Cerebral injury from hypoxemia, whether it is prenatal, intrapartum, or neonatal, is a significant contributor of neurologic morbidity and mortality in neonates. Perinatal asphyxia occurs in approximately 6 per 1000 term live births.1 There is little agreement about what constitutes fetal asphyxia. No single parameter taken in isolation seems to predict the severity of the hypoxic ischemic injury or the long-term neurologic sequelae. Investigators have shown the inadequacy of the Apgar score as a correlate to significant brain insult.2 Fetal acidosis, meconium staining, late decelerations, and respiratory depression are nonspecific markers of neonatal distress. Signs attributed to asphyxia may be due to a neurologically impaired fetus. Fetal acidemia may be induced by parameters other than hypoxemia, and abnormal fetal heart rate patterns may be the result, rather than the cause, of neurologic impairment.
Five principal mechanisms of asphyxia have been described during the antepartum and intrapartum periods: (1) interruption of umbilical circulation, (2) altered placental gas exchange, (3) inadequate maternal perfusion of the placenta, (4) decreased maternal oxygenation, and (5) failure of the infant to transition from fetal to neonatal cardiopulmonary circulation.3 There are many etiologic factors in fetal asphyxia (Table 1). Placental abruption, cord prolapse, and uterine rupture result in an acute, hypoxic-ischemic insult. Partial or intermittent hypoxic-ischemic injury can occur in cases of placental insufficiency. The clinical features of hypoxic-ischemic encephalopathy, however, are nonspecific.
TABLE 1. Clinical Factors Associated With Hypoxic-Ischemic Injury
Maternal hypotension or hypertension
Maternal diabetes
Maternal anemia
Maternal cardiopulmonary disease
Preeclampsia
Intrauterine growth retardation
Umbilical cord compression
Placental abruption, previa, or insufficiency
Altered fetal heart rate or acid-base disturbance
Thick meconium
Infant cardiopulmonary disease
Neurologic signs and symptoms may be observed in the neonate with perinatal asphyxia. It often is difficult to determine the precise timing of insult.4 Whereas an intrapartum event may lead to neurologic signs hours to days later, infants who have sustained a hypoxic insult in utero may have few, if any, clinical abnormalities in the neonatal period. Term infants who suffer an acute asphysic insult resulting in severe long-term neurologic handicaps invariably show a recognizable clinical encephalopathy during the first few days of life.4 A detailed examination will aid in the assessment of severity of hypoxic-ischemic encephalopathy. The spectrum is divided into three classes that have prognostic value.5 Mild encephalopathy is characterized by jitteriness, uninhibited deep tendon reflexes, and hyperalertness. This is not associated with long-term sequelae. Hypotonia, seizures, and lethargy are hallmarks of moderate encephalopathy. The outcome of moderate encephalopathy is more variable. If symptoms persist for longer than 1 week, neurologic sequelae may exist in up to 40%.5 Severe encephalopathy is associated with coma, seizures, brain-stem dysfunction, and increased intracranial pressure. The majority of these infants are affected with long-term sequelae, such as microcephaly, mental retardation, spastic quadriplegia, and seizures.
Radiologic examination helps in the assessment of the extent of cerebral injury. Although ultrasound cannot identify specific patterns of injury, it is a portable tool allowing bedside assessment of germinal matrix and intraventricular hemorrhage. It also can detect changes in echogenicity seen with early periventricular leukomalacia in the premature infant. Computed tomographic (CT) scanning is the investigation of choice in the clinical evaluation of the hypoxic-ischemic brain injury of the term newborn.4 Optimal timing of the scan is debated. It often is obtained during the initial evaluation of the asphyxiated infant, but the maximal extent of tissue damage visualized on CT usually is 2 to 5 days after the insult. CT scanning may identify parasagittal injury, focal ischemia, injury to the basal ganglia, and posterior fossa pathology. Although magnetic resonance imaging is the best technique available for evaluating the anatomic structures of the brain as well as myelination pattern, it is difficult to interpret in the newborn because of the high water content of the brain.6 The brain appears hyperintense on T2-weighted images, making it difficult to distinguish from neuropathic changes due to early ischemic insults.6 An electroencephalogram recording also can aid in the evaluation of an asphyxiated infant. A progression of electroencephalographic patterns correlates with the evolution of the clinical hypoxic-ischemic encephalopathy.4 Often, it shows a discontinuous pattern with marked slowing and reduced amplitude. The low-voltage pattern may deteriorate to an isoelectric tracing that parallels the decline in clinical condition. If there is rapid resolution of the electroencephalogram abnormalities, however, prognostic outcome improves.5
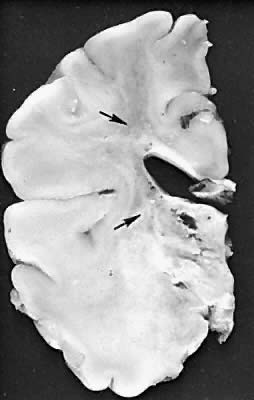
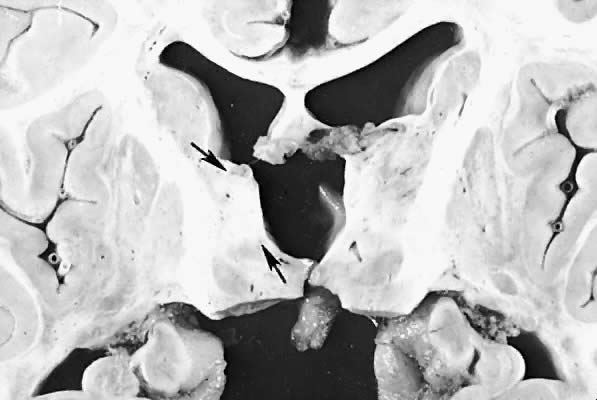
Five major neuropathologic patterns of injury are summarized in Table 2. Distribution of the lesions reflects a combination of regional circulation and metabolic factors that change with the gestational age of the infant. Both the degree of myelination and the presence of excitatory amino acid binding sites also may play a role in the pathophysiology of tissue damage.6 The primary lesions seen on autopsy in term newborns are in the cortex and basal ganglia (Fig. 1). In the premature infant, however, lesions generally occur in the germinal matrix and periventricular regions of the brain (Fig. 2). Efforts to resuscitate the infant should be directed toward adequate ventilation and perfusion, correction of accompanying metabolic acidosis, maintenance of normal blood pressure and glucose, and treatment of seizures. Antidiuretic hormone may be secreted inappropriately after a large cerebral insult, and fluid overload should be avoided. Serum osmolarity and sodium concentration should be monitored to avoid the consequence of cerebral edema and seizures. Although antiedema agents may reduce intracranial pressure, it is unclear whether they improve long-term neurologic sequelae.4 Seizures in the hypoxic-ischemic setting may be refractory to anticonvulsant therapy, but treatment is recommended to reduce the incidence of apnea and hypertension. Handling should be minimized to reduce the incidence of hypoxemic episodes, especially in the premature neonates.
TABLE 2. Neuropathologic Patterns of Hypoxic-Ischemic Injury
Pattern | Distribution |
Selective neuronal necrosis | Hippocampus, cerebellum, brainstem |
Status marmoratus of the basal ganglia | Basal ganglia, thalamus |
Parasagittal cerebral injury | Cerebral cortex, subcortical white matter |
Focal and multifocal brain injury | Cerebral cortex |
|
|