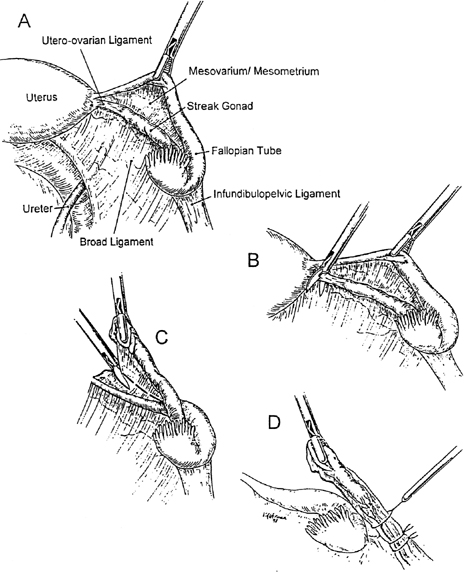
Genital ambiguity may occur in 46,XY individuals having various multiple
malformation syndromes. These include the Meckel-Gruber syndrome, Smith-Lemli-Opitz
syndrome, brachio-skeletal-genital syndrome,17 esophageal-facial-genital syndrome,18 and other disorders. These disorders are usually inherited in either autosomal
recessive or X-linked recessive fashion, and are summarized in Table 1. Many additional syndrome are associated with cryptorchiditism or simple
hypospadias, but these rarely pose diagnostic problems.19 Of relevance as well are existence of various syndromes associated with
XY gender reversal. These are listed in Table 2 and were discussed in Chapter 5-87. These syndromes include campomelic
dysplasia (SOX-9), Denys/Drash and Frasier syndromes (WT-1) autosomal
deletions (1q, 2q, 9p, 10q), and autosomal duplication (1q). In each
condition the phenotype of affected 46,XY individuals is usually complete
gender reversal (female phenotype). However, varied expression exists, some
affected cases may occasionally present as male pseudohermaphrodites (genital
ambiguity). TABLE 2. Multiple Malformation Syndromes Associated with Ambiguous Genitalia
Syndrome | Prominent Features | Etiology |
Ablepharon-macrostomia63 | Absent eyelids, eyebrows, eyelashes, external ears; fusion defects of the
mouth; ambiguous genitalia; absent or rudimentary nipples; parchment
skin, delayed development of expressive language | Autosomal recessive |
Aniridia-Wilms tumor association64,65 | Moderate to severe mental deficiency, growth deficiency, microcephaly, aniridia, nystagmus, ptosis, blindness, Wilms tumor, ambiguous genitalia, gonadoblastoma | Chromosomal or autosomal dominant (WT-1) |
Asplenia-cardiovascular anomalies-caudal deficiency66 | Hypoplasia or aplasia of the spleen complex cardiac malformations, abnormal
lung lobulation, anomalous position and development of the abdominal
organs, agenesis of corpus callosum, imperforate anus, ambiguous genitalia, contractures
of the lower limb | Autosomal recessive |
Beemer34 | Hydrocephalus, dense bones, cardiac malformation, bulbous nose, broad nasal
bridge, ambiguous genitalia | Autosomal recessive |
Deletion 11q67 | Trigonocephaly, flat and broad nasal bridge, micrognathia, carp mouth, hypertelorism, low-set
ears, severe congenital heart disease, anomalies
of limbs, external genitalia | Chromosomal |
Denys–Drash68,69 | Wilms tumor, nephropathy, ambiguous genitalia with 46, XY karyotype | Unknown |
Fraser70,71 | Cryptophthalmia, defect of auricle, hair growth on lateral forehead to
lateral eyebrow, hypoplastic nares, mental deficiency, partial cutaneous
syndactly, urogenital malformation | Autosomal recessive |
Lethal acrodysgenital dysplasia72 | Failure to thrive, facial dysmorphism, ambiguous, genitalia, syndactyly, postaxial
polydactyly, Hirschprung disease, cardiac and renal malformations | Autosomal recessive |
Rutledge73 | Joint contractures, cerebellar hypoplasia, renal hypoplasia, ambiguous
genitalia, urologic anomalies, tongue cysts, shortness of limbs, eye abnormalities, heart
defects, gallbladder agenesis, ear malformations | Autosomal recessive |
SCARF syndrome74 | Skeletal abnormalities, cutis laxa, craniosynostosis, ambiguous genitalia, psychomotor
retardation, facial abnormalities | Uncertain |
Short rib-polydactyly, Majewski type75,76 | Short stature; short limbs; cleft lip and palate; ear anomalies; limb anomalies, including
pre- and postaxial polysyndactyly; narrow thorax; short
horizontal ribs; high clavicles; ambiguous genitalia | Autosomal recessive |
Smith-Lemli-Opitz77,78 | Microcephaly, mental retardation, hypotonia, ambiguous genitalia, and sometimes
gender-reversed abnormal facies | Autosomal recessive (deficiency 7-OH cholesterol dehydrogenase) |
Trimethadione, teratogeneicity1 | Mental deficiency, speech disorders, prenatal onset growth deficiency, brachycephaly, midfacial
hypoplasia, broad and upturned nose, prominent
forehead, eye anomalies, cleft lip and palate, cardiac defects, ambiguous
genitalia | Teratogeneicity |
VATER association79,80 | Vertebral, anal, tracheoesophageal, and renal anomalies; subjects with
ambiguous genitalia as part of the cloacal anomalies | Unknown (if valid entity); alleged progestational teratogeneicity unproved (see
Simpson and Kaufman2) |
Genital–Palato-Cardiac syndrome (Gardner- Silengo-Wachtel)29 | Cleft palate, micrognathia, low-set ears, ventricular septal defect, internal
anomalies; female to male external genitalia with varied gonads (streaks
to testes) | X-linked or autosomal recessive | (Simpson JL, Elias S: Genetics in Obstetrics and Gynecology. Philadelphia, WB
Saunders, 2003.) Two multiple malformation syndromes characterized by genital ambiguity (i.e., Smith-Lemli-Opitz
syndrome and genito-palato-cardiac [Gardner-Silengo-Wachtel] syndrome) are of special interest to obstetrician–gynecologists.
SMITH-LEMLI-OPITZ SYNDROME. In this common autosomal recessive syndrome, 46,XY individuals show genital
abnormalities ranging from hypospadias to genitalia ambiguity. In
Ontario, the incidence has been estimated at 1 per 22,700 among individuals
of European ancestry.20 An estimate of 1 per 20,000 is accepted.21 A characteristic spectrum of dysmorphic features allows diagnosis by experienced
clinicians and geneticists. Mental retardation exists as does
craniofacial dysmorphia characterized by microcephaly, low-set ears, ptosis, anteverted
nares, inner epicanthal folds, broad maxillary ridges
and micrognathia. Syndactly of the third toe is present. Ureteropelvic
junction obstruction and renal anomalies (cystic dysplasia, agenesis, duplication
of kidneys) are also common. Approximately two thirds
have genital problems: usually hypospadias, micropenis, or hypoplastic
scrotum are the manifestations; genital ambiguity is less common. However, a more severe phenotype of Smith-Lemli-Opitz syndrome exists, often
called type II. Here external genitalia may be female (gender reversal).22 Both type I and type II Smith-Lemli-Opitz syndrome, if the distinction
is truly appropriate, are caused by mutation of a gene the product of
which is C7-reductase, the enzyme responsible for converting 7-hydroxycholesteral
to cholesterol.23,24 A defect in exon-intronic splicing is the most common molecular perturbation. Considerable attention is being directed to the feasibility of postnatal
as well as prenatal treatment with a high-cholesterol diet. Given its
low molecular weight, cholesterol should cross the placenta, making
this mode of therapy feasible. During pregnancy, maternal serum estriol
is low to nondetectable,25 making detection possible during maternal serum analyte screening. Maternal
serum analyte screening is based on low maternal serum alpha-fetoprotein (MSAFP; multiple
of mediam [MOM]0.72), μE3 (MOM 0.21) and
human chorionic gonadotropin (hCG; MOM 0.76).21 An algorithm based on performing an invasive procedure when risk is 1:100 will
yield 71% detection at a procedure rate of only 0.44%.21 Definitive prenatal diagnosis is facile with amniotic fluid, based on
molecular studies or presence of the novel compounds dehydroestriol and
dehydropregnanetriol. Diagnosis can even be made in maternal urine for
in normal pregnancies these compounds are undetectable.26 GENITO-PALATO-CARDIAC (GARDNER-SILENGO-WACHTEL) SYNDROME. The existence of a still poorly defined disorder characterized by variable
gender sex reversal and characteristic somatic anomalies is accepted. Several
authors have arrived at this conclusion27,28,29 with the latter best reflecting consensus as to which cases fit within
this spectrum. Greenberg and colleagues29 proposed that this disorder be called palato-genital-cardiac or Gardner-Silengo-Wachtel
syndrome and such appellations are usually applied. In retrospect, Gardner and associates30 reported what is accepted as the first case; however, those authors believed
that oral contraception was causative through teratogenic action. This
diverted attention from the possibility of a newly recognized
syndrome. Silengo and colleagues31 later recounted two cases similar to that of Gardner and colleagues.30 All three were 46,XY with female external genitalia and müllerian
derivatives; gonads were bilateral streaks, but in one case Sertoli
cells were present. Somatic features included cleft palate, micrognathia, other
facial dysmorphias (downward-slanting palpebral features, low-set
ears, anteverted nares, carp mouth), clubfoot, or prominent heels. Bernstein
and colleagues32 followed with a report of four cases, which may or may not have all had
the same disorder. The most thoroughly described cases not only showed 46,XY
gender reversal but also duplication of Xp, in a child and its
aborted fetal sib. Dup (Xp) was present in several normal (XX) females. This
observation served as the basis for postulating that duplication
of Xp (later DAX-1) caused gender reversal (see Chapter 5-86). However, no
other features of adrenal hypoplasia were evident in the case
of Bernstein and colleagues,32 and the somatic features were not those now known to be associated with
DAX-1 duplication. On the other hand, cleft palate, hypertelorism, micrognathia, low-set
eyes, prominent heels, and ventricular septal defect (VSD) were
present. Bernstein and colleagues32 reported three other cases lacking duplication of Xp. These cases showed
cleft palate, microcephaly, low-set ears, and post-axial polydactyly. One
of the three had VSD. External genitalia in all were female, but
their karyotype was not stated. Wolman and colleagues33 described a case with similar somatic findings (cleft palate, micrognathia, clubfeet, thoracic
dysplasia. Gender reversal extended to gonads
showing seminiferous tubules; a uterus was present. Greenberg and colleagues29 reported two 46,XY sibling fetuses who were each aborted following prenatal
detection (ultrasound) of anomalies. One fetus had female external
genitalia and normal ovaries, micrognathia but no cleft palate, low-set
ears, flexion deformities of thumbs and toes, and cardiac defects
that included VSD. The other aborted sib showed not dissimilar somatic
features, plus bilateral cystic kidneys, gastrointestinal defects (agenesis
gallbladder, intestinal rotation, Meckel diverticulum). However, external
genitalia of this sib were male, with first-degree hypospadias
and testes. Beemer and von Ertbruggen34 reported two sibs of consanguineous parents. These sibs may or may not
have had the same disorder.29 In this syndrome, X-linked, or less likely autosomal-recessive, XY gender
reversal syndrome would seem to exist. Defining somatic features include
facial clefts, micrognathia, low-set ears, and other facial dysmorphic
features; cardiac defects such as VSD; flexion deformities; and
various internal anomalies. Not all cases show the same features, and
in some cases Smith-Lemli-Opitz syndrome could be the correct diagnosis. All
cases of genito-palato-cardiac syndrome have been 46,XY and almost
all show female external genitalia. Gonads show unusual variability: streak
gonads, ovary-like gonads comprised of seminiferous tubules
or testes (even in the sib of an XY case who showed ovaries). |