Normal human pregnancy is accompanied by such remarkable physiologic changes that drug disposition and effect may be entirely different from those in nonpregnant patients. These differences are important not only for maternal therapy but also for understanding the effects of fetal drug exposure.
Role of Gender
The effects of the pregnant state on the disposition and action of drugs are superimposed on the changes associated with the female sex. Gender differences on drug disposition in experimental animals has been known for more than 60 years, but it was not until 1993 that the FDA encouraged the inclusion of women in clinical trials.1 There are striking differences in body processes between men and women. Physiologic differences between the sexes may explain variations in the absorption of drugs. Compared to men, women have slower gastric emptying time and prolonged colonic transit time. These differences may be heightened during pregnancy. There are also differences in drug biotransformation.
A multienzyme system is responsible for the degradation of hydrophobic molecules. In a sequential manner, hydrophobic molecules are biotransformed by phase I enzymes and then conjugated by phase II enzymes. The cytochrome P450 superfamily (or CYP) is the major phase I group of isoenzymes. These enzymes are expressed mostly in the liver but also to a lesser extent in other tissues (e.g., intestine). The expression pattern of different CYP isoforms differs in the sexes. For example, the cytochrome P450 CYP3A4 is more active in women than in men.2 Theophylline and acetaminophen, which are metabolized by CYP3A4, are eliminated faster by women. Other drugs, such as diazepam, caffeine, and some anticonvulsants, metabolized by CYP2C19 or CYP1A2 appear to be metabolized faster in men than in women.3 Gender differences in drug biodisposition has been linked to variations in sex hormones.
There are also sex differences in the sensitivity to drugs .4 Opioids such as pentazocine show a greater drug response in women, whereas ibuprofen produces a better response in men.5,6 In addition, there are gender differences in the incidence of adverse drug reactions. For example, drug-induced torsades des pointes and the cough induced by angiotensin-converting enzyme inhibitors occur more commonly in women.7,8
Mother-Fetus: A Two-Compartment System
A fundamental aspect of fetal pharmacology is that of fetal dose. The amount and rate of transfer of drugs to the fetus determine the presence or absence of pharmacologic or toxic effects.
With the rare exception of drugs injected directly into the fetal compartment, the path a drug must take from its administration to the mother is across the maternal organism to its site of action in the fetus.
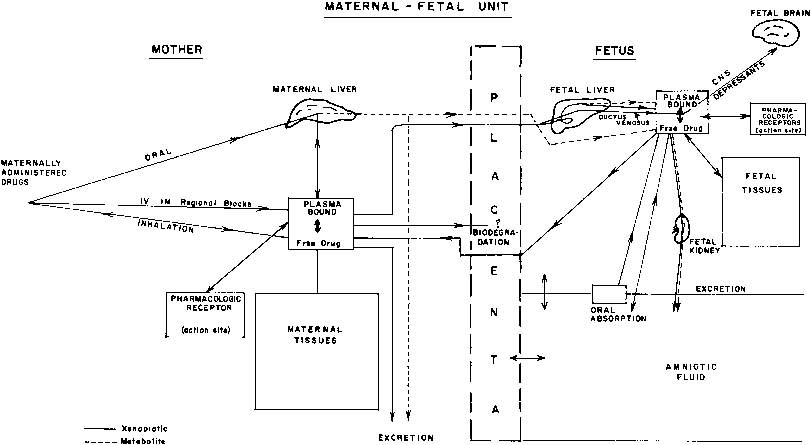
This multicompartment system is especially complicated because it does not represent a constant relationship but one that is continuously changing throughout pregnancy (Fig. 1).
|
THE MOTHER
Absorption
The gastrointestinal absorption of drugs has not been systematically studied during pregnancy. Reports in the literature suggest that a generalized malabsorption state may be induced or exacerbated during pregnancy.9,10 Both gastric emptying time and gastrointestinal transit times are prolonged, probably due to the high levels of progesterone. There is also indirect evidence that absorption of certain compounds such as digitoxin, salicylamide, and phenytoin may be delayed in pregnant patients.11–13
It is possible that the confluence of different physiologic alterations during pregnancy may potentiate the effect of individual changes. For example, the increased residence time due to the decrease in intestinal motility could lead to a decreased bioavailability because of an increase in gut metabolism. The latter may occur if the activity of gut CYP3A4 mirrors the increase in the activity of hepatic CYP3A4 that has been documented during pregnancy.
The complexities of performing bioavailability studies in pregnancy can be simplified with the use of stable isotopes. The intravenous injection of a stable isotope-labeled drug coupled with the concomitant oral administration of unlabeled drug permits the simultaneous determination of both drug profiles and therefore minimizes the variability associated with two separate studies.14 The normal circulatory adjustments that occur during pregnancy are therefore likely to influence the extent of drug absorption. These adjustments can be expected to be more influential toward the end of pregnancy. Indeed, the absorption rate of meperidine after intramuscular administration has been found to be slower in women during labor than in nonpregnant controls.15,16 On the other hand, circulatory changes are on occasion pharmacologically advantageous. Studies have shown that pregnant women at term require 40% to 50% less drugs for peridural block because at term the capacity of the peridural space is reduced by the physiologic engorgement of the internal vertebral plexus.
Distribution
The remarkable changes in the volume of water and composition of body compartments, coupled with the hemodynamic adjustments that occur during pregnancy, set a background for drug distribution quite different from that present in nonpregnant persons. Dilutional hypoalbuminemia, especially in the last trimester, is mainly responsible for a decrease in drug-binding capacity and a consequent increase in body distribution.17 As a corollary, the acceptable therapeutic range of total plasma concentration of drugs exhibiting decreased protein binding (i.e., phenytoin) is lower than in the nonpregnant state. The great interindividual variability in the distribution of drugs such as meperidine given during labor may be attributed to variations in the hemodynamic makeup of different women.18
Metabolism
The rate and extent of drug biotransformation depend on hepatic blood flow, activities of drug-metabolizing enzymes systems, and hormonal influence. Hepatic blood flow measured in absolute terms does not appear to be altered during pregnancy. Proportionally, however, the percentage of cardiac blood flow reaching the liver is decreased.19
Knowledge of drug metabolism during pregnancy and in women in labor is scanty and far from satisfactory. The sex hormone changes characteristic of pregnancy and placental hormones influence hepatic drug-metabolizing enzymes activities during gestation. Clinical studies using metabolic probes have shown that CYP1A2, xanthine oxidase, and N-acetyltransferase activities are decreased, but CYP3A4 is increased during pregnancy.20 N-acetyltransferase activity, measured using caffeine as a probe, has been found to be reduced by about 30% in late pregnancy compared to the enzyme activity during the puerperium.20 A differential effect on the activity of CYP2D6 was reported in pregnant subjects. In both homozygous and heterozygous fast metabolizers, CYP2D6 activity increased, whereas the activity of this polygenic enzyme decreased in homozygous poor metabolizers.21 Demethylation of meperidine occurs less readily in women in labor.22 Conversely, other metabolic pathways may be inducible. The reported increase in plasma clearance of phenytoin and phenobarbital during pregnancy has been attributed to an increase in metabolic rate.23 The increased microsomal oxidation of carbamazepine and phenytoin during pregnancy is not associated with a proportional increase in the subsequent hydroxylation and glucuronidation of the initial metabolite.24
Glucuronide conjugation of endogenous substances and xenobiotics (compounds foreign to the body) is inhibited during late pregnancy. The inhibition of glucuronosyltransferase activity may be related to the high tissue levels of progesterone and pregnanediol. Glucuronidation of salicylamide has been found to be depressed in parturients. There is indirect evidence, however, of increased glucuronidation of other substrates, such as zidovudine.25 There is no information on the sulfation of drugs during pregnancy.
Excretion
Although almost all maternal physiologic systems undergo adjustments during pregnancy, perhaps the greatest upheaval is seen in the renal system. The profound changes in function are likely to affect the renal excretion of drugs. The increase in glomerular filtration observed during pregnancy is counterbalanced to a significant extent by modifications in tubular reabsorptive capacity. Of greatest importance from the pharmacokinetic standpoint are diurnal variations in function.
These factors are likely to markedly influence the “dosing” of drugs transferred to the fetus. Although for the purpose of this discussion, absorption, distribution, and elimination of drugs in the maternal organism have been considered separately, the interplay among these factors determines the time course of maternal plasma levels. Little is known about the extent to which complications of pregnancy modify drug disposition in the mother.
ROLE OF THE PLACENTA
For years the placenta has been regarded as the somewhat passive and inert barrier to the transfer of drugs between the fetal and maternal compartments. However, recent work has uncovered a bewildering number of complex functions affecting both maternal and fetal physiology. Two aspects of placental pharmacology assume equal importance: the transfer and disposition of xenobiotics reaching the organ from the maternal and fetal side, and the biodegradation properties affecting xenobiotics or being affected by them. The three major factors affecting drug transfer across the placenta are physiochemical characteristics of the compound, pharmacologic properties of the placental tissue, and maternal and fetal placental blood flow.
Physiochemical characteristics include molecular weight, lipid solubility, degree of ionization, molecular configuration, and tissue binding protein properties. Generally, lipophilic substances and compounds with low molecular weight tend to diffuse rapidly into the fetal circulation. Poorly ionized drugs at physiologic pH, such as thiopental, reach the fetal circulation quite rapidly. Certain compounds, such as the sympathomimetic agents, salbutamol, ritodrine, and norepinephrine, appear to have a low transfer rate despite their small molecular weights (170 to 290). Still, sufficient amounts of both salbutamol and ritodrine are transferred to produce fetal tachycardia.26,27
The limited transfer rate of norepinephrine may be attributed in part to biodegradation by placental tissue. The purported placental impermeability to polar compounds is relative rather than absolute. The high lipid solubility of certain compounds, such as salicylates, allows for rapid transfer despite being almost 100% ionized at physiologic pH. Xenobiotics cross the placenta by different transfer mechanisms: simple diffusion, facilitated diffusion, active transport, and pinocytosis. The metabolic conversion by the placenta of one compound into another compound that in turn may be transferred cannot be discounted. Most drugs cross the placenta by simple diffusion at a rate that is directly related to the difference between the maternal and fetal blood concentrations. Recent studies have shown that the syncytiotrophoblast expresses membrane proteins that act as drug transporters.28 P-glycoprotein, an ATP-dependent drug efflux pump, is present in the brush border of the syncytiotrophoblast.29 Drug transport by P-glycoprotein is unidirectional from the fetal to the maternal side and thus protects the fetus from toxic compounds. A wide variety of drugs are substrates for this transporter (e.g., digoxin, verapamil, chemotherapeutic agents). Transporters in the opposite direction have not been sufficiently characterized.
The placenta undergoes continuous structural changes during its life span that are likely to significantly affect rates of drug diffusion. Studies in the pregnant rodent seem to indicate that drug transfer is lowest in midgestation and peaks at the beginning and end of pregnancy.30 There is little information on the effect of placental aging on drug transfer in normal human pregnancy, let alone those changes that occur during abnormal conditions, when drugs are most often prescribed. The relative maternal and fetal blood flow through the placenta is of paramount importance in determining the rate of drug transfer from mother to fetus and vice versa.
Adequate measurements of uterine blood flow are flawed by technical difficulties. Despite this, several studies have shown an increase in uterine flow per kilogram of uterine weight toward term. When data are analyzed in terms of uterine blood flow per kilogram of fetus, however, a decrease is demonstrated at term. The time course of uterine and fetal plasma concentrations usually follows the following pattern:
- Establishment of a maternal—fetal concentration gradient
- Equilibration phase, in which the highest fetal drug concentration will
depend on the placental factors discussed above
- Fetal drug elimination phase. During this period, the combined effects
of maternal drug biodegradation and elimination lower maternal drug concentrations, creating
a fetal—maternal gradient and reversing the
direction of drug transfer across the placenta.
Delivery can occur at any point during this sequential pattern, and its timing will determine the amount of drug present and the ability of the newborn to handle xenobiotics. Many factors can influence maternal and fetal hemodynamics, thereby disturbing maternal and fetal drug distribution. Those affecting maternal hemodynamics are briefly reviewed here. A decrease in uteroplacental blood flow may be secondary to vasoconstriction of myometrial arterioles or obstruction of uterine venous outflow. The amount of drug transfer to the fetus, especially after a single intravenous pulse injection, will vary depending on the type of blood flow obstruction and the temporal relationship between drug administration and the onset of uterine hypoperfusion. For drugs given before the onset of uterine blood flow obstruction, myometrial arteriole vasoconstriction will tend to protect the fetus, whereas venous obstruction, by allowing a longer period of placental residency time, will result in increased fetal drug extraction. Alterations in uterine blood flow of particular interest are those related to abnormal labor, excessive uterine activity (spontaneous or oxytocin-induced), vasoactive drugs, or vena cava compression and supine hypotension, as may occur at the time of removal of amniotic fluid.
Pathophysiologic conditions such as preeclampsia, hypertension, and diabetes, which may be associated with impaired uteroplacental blood flow, can be expected to decrease drug transfer across the placenta. On the other hand, these pathophysiologic conditions often are associated with profound fetal hemodynamic changes that favor drug distribution to the fetal brain.
The demonstration that the human placenta is capable of metabolizing xenobiotics spurred a burst of investigative activity. Placental CYP1A1 is inducible by maternal smoking. CYP4B1 and CYP19 may contribute to the metabolism of some drugs.31 The picture emerging from the available research, however, indicates that although the placenta is a major metabolic organ for the biotransformation of endogenous substances, especially steroidal hormones, its contribution to the overall degradation of drugs during pregnancy is quantitatively meager.32 The demonstration that foreign organic substances could undergo oxygenation in human placental tissues raises the possibility that xenobiotics and endogenous steroids might share common biotransformation reactions. The balance of present evidence, however, refutes this contention and supports the existence of separate P450 species of isoenzymes for the catalysis of xenobiotic and steroidal hydroxylation reactions. The discovery that the placental mono-oxygenase activities are inducible by maternal cigarette smoking and not by other inducers is of considerable interest in this regard.33 The placental tissue of smokers contains bioactivating enzymes that catalyze the formation of metabolites that covalently bind to DNA34 or produce mutations in Salmonella typhimurium.35 It remains a challenge for researchers in the next decade to determine whether the demonstrated bioactivating capacity of the placenta allows the formation of reactive metabolites of chemical carcinogens and mutagens that could damage the embryo or the fetus.
THE FETUS
The study of the pharmacologic and toxic effects of drugs on the human fetus has been limited by lack of accessibility and by societal and ethical constraints against human fetal research. The effects of drugs given during the first trimester are not considered in this review.
During its intrauterine existence, the fetus may be exposed to a multitude of chemicals, including drugs, herbal medicines, alcohol, caffeine, tobacco, and environmental pollutants. The available data have been gathered by different approaches: (1) in vitro studies of drug metabolism using subcellular fractions (e.g., liver, adrenal glands) or isolated hepatocytes from aborted fetuses; experiments with isolated cells yield more meaningful information than those performed with subcellular fraction of tissues because the enzyme system operates under more physiologic conditions; (2) studies of drug disposition or distribution in fetuses with lethal malformations (e.g., anencephaly) or stillbirths; and (3) postnatal pharmacokinetic studies of drugs transferred in utero before birth. Postnatal in vivo studies using plasma elimination curves are useful for analysis of drug distribution and effects of different dose-delivery intervals. They are, however, of limited value for studies of drug metabolism because metabolites may be present due to transfer across the placenta (see Fig. 1), and there is a marked shift in the balance between metabolic and excretory pathways of elimination immediately after delivery.
The late gestational period, of particular interest for this review, is also the time of fetal life that is largely beyond the reach of investigation. The study of preterm stillborn infants is of limited value because of the pathologic circumstances under which these studies are performed. Pregnant sheep and primate models have significantly contributed to the present understanding of fetal drug disposition.
Distribution
The fetal circulation is so unusual that it greatly modifies drug distribution. Xenobiotics enter the fetus mostly through the umbilical vein. Most (60% to 80%) of the umbilical vein flow perfuses the liver; the remainder is shunted to the inferior vena cava by way of the ductus venosus. Dilution of umbilical venous blood in the right atrium and shunting across the foramen ovale and ductus arteriosus into the systemic circulation significantly affect fetal drug distribution. Fetal hepatic blood flow is quite variable and can be markedly reduced by hypoxia. Under these circumstances, drugs bypass the liver and can reach high concentrations in the fetal brain and other tissues. The physiologic characteristics of the fetal circulation are such that even under normal circumstances, the brain of the fetus receives a larger share of the cardiac output than the brain at any other period in life. Redistribution of the cardiac output away from nonessential vascular beds during asphyxia may have a profound effect on drug distribution to the fetal brain and heart. The recent discovery of the lack of autoregulation of the cerebral circulation in preterm infants may render this organ even more vulnerable to large infusions of drugs under hypoxic conditions.
The composition of fetal blood itself may produce changes in drug distribution that are clinically significant. Fetal serum proteins usually bind drugs to a lesser degree than adult proteins.36,37 This results in an increase in the proportion of unbound free drug that is responsible for pharmacologic effect. This difference in binding affinity is not universal. Neonatal and adult sera bind digoxin to a similar extent, and fetal red cells take up more trichloroethylene than maternal erythrocytes. The reduced plasma protein binding of various drugs is due to a combination of factors: reduced total plasma protein, persistence of fetal albumin with lower binding affinity for drugs, lower concentration of immunoglobulins, and competitive binding of endogenous substances and compounds of maternal origin.
The kinetics of drug distribution to various tissues and body compartments is profoundly affected by the remarkable changes in fetal body composition that occur during pregnancy. Total body water content decreases from about 94% of total body weight at 16 weeks of gestation to about 76% at term. A progressive decrease in extracellular water is responsible for the change. Fat, which is virtually absent in fetuses weighing less than 1000 g, is accrued during the last trimester. Even at term, fat tissue is relatively scarce (15% of body weight) and contains more water than similar tissue from older individuals. The relative paucity of fat tissue limits the distribution in fetal tissues of lipid-soluble compounds such as barbiturates, general anesthetics, and diazepam. The changes in the composition of individual organs and their relative contribution to the overall body composition further complicate the understanding of drug distribution in the fetus. In relation to total body mass, the newborn has less skeletal muscle and greater brain and liver tissue than the adult. The composition of fetal brain tissue is also quite different from adults in that its myelin content is low and its water content is relatively large. These compositional changes, coupled with high cerebral blood flow with preferential perfusion of brain stem structures, are likely to lead to different distribution of flow-dependent lipophilic compounds in the fetal brain.
Amniotic Fluid Dynamics
Fetal drug clearance depends on the combined contribution of fetal renal excretion and hepatic metabolism. The interposition of the amniotic fluid compartment between mother and fetus adds an element of complexity to the distribution and disposition of drugs reaching the fetus.
Although the possibility of direct maternal amniotic fluid exchange cannot be excluded, if it occurs it is likely to be small. Most exchange occurs through the fetus. In this regard the amniotic fluid compartment should be considered as one of the fetal excretory pathways, but because of fetal swallowing, recirculation is likely to occur. Sequestration of drugs in the fetal gut must also be considered. Recent interest in fetal therapeutics has raised the possibility of amniotic fluid infusion of drugs. Although promising, studies are still preliminary and in the animal testing stage.38 The limited data available suggest that water-soluble compounds enter the fetal compartment slowly, probably by fetal swallowing. In contrast, lipid-soluble compounds introduced into the amniotic sac reach the fetus much more rapidly and in significant amounts. It has been speculated that lipid-soluble solutes enter the fetus through the skin or the fetal surface of the placenta, or both. Systematic studies are needed to clarify the various factors determining drug absorption by this route.
Metabolism of Xenobiotics
Research during the past 10 years has clearly demonstrated that the human fetus is endowed with a well-developed system of xenobiotic metabolizing enzymes.39 Enzymatic activities, however, are significantly reduced in comparison with adult values but appear to increase with advancing fetal and postnatal age. The most significant pathways of drug biotransformation are oxidative reactions. The enzymatic systems that are located in the microsomal fraction of hepatocytes also catalyze the biotransformation of fatty acids, bile acids, and steroid hormones. It is likely that endogenous substances have a much higher affinity for the terminal oxidase in the microsomal system than do xenobiotics. The different components of the microsomal oxidizing system that function as an electron transport chain are present in the human fetal liver at levels one-fifth to four-fifths those of the adult. Cytochrome P450, the last component in the chain, is already present in the human fetal liver by the latter part of the first trimester.40 Studies of postmortem liver samples have shown the total P450 content remains stable from the first trimester of pregnancy in the fetus until the first year of postnatal life.41
Recent studies comparing cytochrome P450-dependent enzyme activities of adult liver, placenta microsomes, and fetal liver suggest that the differences between these tissues are due to the existence of different tissue-specific isoenzymes.42 CYP2C proteins are absent from the fetal liver. Hydroxylation of tolbutamide and demethylation of diazepam depend on CYP2C activity. The P450 subfamily CYP3A includes three isoforms:CYP3A4, CYP3A5, and CYP3A7. CYP3A7 is mostly expressed in the fetal liver and is replaced at birth by CYP3A4.43 CYP1A2 and CYP2D6 are not expressed in the fetus. The N-demethylation of caffeine and theophylline is particularly deficient, probably because CYP1A2 is involved in their metabolism.44
Knowledge of the fetal ontogenic profiles of CYP proteins is an important step toward the estimation of risk associated with fetal drug exposure. Substrates that have been metabolized by phase I (e.g., CYP proteins) are further biotransformed by phase II conjugation drug-metabolizing enzymes. Among synthetic phase II reactions, sulfate conjugation and glycine conjugation are especially efficient and approach rates found in adults. It has been suggested that these well-developed reactions compensate for the deficiency in glucuronic acid conjugation by glucuronyl transferases (UGTs) in fetal life. The large amounts of sulfate conjugates of steroids in human fetal tissues support this hypothesis. UGTs consists of at least 18 different isoforms. UGT1A1 is responsible for the conjugation of bilirubin, steroids, and several drugs. UTG2B7 is the most important isoform responsible for the metabolism of morphine, opioid derivatives, lorazepam, and nonsteroidal anti-inflammatory agents.45 Although UGTs enhance renal excretion of hydrophilic intermediates, glucuronide metabolites may be potentially toxic. For example, glucuronide metabolites of morphine are active (morphine-6-glucuronide is 100 times more potent than morphine and morphine-3-glucuronide is neuroexcitatory). It has been shown that fetal baboons conjugate morphine at both 3-OH and 6-OH positions.46 The formation of zidovudine-glucuronide during fetal infusions in baboons also gives credence to a significant contribution of fetal glucuronidation to the nonplacental clearance of both drugs. Biochemical assays of UGTs in fetal hepatic tissues have shown limited activity (less than 20% of adult activity).47 Studies are needed to compare in vitro UGT activities with rates of metabolite formation in fetal animals. Further, because UGTs are inducible, patterns of their fetal expression may vary during pregnancy.
Molecular and genetic tools are now available to probe for specific CYPs and UGTs and to determine factors responsible for their expression in the fetus. A major goal for future research is the quantification of the nonplacental clearance attributable to fetal metabolism. The possibility of increased concentration of drugs or their metabolites that may even exceed maternal concentrations need to be considered, particularly when placental transfer back to the mother is compromised. Fetal consequences may ensue if toxic active metabolites are increased.48 Alternatively, fetal drug concentrations may be decreased by fetal metabolism. This may be of significant concern if the fetus is the target of therapy, as in the prevention of HIV perinatal transmission (e.g., zidovudine therapy).
The low levels of blood esterase activities in preterm infants partially explain the cardiac and respiratory depression observed at birth when local anesthetics containing ester bonds are used during labor and delivery.
During recent years, extrahepatic sites for xenobiotic metabolism have attracted considerable interest. Present knowledge is preliminary and allows only tentative conclusions. Considerable activity toward some substrates has been found in the adrenal, pancreas, and gonad tissue of the human fetus. Activation of some compounds such as diethylstilbestrol and aromatic hydrocarbons has been related to these findings.49 Inducibility of fetal biotransformation reactions remains a fascinating but modestly understood subject. Xenobiotics, including alcohol administered in large doses and for prolonged periods during the first half of pregnancy, may induce drug-metabolizing enzymes in human fetal liver.50 Pharmacokinetic studies of diazepam and phenytoin in newborns who were exposed in utero support the concept of transplacental induction by anticonvulsants.51,52
Excretion
Although the placenta is a major excretory organ, secondary excretory routes may affect the residency time of drugs in the fetal compartment. Xenobiotics or their metabolites may be excreted by the fetal kidneys. Fetal urination contributes significantly to amniotic fluid formation. Hypotonic urine has been obtained from the fetal bladder as early as 12 weeks of gestation. It has been estimated that close to term, the fetus produces 600 to 800 mL/day of very hypotonic urine (80 to 140 mOsm/kg water). Water-soluble lipid-insoluble drugs such as antibiotics may be excreted in significant amounts in the fetal urine during the last trimester. The high rate of urine production and high fetal urine concentration of these drugs account for peak amniotic fluid concentrations that are higher than maternal or fetal plasma. A trapping effect of these compounds in the amniotic fluid after excretion by the fetal kidney occurs because equilibration with either fetal or maternal compartments is likely to be a very slow process. Indeed, there are many examples of compounds that accumulate in the amniotic fluid by this mechanism. Ampicillin, penicillin, kanamycin, gentamycin, sulfonamides, methicillin, and some of the cephalosporins behave in this manner.
Evidence supporting this mechanism comes from acute experiments in late pregnancy. When ampicillin was given by pulse intravenous injection to the mother, peak levels in fetal serum were demonstrated 30 to 60 minutes after injection, and prolonged peak concentrations were found in the amniotic fluid 6 to 12 hours later. When the fetus was dead, however, only insignificant amounts of ampicillin could be demonstrated in the amniotic fluid.53 The fetal skin may play a role as a minor fetal excretory pathway, particularly during the second trimester. Experiments using nitrous oxide in primates provide support for this excretory mechanism.54 The recent demonstration of a sizable tidal flow of amniotic fluid through the fetal lungs in relation to fetal respiratory movements during the last trimester raises the intriguing possibility of drug exchange at this level.55 Finally, fetal swallowing may allow certain drugs to be recirculated. The early maturation of the intestinal enzyme glucuronidase may make it possible to hydrolyze conjugated compounds excreted by the fetal kidney and permit their intestinal reabsorption.
Pharmacologic and Toxic Effects in the Fetus
Drugs exert their effect at receptor sites. Information concerning the pattern of development of drug receptors during fetal life remains sketchy, fragmentary, and for the most part unknown. From studies of the functional development of individual organs in the fetus, it can be deduced that maturation of pharmacologic receptors is likely to proceed at different rates. With the notable exception of the antepartum administration of glucocorticoids for the prevention of hyaline membrane disease, pharmacologic manipulation of the fetus remains a hope for the future. Our present concerns deal with toxic and teratologic effects. There are three successive periods in fetal development for drug-related teratologic and toxic effects: (1) fertilization and implantation (days 0 to 17): at this stage a fetotoxic agent can cause death of the embryo; (2) organogenesis (days 18 to 55): this is the most sensitive period for development of malformations; and (3) fetal period (56 days to birth): in this stage, drugs can decrease cell size and number or affect the organization of the cerebral cortical layers.
Drugs may affect the fetus directly or indirectly by changes in maternal nutrient delivery to the fetus, changes in placental circulation, or alterations in maternal glucose homeostasis. Both acute and chronic drug exposures can cause adverse effects.
Acute toxicity occurs mostly during labor and delivery, when the fetus abandons its major drug excretory organ, the placenta. Rarely has fetal poisoning been the result of attempted suicide in the mother, ingestion of toxins unknown to the mother, or mistaken medications. The overwhelming concern, however, centers on the effect of drugs and environmental chemicals during pregnancy. Distinction between toxic and teratologic effects seems arbitrary and unwarranted. Traditionally, teratologic effects have been defined as congenital anatomic malformations secondary to deleterious agents administered during the first trimester of pregnancy. This definition is misleading and too confining. It is now widely acknowledged that drugs and other agents taken at any time during gestation can produce different types of developmental defects, including prenatal or postnatal growth retardation, distortion of cell architecture of the cerebral cortex, postnatal functional or behavioral disorders, and prenatal or postnatal neoplasms. The concept of developmental toxicity is preferred because it is more encompassing and does not presuppose the presence of anatomic defects at birth. In fact, drug effects may not be seen until many years after birth (e.g., adenocarcinoma of the vagina after intrauterine exposure to diethylstilbestrol).
The fear of fetal toxicity often results in undertreatment, which in turn may complicate the interpretation of an abnormal outcome. It is known that chronic exposure to tricyclic antidepressants may lead to fetal growth deficits. Likewise, women with symptoms of depression have an increased risk of delivering a low-birthweight infant.
A similar situation occurs with the treatment of chronic hypertension. Prematurity and low birthweight may result from the placental vascular changes associated with hypertension or may result from chronic exposure to some antihypertensive drugs.
A number of factors complicate the interpretation of the available data on the fetal consequences of drug exposure during pregnancy The determination of drug exposure by retrospective recall is known to be associated with a high error rate. Multiple drug exposures are often not recorded; drug—drug interactions are difficult to determine; there is an overreliance on the clinical significance and the predictive value of animal studies; and there is a paucity of long-term studies.