Neonate | Female or ambiguous | Female | Ambiguous |
Puberty | Variable virilization male-like breasts | Female habitus, decreased axillary/public hair, female breast development | Variable virilization |
Internal structures | Tests, wolffian derivatives (epididymides, vas deferens, seminal vesicles), no müllerian derivatives (uterus, fallopian tubes) | Testes (variable location), no wolffian derivatives, no müllerian derivatives | Testes, no wolffian derivatives, no müllerian derivatives |
GENITAL DIFFERENTIATION
If the embryo, specifically the gonadal stroma, is 46,XY, the indifferent gonads will develop into testes. This process begins approximately 43 days after conception. Testes become morphologically identifiable 7–8 weeks after conception (9–10 gestational or menstrual weeks).
Sertoli cells are the first cells to become recognizable in testicular differentiation. These cells organize the surrounding cells into tubules. Both Leydig cells3 and Sertoli cells4 function in dissociation from testicular morphogenesis, which is consistent with these cells' directing gonadal development, rather than the converse. These two cells secrete hormones that direct subsequent male differentiation (Fig. 1).
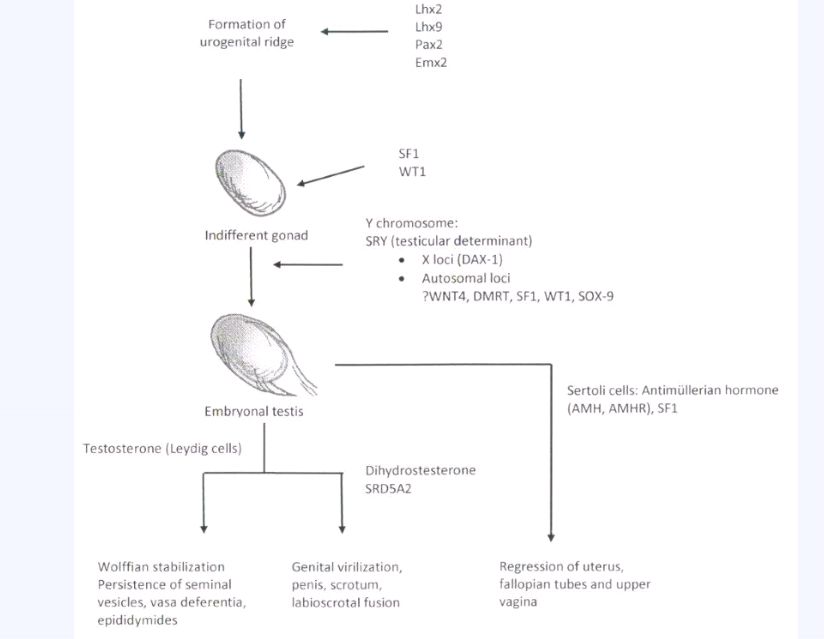 Fig. 1. Schematic diagram illustrating embryonic differentiation in the normal male. The role of certain genes in formation of the urogenital ridge and indifferent gonad have been deduced in rodents but not necessarily confirmed in humans. Known human genes are designated by standard nomenclature. (Reproduced with permission from Simpson JL: Mammalian Sex Determination, Encyclopedia of Life Sciences, 2008, John Wiley and Sons, Ltd. published online March 2008, doi: 10.1002/9780470015902.a0001886.pub2.)
Fig. 1. Schematic diagram illustrating embryonic differentiation in the normal male. The role of certain genes in formation of the urogenital ridge and indifferent gonad have been deduced in rodents but not necessarily confirmed in humans. Known human genes are designated by standard nomenclature. (Reproduced with permission from Simpson JL: Mammalian Sex Determination, Encyclopedia of Life Sciences, 2008, John Wiley and Sons, Ltd. published online March 2008, doi: 10.1002/9780470015902.a0001886.pub2.)
Fetal Sertoli cells produce antimüllerian hormone (AMH), which is also called müllerian-inhibiting substance (MIS). This glycoprotein diffuses locally to cause regression of müllerian derivatives (uterus and fallopian tubes). Antimüllerian hormone production and müllerian duct regression occurs by fetal age of 8 weeks, before secretion of testosterone and stimulation of the wolffian ducts.
Fetal Leydig cells produce an androgen testosterone, the function of which is to stabilize wolffian ducts and to permit differentiation of the vas deferens, epididymides, and seminal vesicles.
Testosterone secreted by the fetal testes is also converted to dihydrotestosterone by 5α-reductase, an enzyme in the primordia of the external genitalia. Acting locally, dihydrotestosterone stimulates differentiation of the glans penis and corpora cavernosa from the genital tubercle; the corpus spongiosum (which surrounds the penile urethra) from the urethral folds; and the scrotum from the labioscrotal swellings (Fig. 2).5 Dihydrotestosterone also stimulates the formation of the prostate and Cowper's glands. Testosterone alone cannot accomplish these steps, although it can produce virilization at puberty.
|
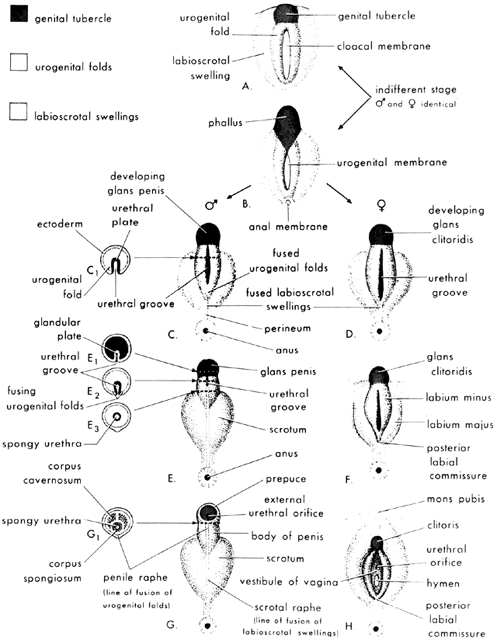
In the absence of testosterone, dihydrotestosterone, and antimüllerian hormone, the following occur: (1) the wolffian ducts regress; (2) the müllerian ducts develop into the uterus, fallopian tubes, and upper vagina; and (3) the external genitalia develop along female lines. The genital tubercle gives rise to a clitoris, the urethral folds to the labia minora, the labioscrotal swellings to the labia majora, and the urogenital sinus to the lower two thirds of the vagina and to Bartholin's and Skene's glands (see Fig. 2).5
Differentiation of the external genitalia begins in the fetus approximately 7 weeks after the last menstrual period, at which time the genital tubercle first becomes evident. Genital differentiation is completed by 14 weeks in the female fetus and by 16 weeks in the male fetus. Thus, pseudohermaphroditism arises as the result of an abnormality in genital differentiation before this time. In the female fetus, clitoral hypertrophy, labioscrotal fusion, and posterior displacement of the urethral orifice creating a urogenital sinus can occur by 14 weeks; clitoral hypertrophy is the only genital abnormality that can develop after 14 weeks' gestation. In the male fetus, hypospadias and undescended testicles can occur by 16 weeks; undescended testes is the only abnormality that can develop after 16 weeks' gestation. It is normal for premature infants to be born with undescended testes; in these infants, descent will occur in the first several weeks after birth.
ANDROGEN BIOLOGY
Secreted by the testes, testosterone is the principal androgen in adult and fetal male plasma. Bound to testosterone-binding globulin (sex hormone-binding globulin) and albumin, testosterone can be converted to dihydrotestosterone and to estradiol.
Testosterone is converted to estradiol by the enzyme aromatase. In normal men, the ratio of production of testosterone to estradiol is 100:1. An excess in absolute or relative estrogen, via either an increase in estrogen production or a decrease in testosterone (synthesis or action), will result in feminization.
Testosterone is converted to dihydrotestosterone by 5α-reductase. In addition to its effects on C19 androgen metabolism, 5α-reductase affects C21 steroid metabolism. However, only the C19 androgen metabolic changes are clinically relevant.6 5α-Reductase is a nicotinamide adenine dinucleotide phosphate (NADPH)- dependent, non-P450 enzyme associated with both the nuclear membrane and the endoplasmic reticulum membrane. 5α-Reductase activity has been demonstrated in tissue of the urogenital sinus, urogenital swellings, and urogenital tubercle, but not in that of the wolffian duct anlage.7 Two isozymes of 5α-reductase are recognized: 5AR-1 and 5AR-2. These isozymes each contain approximately 250 amino acids.8 The 5AR-1 isozyme has an optimal pH range of 7.5–8.5, whereas 5AR-2 has an optimal pH of 5.5.95AR-2 is responsible for at least some forms of male pseudohermaphroditism.
5α-REDUCTASE DEFICIENCY
Some persons who are genetically male have ambiguous external genitalia at birth, and proceed to virilize like normal males at puberty. The external genitalia of these male subjects consist of a phallus that bears a closer resemblance to a clitoris than to a penis, a perineal urethral orifice, and usually a separate blindly ending perineal orifice that resembles a vagina (pseudovagina)10 (Fig. 3).11 Testes are relatively normal in size and secrete testosterone in normal amounts. At puberty, affected males undergo phallic enlargement, increased facial hair, muscular hypertrophy, voice deepening, and no breast development. This phenotype is now known to be due to 5α-reductase deficiency. This is consistent with observations that virilization of the external genitalia during embryogenesis requires dihydrotestosterone, but wolffian differentiation requires only testosterone.10 Newborns with proven 5α-reductase deficiency present with ambiguous external genitalia, bilateral testes, and normally virilized wolffian structures that terminate in a vagina.
|
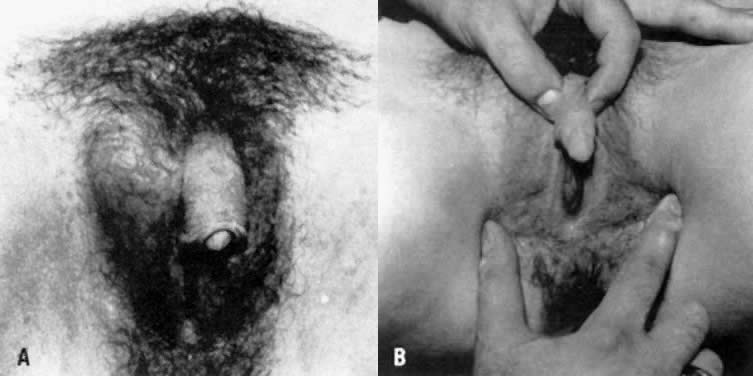
Hormone levels
Serum testosterone levels are not only normal (i.e. male) but increase in response to human chorionic gonadotropin (hCG), indicating normal hypothalamic–gonadal feedback control.12 Dihydrotestosterone levels are decreased and, predictably, the testosterone-to-dihydrotestosterone ratio (T:DHT) increased. Luteinizing hormone levels are normal or slightly increased. Estradiol levels are in the normal male range.7, 12
Genetics and genetic diagnosis
Initially called “pseudovaginal perineoscrotal hypospadias (PPSH)”, inheritance of the phenotype was shown to be autosomal recessive.13 Studies in the Dominican Republic later verified that 5α-reductase deficiency was also an autosomal recessive trait.14 Familial aggregates (sibships) of 5α-reductase deficiency (PPSH phenotype) have also been observed in the United States (black and white populations), Northern Europe, Turkey,15 Latin America,16 and elsewhere. The disorder is silent in females, who have not only a normal phenotype but a normal reproductive history.17
As noted above, two 5α-reductase proteins, 5AR-1 (SRD5A1) and 5AR-2 (SRD5A2), have been identified and their respective genes have been cloned.18 Both genes consist of five exons and four introns; the two isozyme genes share 50% of their sequence identity.8 The 5AR-1 gene is located on the short arm of chromosome 5 (5p15), whereas the 5AR-2 gene is located on chromosome 2 (2p23).18 Only the type II isozyme is expressed in gonads, and predictably 5AR-2 has been shown responsible for the 5α-reductase deficiency syndrome.19 Several investigators have identified mutations in 5AR-2 affecting 22 ethnic groups.
Missense mutations are common20 and different ethnic groups typically show different mutations. These are scattered among the five exons, presumably reflecting founder effects (Fig. 4). Within a given ethnic groups, a sentinel mutation is usually observed, typically homozygous and presumably reflecting unwitting parental consanguinity. Deletions are reported21 and cases from Papua New Guinea are usually so characterized. Molecular studies may be exploited for prenatal genetic diagnosis and genetic counseling, especially after one affected case has been detected in a kindred.
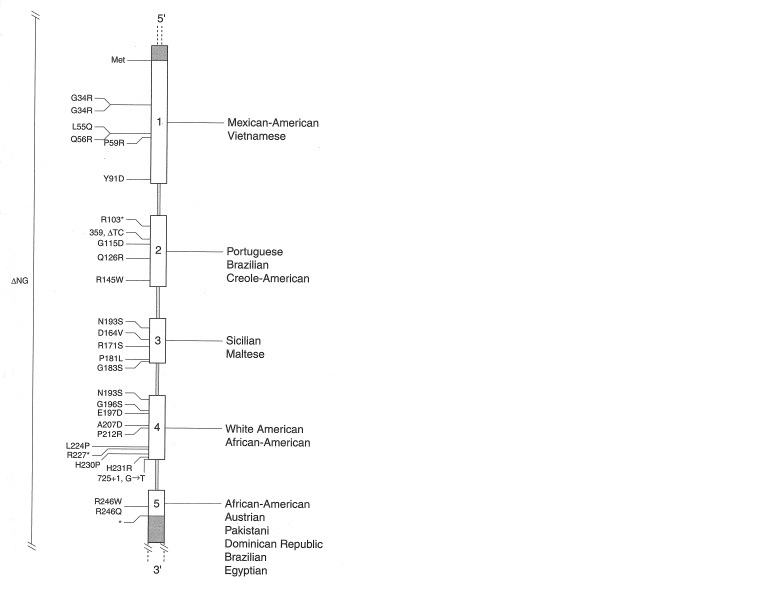 Fig. 4. Mutation in the steroid 5α-reductase 2 gene and protein. A schematic diagram of the 5α-reductase gene is shown in the middle. On the left are the location of 28 different mutations, and the predominant ethnic group on the right. ΔNG (far left) represents the deletion of the gene in the New Guinea cohort. (Data from Wilson JD, Griffin JE, Russell DW.22 Steroid 5α-reductase 2 deficiency. Endocr Rev 14:577-593.)
Fig. 4. Mutation in the steroid 5α-reductase 2 gene and protein. A schematic diagram of the 5α-reductase gene is shown in the middle. On the left are the location of 28 different mutations, and the predominant ethnic group on the right. ΔNG (far left) represents the deletion of the gene in the New Guinea cohort. (Data from Wilson JD, Griffin JE, Russell DW.22 Steroid 5α-reductase 2 deficiency. Endocr Rev 14:577-593.)
Like many other disorders of sex differentiation, the molecular heterogeneity precludes much diagnostic assistance at this time. Although screening at the molecular level is not feasible on a large scale, diagnosis via linkage analysis or direct mutation detection should be feasible in families where probands (and the mutation or linkage polymorphisms) have already been identified. Prenatal diagnosis of 5α-reductase deficiency has not been reported to date, but both this and postnatal diagnosis should be possible molecularly if mutations have been identified in both parents.
Clinical and endocrine diagnosis
5α-reductase deficiency can be diagnosed during infancy or at puberty. Neonates would typically present with ambiguous genitalia, palpable testes and a vagina or pseudovagina. At puberty, patients often present with virilizing features, including phallic enlargement, increased body hair growth, and voice deepening. Affected males have a 46,XY complement, normal serum testosterone levels, and an increased T:DHT ratio.23 Differential diagnosis includes abnormalities of testosterone biosynthesis and partial (incomplete) androgen insensitivity syndrome. In infants, baseline testosterone and dihydrotestosterone levels are so low that it may be difficult to distinguish between normal and affected infants; however, diagnosis is readily achieved via hormonal assays (Fig. 5). The elevated T:DHT ratio becomes further accentuated after administration of either hCG or testosterone propionate,10 as does the ratio of the respective urinary metabolites of testosterone and dihydrotestosterone (i.e. etiocholanolone/androsterone).7, 24
|
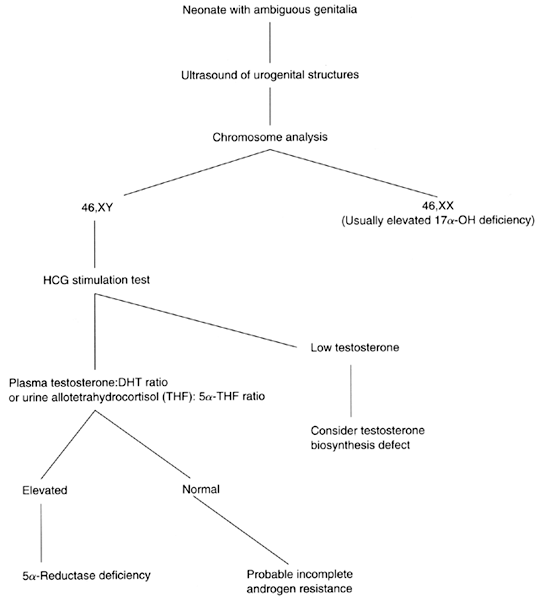
The hCG stimulation test requires that prestimulation plasma and urinary steroid levels be obtained. The patient is then given 2000 units hCG daily for 3 days. Alternatively, a single dose of 5000 units can be administered. Plasma and urinary steroid levels are obtained 24 hours after that last hCG dose. Normal infants (2 weeks to 6 months of age) have a mean prestimulation plasma T:DHT ratio of 3.9 ± 2.7 (SD) and a mean poststimulation ratio of 4.8 ± 2.7 (SD). In one study, infants with 5α-reductase deficiency (3 weeks to 3 months of age) had a prestimulation T:DHT ratio of 14:31.8 and a poststimulation ratio of 20:60.25, 24 One report described a Pakistani boy with 5α-reductase deficiency who had a normal plasma T:DHT ratio at 3 days of age, but an abnormal ratio at 9 months of age; however, the urinary metabolite ratios were abnormal at 3 days of age, which is indicative of 5α-reductase deficiency. The authors suggested that both plasma and urinary metabolite studies be performed, and that these studies be repeated at later ages.26
5α-Reductase activity is higher in some tissues than in others, which is why it is preferable to assay cells derived from genital tissue (e.g. foreskin). There is, however, considerable variability in 5α-reductase activity among control genital tissue, with near-overlap between controls and persons recognized on other grounds to have 5α-reductase deficiency. Thus 5α-reductase activity in cultured genital fibroblasts excludes the diagnosis of 5α-reductase deficiency, but absence of 5α-reductase offers less confidence in confirming this diagnosis.
Clinical heterozygote detection is difficult, at best. One study found obligate heterozygotes of two affected children to have urinary metabolite ratios, including etiocholanolone-to-androsterone, within the normal adult range; however, the ratio of 11β-hydroxyetiocholanole to 11β-hydroxyandrosterone was slightly increased in the two fathers.24
Management
Most patients with 5α-reductase deficiency are raised as females. Such persons require removal of the testes to prevent further virilization and to reduce the risk of tumors. At puberty, estrogen treatment is needed to produce feminization. Any of several forms of estrogen (e.g. conjugated estrogen, ethinyl estradiol, micronized estradiol, piperazine estrone sulfate) may be used. Because these patients do not have a uterus, one can prescribe continuous estrogen therapy (i.e. without progesterone). The clinician must tailor the estrogen dosage to the individual patient, balancing the benefits (e.g. feminization, prevention of osteoporosis) against the side-effects (e.g. hypercoagulability, liver and gallbladder disease). Creation of an adequate vagina can be achieved via medical (use of vaginal dilators) or surgical means.
Some patients with 5α-reductase deficiency who are raised as males may require androgen supplementation for virilization. Because oral dihydrotestosterone is currently not available, supraphysiologic levels of testosterone must be administered. Androgens that do not require 5α-reductase, such as 19 nortestosterone, also can be given.12 The long-term effects of such high levels of androgens are not known. In addition to receiving androgen treatment, these boys must undergo urologic reconstructive surgery. Odame and colleagues26 reported on the application of topical dihydrotestosterone cream to the external genitalia before urologic reconstructive surgery. The dihydrotestosterone cream increased the virilization of the external genitalia, which facilitated the surgery.
ANDROGEN INSENSITIVITY SYNDROME
Formerly known as “testicular feminization syndrome,” androgen insensitivity syndrome (AIS) is an X-linked disorder in which a 46,XY shows a female phenotype. The prevalence of complete AIS has been reported to be 1 in 60,000.12, 27 Diagnosis is usually not made until puberty, at which time normal linear growth and normal breast development have occurred, but menarche has not. Despite pubertal feminization, some persons with androgen insensitivity show clitoral enlargement and labioscrotal fusion. The term partial (incomplete) androgen insensitivity (formerly incomplete testicular feminization) is applied to these patients. Both complete and partial androgen insensitivity are inherited in an X-linked recessive fashion, and both involve the same gene; however, the two disorders are considered distinct because they clearly breed true in a given family.
THE ANDROGEN RECEPTOR
The androgen receptor is a member of the superfamily of transcription regulators. This receptor regulates transcription of androgen-responsive genes. Located intracellularly, the receptor is inactive unless bound to an androgen (testosterone or dihydrotestosterone). The receptor–androgen complex then governs DNA transcription in the cell nucleus of target tissues. Both testosterone and dihydrotestosterone bind to the same receptor, but they produce different physiologic effects. Testosterone regulates secretion of luteinizing hormone by the hypothalamic–pituitary axis and stimulates differentiation of the wolffian ducts to form the epididymides, vas deferens, and seminal vesicles. As noted above, dihydrotestosterone is required for male differentiation of the external genitalia (penis, scrotum), urethra, and prostate during embryogenesis. Dihydrotestosterone also contributes to male virilization at puberty.9 A partial explanation for the different physiologic effects of these two hormones is the fact that their interaction with the androgen receptor differs, dihydrotestosterone having a greater binding affinity to the receptor than testosterone. The testosterone–receptor complex is thus less stable and has a shorter half-life than the dihydrotestosterone–receptor complex. In addition, it is thought that the dihydrotestosterone–receptor complex is transformed to the DNA-binding state and thus activates the targeted gene more efficiently, producing an amplification of the androgenic signal.12, 28
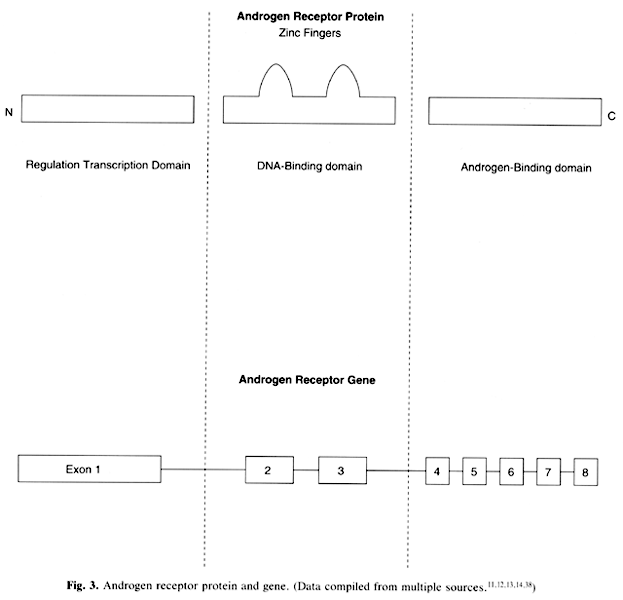
The androgen-receptor protein, which is 902–919 amino acids in length, consists of three domains (Fig. 6) (eight exons).25, 27, 29, 30 The amino-terminal domain regulates transcription and includes motifs (21 glutamine residues, 8 proline residues, 24 glycine residues) that vary in length in the normal population (i.e. are polymorphic).31, 32 Interestingly, an increase in the glutamine region, corresponding to an increase in CAG repeats, has been identified in Kennedy's disease (X-linked spinal and bulbar muscular atrophy).33 The central or DNA-binding domain consists of 66–68 amino acids and forms two zinc fingers. This DNA-binding domain interacts with hormone-responsive elements of the target genes. These hormone-responsive elements are usually located 5' to the gene and consist of 15 nucleotides forming a palindromic sequence.31 The C-terminal domain, composed of approximately 250 amino acids, is the site of androgen binding. As discussed above, the greater stability of the dihydrotestosterone–receptor complex is probably why dihydrotestosterone is a more potent androgen than testosterone.30
|
COMPLETE ANDROGEN INSENSITIVITY SYNDROME
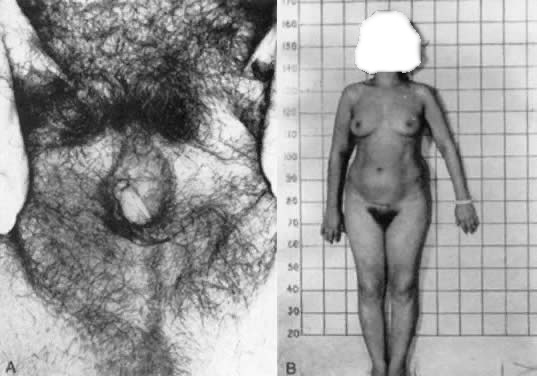
Persons with complete androgen insensitivity syndrome (CAIS) may appear quite normal and show excellent breast development, and most are similar in appearance to unaffected females in the general population. Their breasts contain normal ductal and glandular tissue, but their areolae are often pale and underdeveloped. Pubic and axillary hair are usually sparse, but scalp hair is normal. The vagina terminates blindly. Sometimes vaginal length is shorter than usual, presumably because müllerian ducts fail to contribute to formation of the vagina. Rarely, the vagina is only 1–2 cm long or represented merely by a dimple (Fig. 7).35
|

In CAIS, testes are usually normal in size and located in the abdomen, inguinal canal, or labia (i.e. anywhere along the path of embryonic testicular descent). If present in the inguinal canal, testes can produce inguinal hernias. It may therefore be worthwhile to determine cytogenetic status of prepubertal girls with inguinal hernias, although most will be 46,XX.
Height is slightly increased over that of normal women, but unremarkable compared with 46,XY males. In a Brazilian cohort (25 subjects036 adult height ranged from −1.35 SD to 0.59 SD using normal male standards, i.e., taller than 46,XX females, and shorter than normal 46,XY males. Presumably the increased height reflects the influence of the Y chromosome. Consistent with this is the impression expressed by many clinicians that the hands and feet of these women are relatively large compared with those of normal women.
The frequency of gonadal neoplasia is increased, but probably less so than once believed. In 1953, Morris and Mahesh37 stated that 22% of affected patients had neoplasia. The actual risk is probably no greater than 5%.38, 39 Most investigators now agree that the risk of neoplasia is low before 25–30 years of age. Benign tubular adenomas (Pick's adenomas) are especially common in postpubertal patients, probably as a result of increased secretion of luteinizing hormone. The pathogenesis of CAIS involves end-organ insensitivity to androgens.
Neither a uterus nor fallopian tubes are ordinarily present. Occasionally, fibromuscular remnants, rudimentary fallopian tubes, or rarely even a uterus are detected.40, 41, 42 The absence of müllerian derivatives is expected because antimüllerian hormone, which is secreted by the fetal Sertoli cells, is not an androgen; therefore, müllerian regression is expected to occur in males with androgen sensitivity, just as in normal males. The only other condition in which a uterus is absent in a phenotypic female is müllerian aplasia, which is readily distinguishable on the basis of pubic hair and a 46,XX complement.
Hormone levels
Serum testosterone and dihydrotestosterone levels are normal or elevated in AIS. Luteinizing hormone and estrogen are elevated, suggesting abnormal gonadal–hypothalamic feedback. Consistent with this, Leydig cells are hyperplastic.10 Androgen binding is absent, decreased, or qualitatively abnormal. In a study of androgen binding of genital skin fibroblasts from 42 patients with CAIS, 24 (57%) showed absent androgen binding, 15 (36%) decreased or qualitatively abnormal androgen binding, and three (7%) ostensibly normal androgen binding.9
Diagnosis
Patients with AIS have normal female external genitalia, feminize at puberty (develop breasts), and show primary amenorrhea. History may elicit prior inguinal hernias. Physical examination may reveal a shortened and blindly ending vagina, as well as an absent uterus and cervix (Fig. 8).43 An absent uterus and ovaries is found on ultrasound examination, and chromosome analyses reveal a 46,XY complement. Receptor studies or DNA studies are not routinely available except through laboratories involved with AIS research, and thus are not considered obligatory.
|
Prenatal diagnosis for AIS can be achieved by the use of DNA obtained from chorionic villi sampling or amniocentesis. Direct mutation analysis or restriction fragment length polymorphism (RFLP) analysis can be used if the molecular defect is known.44
Management
Treatment is straightforward. Affected persons are raised as female and act female. As previously mentioned, affected persons are at increased risk for gonadal neoplasia, and an orchiectomy is eventually needed. It is acceptable to leave the testes in situ until after pubertal feminization, but most surgeons would perform orchiectomies if herniorrhaphies prove necessary before puberty. There may also be psychologic benefit in prepubertal orchiectomies. Inguinal or intra-abdominal testes can sometimes be removed laparoscopically.45
After orchiectomy, estrogen replacement is necessary. Because these patients do not usually have a uterus, continuous therapy with conjugated estrogens or other estrogen forms may be prescribed. As mentioned above, the clinician must tailor the estrogen dosage to the individual patient, balancing its benefits against its side-effects. Vaginoplasty is rarely necessary, but occasionally dilators may be required to increase vaginal length.
PARTIAL ANDROGEN INSENSITIVITY SYNDROME
Partial AIS (PAIS) is the result of a mutation of the same androgen-receptor gene as is involved in complete AIS.46, 47 Persons with partial AIS (incomplete testicular feminization) feminize (i.e. exhibit breast development) despite having external genitalia characterized by phallic enlargement and partial labioscrotal fusion. Both PAIS and CAIS share the following features:
Bilateral testes with similar histologic findings
No müllerian derivatives
Pubertal breast development
Lack of pubertal virilization
Normal (male) plasma testosterone
The pathogenesis of PAIS logically would appear to involve a decreased number of receptors or qualitative defects in the androgen receptors.44, 48, 49, 50, 51, 52 Complete absence of receptors has been observed, but this is more likely to be associated with CAIS. Surprisingly, poor correlation exists between receptor levels (or androgen-binding affinity) and the degree of masculinization, nor are precise correlations evident between a specific mutation and the phenotype. Irrespective, the clinical significance of PAIS is that this disorder must be excluded before the decision is made to rear a child as a male. Presence of androgen receptors and demonstration of response to exogenous androgen is therefore necessary to exclude the diagnosis of partial androgen insensitivity.
Diagnosis
Infants with PAIS usually present with ambiguous genitalia. Adrenal 21-OH deficiency can be excluded readily by chromosome studies and 17α-OH progesterone levels.53 Ultrasound can aid in the evaluation of internal genitalia. Serum hormone levels are assessed after hCG is administered; the presence of normal testosterone excludes the presence of a defect in testosterone biosynthesis. An elevated T:DHT ratio indicates 5α-reductase deficiency. If a normal T:DHT ratio is present in an XY person with genital ambiguity, the clinician should consider the diagnosis of PAIS. Prenatal diagnosis for CAIS and PAIS is now possible with molecular analyses of the androgen-receptor gene from trophoblastic or amnionic DNA, if the molecular defect is known.
Management
Treatment is difficult. If external genitalia are characterized by other than simple hypospadias, a female sex of rearing is preferable. These patients require orchiectomy and estrogen replacement (as with CAIS patients). If no androgen receptors are present, one must assess the degree of androgen response in infants before considering male sex of rearing. Patients raised as males require androgen replacement, which may or may not be efficacious.54 Patients raised as males often require multiple urogenital reconstructive surgeries, sometimes still with poor results.54 Furthermore, little information is available on the long-term success of the corrective surgery and masculinization, sexual performance, and fertility of these patients.55 Overall, it is preferable for these patients to be raised as females.
MOLECULAR STUDIES IN CAIS AND PAIS
Perturbation of the androgen receptor gene located on Xq11 is responsible for the phenotypes described (CAIS, PAIS). As an X-linked recessive condition that is genetically lethal, one third of all cases would be predicted to be new (sporatic) mutations. Indeed, eight of 30 (30%) patients studied by Hiort and coworkers56 were the result of a new mutation. A few heterozygous women show decreased pubic hair and delayed puberty.
The gene is approximately 90,000 base pairs (bp) long but codes for fewer than 2000 amino acids. There are eight exons (Fig. 9). Exon 1 confers regulatory function. The DNA-binding domain of amino acids 552–616 is encoded by exon 2 and part of exon 3. The latter part of exon 3 and part of exon 4 encode bipartite nuclear localization (617–636 amino acids). The C-terminal region of 250 amino acids extends over exons 4–8 and constitutes the androgen-binding domain.57 The gene contains two regions of homopolymeric amino acid "repeats" of varying size: one polyglutamine (9–36 amino acids) and the other polyglycine (10–31 acids). Many different mutations have been reported,57 tabulated in a registry maintained at McGill University (email:mc33@musica.mcgill.ca or Website: www.mcgill.ca).
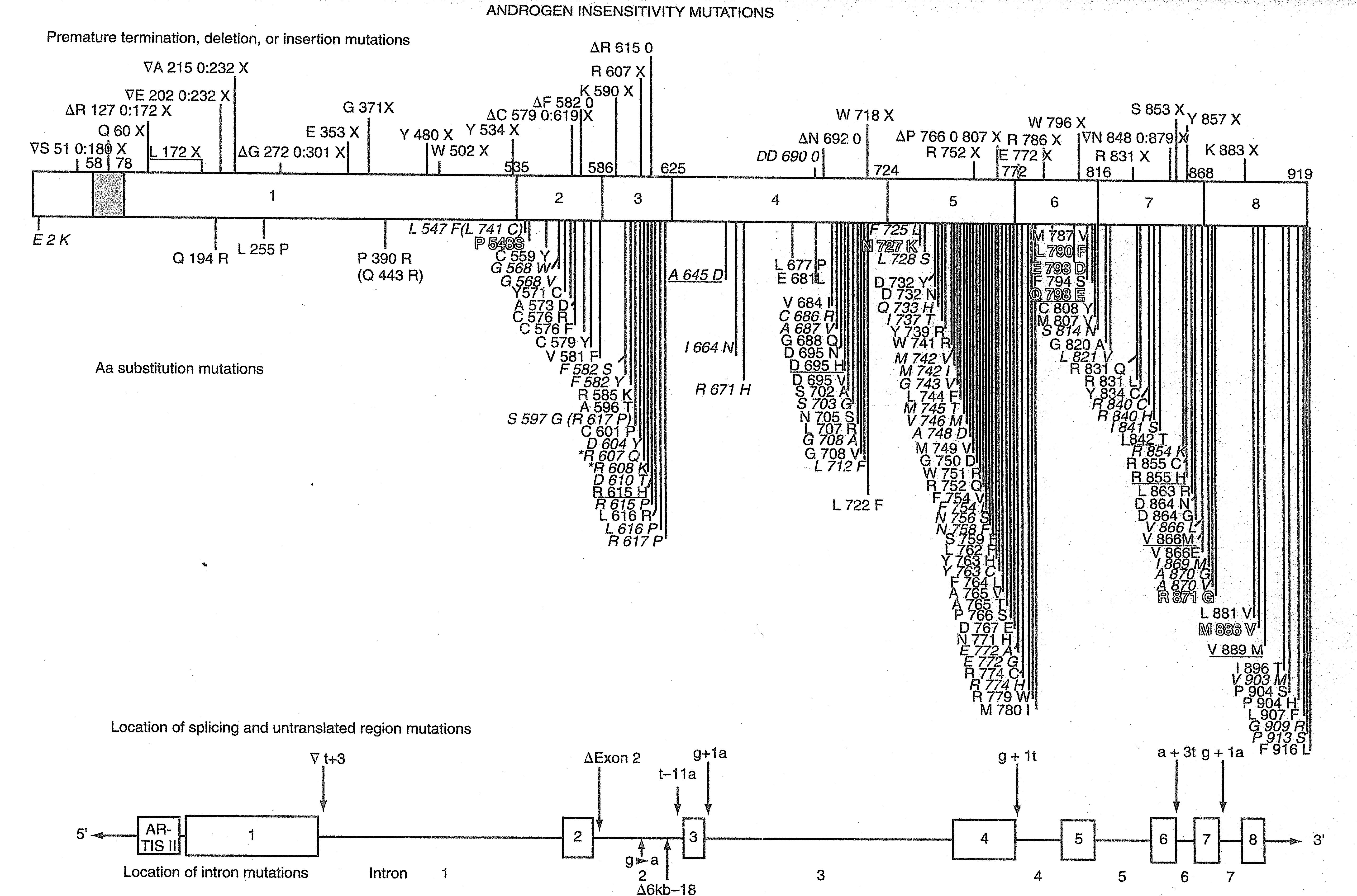 Fig. 9. Structure of the androgen receptor (AR) indicating the location of all exons and intron androgen insensitivity syndrome mutations as of 1999. Mutations in normal type indicate complete androgen insensitivity (CAI). Mutations in italic indicate partial androgen insensitivity (PAI). Mutations in outline denote mild androgen insensitivity (MAI). Mutations that are underlined cause both CAI and PAI. Mutations in outline and underlined cause PAI and MAI. Mutations in italic and underlined occur in both normal individuals and those with PAI. (Data from Gottleib B, Pinsky L, Beitel LK et al.58 Androgen insensitivity. Am J Med Gen [Semin Med Genet] 89:210-217.)
Fig. 9. Structure of the androgen receptor (AR) indicating the location of all exons and intron androgen insensitivity syndrome mutations as of 1999. Mutations in normal type indicate complete androgen insensitivity (CAI). Mutations in italic indicate partial androgen insensitivity (PAI). Mutations in outline denote mild androgen insensitivity (MAI). Mutations that are underlined cause both CAI and PAI. Mutations in outline and underlined cause PAI and MAI. Mutations in italic and underlined occur in both normal individuals and those with PAI. (Data from Gottleib B, Pinsky L, Beitel LK et al.58 Androgen insensitivity. Am J Med Gen [Semin Med Genet] 89:210-217.)
Mutations can be found in most cases of CAIS, and in about 75% of PAIS (15). Exon 1 encompasses half of the AR gene, but has the fewest recognized mutations (10%). Most mutations have been found in exons 2 and 3 (the DNA-binding domain). The phenotype may be either CAI or PAI. The most common sites of mutation lie in exons 5– to 8, the androgen-binging domain. Large deletions and mutations resulting in premature termination (stop codon) predictably cause CAIS, PAIS or MAIS in seemingly random fashion. Presumably, some point mutations are compatible with production of limited quantities of functional androgen receptors, although the receptor may be unstable or characterized by poor binding.59 That a given mutation may be associated with either PAIS or MAIS60 could suggest modifying genes.
REFERENCES
Simpson JL: Disorders of the gonads, genital tract, and genitalia. In Rimoin DL, Connor JM, Pyeritz RE (eds.): Principles and Practice of Medical Genetics, 6th edn. New York, Churchill-Livingstone, 2011; in press |
|
Simpson JL, Elias S: Genetics in Obstetrics and Gynecology. 3rd ed., Philadelphia, W.B, Saunders, 2003 |
|
Patsavoudi E, Magre S, Castinior M et al: Dissociation between testicular morphogenesis and functional differentiation of Leydig cells. J Endocrinol 105: 235, 1985 |
|
Magre S, Jost A: Dissociation between testicular morphogenesis and endocrine cytodifferentiation of Sertoli cells. Proc Natl Acad Sci USA 81: 7831, 1984 |
|
Moore KL, Persaud TVN: The Developing Human: Clinically Oriented Embryology, p 291. Philadelphia, WB Saunders, 1993 |
|
Imperato-McGinley I: 5α-Metabolism in finasteride-treated subjects and male pseudohermaphrodites with inherited 5α-reductase deficiency. Eur Urol 20 (suppl 2): 78, 1991 |
|
Imperato-McGinley I: 5α-Reductase deficiency: Human and animal models. Eur Urol 25 (suppl 1): 20, 1994 |
|
Wigley WC, Prihoha JS, Mowszowicz I et al: Natural mutagenesis: Study of the human steroid 5α-reductase 2 isozyme. Biochemistry 33: 1265, 1994 |
|
Wilson JD: Syndromes of androgen resistance. Biol Reprod 46: 168, 1992 |
|
Simpson JL, Elias S: Genetics in Obstetrics and Gynecology, 3rd edn. pp 148–156. Philadelphia, WB Saunders, 2003 |
|
Opitz JM, Simpson JL, Sarto GE et al: Pseudovaginal perineoscrotal hypospadias. Clin Genet 3: 1, 1972 |
|
Griffin JE, McPhaul MJ, Russell DW, Wilson JD: The androgen resistance syndromes: 5α-Reductase 2 deficiency, testicular feminization, and related disorders. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds): The Metabolic and Molecular Bases of Inherited Disease, 7th ed, pp 2967–2991. New York, McGraw-Hill, 1994 |
|
Simpson JL, New M, Peterson RE, German J: Pseudovaginal perineoscrotal hypospadias (PPSH) in sibs. Birth Defects 7 (6): 140, 1971 |
|
Imperto-McGinley J, Gurrero L, Gautier T, Peterson RE: Steroid 5α-reductase deficiency: An inherited form of male pseudohermaphroditism. Science 186: 1213, 1974 |
|
Akgun S, Ertel NH, Imperato-McGinley J et al: Familial male pseudohermaphroditism due to 5α-reductase. Am J Med 81: 267, 1974 |
|
McKusick VA: Mendelian Inheritance in Man, 10th ed, p 1160. Baltimore, The Johns Hopkins University Press, 1992 |
|
Wilson JD, Griffin JE, Russell DW: Steroid 5α-reductase 2 deficiency. Endocr Rev 14: 577, 1993 |
|
Thigpen AE, Davis DL, Milatovich A et al: Molecular genetics of steroid 5α-reductase 2 deficiency. J Clin Invest 90: 799, 1992 |
|
Labrie F, Sugimoto Y, Luu-The V et al: Structure of human type II 5α-reductase gene. Endocrinology 131: 1571, 1992 |
|
Thigpen AE, Davis DL, Milatovich A et al: Molecular genetics of steroid 5 alpha-reductase 2 deficiency. J Clin Invest. 1992 Sep;90(3):799-809. |
|
Andersson S, Berman DM, Jenkins EP et al: Deletion of steroid 5 alpha-reductase 2 gene in male pseudohermaphroditism. Nature. 1991 Nov 14;354(6349):159-61. |
|
Simpson JL, Rebar RW: Normal and abnormal sexual differentiation and development. In Becker KL, Bilezikian JP, Bremner WJ et al (eds): Principles and Practice of Endocrinology and Metabolism, 2nd ed, pp 788–822. Philadelphia, JB Lippincott, 1995 |
|
Imperato-McGinley J, Gautier T, Pichardo M, Shackleton C: The diagnosis of 5α-reductase deficiency in infancy. J Clin Endocrinol Metab 63: 1313, 1986 |
|
Lumbroso S, Lobaccaro JM, Belon C et al: A new mutation within the deoxyribonucleic acid-binding domain of the androgen receptor gene in a family with complete androgen insensitivity syndrome. Fertil Steril 60: 814, 1993 |
|
Odame I, Donaldson MDC, Wallace AM et al: Early diagnosis and management of 5α-reductase deficiency. Arch Dis Child 67: 720, 1992 |
|
Kupfer SA, Quigley CA, French FS: Male pseudohermaphroditism. Semin Perinatol 16: 319, 1992 |
|
Pinsky L, Kaufman M, Levitsky LL: Partial androgen resistance due to a distinctive qualitative defect of the androgen receptor. Am J Med Genet 27: 459, 1987 |
|
Deslypere J-P, Young M, Wilson JD, McPhaul MJ: Testosterone and 5α-dihydrotestosterone interact differently with the androgen receptor to enhance transcription of the MMTV-CAT reporter gene. Mol Cell Endocrinol 88: 15, 1992 |
|
Batch JA, Williams DM, Davies HR et al: Role of the androgen receptor in male sexual differentiation. Horm Res 38: 226, 1992 |
|
Janne OA, Palvimo JJ, Kallio P et al: Androgen receptor and mechanism of androgen action. Ann Med 25: 83, 1993 |
|
Chong C, Kokontis J, Liao S: Structural analyses of cDNA and amino acid sequences of human and rat androgen receptor. Proc Natl Acad Sci USA 85: 7211, 1988 |
|
La Spada AR, Wilson EM, Lubahn DB et al: Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352: 77, 1991 |
|
Sultan C, Lobaccaro JM, Belon C et al: Molecular biology of disorders of sex differentiation. Horm Res 38: 105, 1992 |
|
Simpson JL: Disorders of Sexual Differentiation. New York, Academic Press, 1976 |
|
Arnhold IJ, Melo K, Costa EM et al: 46,XY disorders of sex development (46,XY DSD) due to androgen receptor defects:androgen insensitivity syndrome. Adv Exp Med Biol. 2011;707:59-61. |
|
Morris JM, Mahesh VB: Further observations on the syndrome “testicular feminization.” Am J Obstet Gynecol 87: 731, 1953 |
|
Simpson JL, Photopulos G: The relationship of neoplasia to disorders of abnormal sexual differentiation. Birth Defects 12: 15, 1976 |
|
Verp MS, Simpson JL: Abnormal sexual differentiation and neoplasia. Cancer Genet Cytogenet 25: 191, 1987 |
|
Ulloa-Aguirre A, Mendez JP, Angeles A et al: The presence of müllerian remnants in the complete androgen insensitivity syndromes: A steroid hormone-mediated defect? Fertil Steril 45: 302, 1986 |
|
Heller DS, Ranzini A, Futterweit et al: Müllerian remnants in complete androgen insensitivity syndrome. Int J Fertil 37: 283, 1992 |
|
Swanson ML, Coronel EH: Complete androgen insensitivity with persistent müllerian structures: A case report. J Reprod Med 38: 565, 1993 |
|
Park IF, Jones HW: Familial male hermaphroditism with ambiguous genitalia. Am J Obstet Gynecol 108: 1197, 1970 |
|
McPhaul MJ, Marcelli M, Aoppi S et al: Genetic basis of endocrine disease 4: The spectrum of mutations in the androgen receptor gene that causes androgen resistance. J Clin Endocrinol Metab 76: 17, 1993 |
|
Lee RM, Peterson CM, Kreger DO et al: Digital blunt dissection technique to assist laparoscopic gonadectomy in inguinally located/adhered gonads. J Laparoendosc Surg 3: 229, 1993 |
|
DeBells A, Quigley CA, Marschke KB et al: Characterization of mutant androgen receptors causing partial androgen insensitivity syndrome. J Clin Endocrinol 78: 513, 1994 |
|
Lumbroso S, Lobaccaro JM, Belon C et al: Molecular prenatal exclusion of familial partial androgen insensitivity (Reifenstein syndrome). Eur J Endocrinol 130: 327, 1994 |
|
Zoppi S, Wilson CM, Harbison MD et al: Complete testicular feminization caused an amino-terminal truncation of the androgen receptor with downstream initiation. J Clin Invest 91: 105, 1993 |
|
Wilson JD, Harrod MJ, Goldstein JL et al: Familial incomplete male pseudohermaphroditism, type I. N Engl J Med 290: 1097, 1974 |
|
Wilson JD, Carlson BR, Weaver DD et al: Endocrine and genetic characterization of cousins with male pseudohermaphroditism: Evidence that the Lubs phenotype can result from a mutation that alters the structure of the androgen receptor. Clin Genet 26: 363, 1984 |
|
Sultan C, Lumbroso S, Poujol N et al: A single-base substitution in exon 6 of the androgen receptor gene causing complete androgen insensitivity: The mutated receptor fails to transactivate but binds to DNA in vitro. Human Mol Genet 2: 1809, 1993. |
|
Sultan C: Androgen receptors and partial androgen insensitivity in male pseudohermaphroditism. Ann Genet 29: 5, 1986 |
|
Hughes IA, Williams DM, Batch JA et al: Male pseudohermaphroditism: Clinical management, diagnosis and treatment. Horm Res 38 (suppl 2): 77, 1992 |
|
Williams DM, Patterson MN, Hughes IA: Androgen insensitivity syndrome. Arch Dis Child 68: 343, 1993 |
|
Shah R, Wooley MM, Costin G: Testicular feminization: The androgen insensitivity syndrome. J Pediatr Surg 27: 757, 1992 |
|
Hiort O, Sinnecker GH, Holterhus PM et al: Inherited and de novo androgen receptor gene mutations: investigation of J Pediatr. 1998 Jun;132(6):939-43. |
|
Gottlieb B, Trifiro M, Lumbroso R et al: The androgen receptor gene mutations database. Nucleic Acids Res. 1996 Jan 1;24(1):151-4. |
|
McPhaul MJ, Marcelli M, Zoppi S et al: Genetic basis of endocrine disease. 4. The spectrum of mutations in the androgenreceptor gene that causes androgen resistance. J Clin Endocrinol Metab. 1993 Jan;76(1):17-23. |
|
Gubbay J, Collignon J, Koopman P et al: A gene mapping to the sex-determining region of the mouse Y chromosome is amember of a novel family of embryonically expressed genes. Nature. 1990 Jul 19;346(6281):245-50. |