Basic Genetics for Maternal Fetal Medicine
Authors
INTRODUCTION
“The double helix is indeed a remarkable molecule. Modern man is perhaps 50,000 years old, civilization has existed scarcely 10,000 years, and the United States for just over 200 years; but DNA and RNA have been around for at least several billion years. All that time the double helix has been there, and active, and yet we are the first creatures on Earth to become aware of its existence”
Francis Crick
Modern clinical genetics is a highly specialized and complex discipline, built on a foundation of many decades of academic and clinical research. Here we aim to give a brief history of the development of clinical genetics, highlighting a number of key contributions and experiments. We also aim to outline the current technologies used in clinical practice.
AGRICULTURE
The principles of heredity have been recognized and manipulated by humans for millennia.1 Archeological and genetic evidence suggests that agriculture began independently in several parts of the world simultaneously between 10,000 and 8000BC, with selective breeding of maize in Mesoamerica, rice in the Yangtze River region of China, potato and manioc in the lowlands of South America, and wheat, flax and chick pea in the fertile crescent area surrounding the Euphrates and Tigris rivers, in modern day Iraq.2 The domestication of animals may predate agriculture, with recent studies dating the domestication of the dog (Canis lupus familiaris) to 13,000BC.3 This period in human history, termed the early Neolithic period, represents a transition from a nomadic hunter gatherer society, dependent on wild resources, to more stable populations with greater security of nutrition,2 with humans exerting control over the natural environment.
Charles Darwin recognized the process of ‘accumulative selections’ of natural variation by human intervention, suggesting that domestication represents an example of selection in action:4
“We can not suppose that all the breeds were suddenly produced as perfect and as useful as we now see them; indeed, in many cases, we know that this has not been their history. The key is man's power of accumulative selection: nature gives successive variations; man adds them up in certain directions useful to him”
Charles Darwin
However, it was not until the 19th century that the principles of heredity were systematically interrogated.
MENDELIAN GENETICS
Gregor Mendel was an Augustan Friar and Abbot of Saint Thomas Abbey in Brno. Between 1856 and 1863 he undertook a detailed and systematic research program of plant hybridization using the pea plant (Pisum sativum). Although his research was published in 1866, its significance was not recognized until after his death, and he is posthumously considered to be the founder of classical genetics.5
Mendel selected the pea plant, as it has a range of differentiating characteristics which can easily be identified. Furthermore, peas can either self-pollinate or cross-pollinate, allowing Mendel to form hybrids between plants. The pea plant also possesses a floral structure which reduces the likelihood of accidental impregnation by foreign pollen, which Mendel realized would invalidate his results.6
Mendel began by identifying seven discrete characteristics (termed ‘phenotypes’) in different seed lines to study for his experiments. He then self-crossed these lines to ensure that he had ‘pure lines’ for each characteristic, in which he was confident that the characteristic would be expressed in the following generation on self-fertilization.
Mendel’s most evocative experiment was to cross plants with purple and white flowers. He found that the progeny of these crosses were all an identical color to the parental purple flower. However, when this generation of purple flowered plants were crossed he found a three to one ratio of purple to white flowers (of 929 pea seeds from this experiment, 705 yielded purple flowers and 224 yielded white flowers). Mendel realized that some of the purple flowers retained the potential to generate white flowers in future generations, and the purple characteristic was dominant over the white characteristic. From his experiment’s he was able to postulate the following laws.
- The law of segregation: the alleles (i.e. alternative forms of a gene) segregate from each other so that each gamete (the organisms reproductive cell) carries only one allele for each gene;
- The law of independent assortment: genes for different traits segregate independently during the formation of gametes;
- The law of dominance: some alleles are dominant while others are recessive; an organism with at least one dominant allele will display the effect of the dominant allele.
Mendel’s laws allow the modelling of how binary characteristics are inherited. At the time it was not known what substance carried hereditary information, and Mendel referred to “factors” which create the characteristic. It was not until the mid-20th century that the nature of hereditary material was confirmed.
DNA AS HEREDITARY MATERIAL
The first reported isolation of DNA is dated to 1871.7 A medical graduate named Freidrich Miescher working at the University of Tübingen performed experiments on human leukocytes from surgical bandages:8
“I had set myself the task of elucidating the constitution of lymphoid cells. I was captivated by the thought of tracking down the prerequisites of cellular life on this simplest and most independent form of animal cell”
Freidrich Miescher
Miescher identified an unknown substance, which could be precipitated in acidic solutions, and dissolved in weak alkali solutions. He found it resistant to protease digestion, and to be rich in phosphorous. Miescher termed this substance ‘nuclein’, as he had derived it from the nucleus which he had separated from the cell with dilute hydrochloric acid.8
The term ‘nucleic acid’ was first used by a student of Miescher’s, Richard Altmann, in his publication of 1889: Ueber Nucleinsäuren. Altmann isolated ‘nuclein’ from salmon sperm, and was then able to further purify this by separating it from associated proteins. He noted that it had acidic properties, and termed this substance ‘nucleic acid’ on this basis.9 Miescher initially speculated this novel substance may be involved in the transmission of hereditary traits; however, he later abandoned this idea, and the role of nucleic acids in biology remained obscure.
Between 1874 and 1876 the German Anatomist Walther Flemming was undertaking detailed research into the division of animal cells, which would ultimately lead to further insights as to the nature of hereditary material.10 Flemming carefully recorded nuclear events during cell division, and was able to develop dyes which stained ‘thread-like’ structures within the nucleus, which he termed ‘chromatin’:11
“We designate that substance, in the nucleus which upon treatment with dyes known as nuclear stains does absorb the dye… In nuclear division it accumulates exclusively in the thread figures”
Walther Flemming
He was able to record in detail the changes that occur in chromatin cell division, and termed these changes ‘mitosen’, which were beautifully illustrated in his publication of 1882, Zellsubstanz, Kurn und Zellteilung (Figure 1). These stainable nuclear tracts were termed ‘chromosomes’, meaning colored bodies, by Flemming’s colleague, Heinrich Wilhelm von Waldeyer, in 1888.12 However, it was only with the rediscovery of Mendel’s work in 1900, and its translation into English in 1902 by William Bateson that Mendel’s ideas became widely accessible, and chromosomes and inheritance were linked. Two scientists, Theodor Boveri and Walter Sutton, working independently are widely credited with developing the chromosome theory of inheritance.13
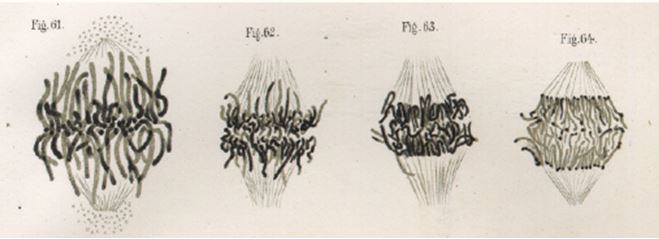 Figure 1 The stained “thread figures”, recognizable as chromosomes attached to spindle fibers during the anaphase stage of cell division. Figure taken from Walther Flemming’s Zellsubstanz, Kurn und Zellteilung of 1882.11
Figure 1 The stained “thread figures”, recognizable as chromosomes attached to spindle fibers during the anaphase stage of cell division. Figure taken from Walther Flemming’s Zellsubstanz, Kurn und Zellteilung of 1882.11
Theodor Boveri, a German zoologist, undertook a series of experimental manipulations of sea urchin eggs. Sea urchin zygotes (the fertilized cell formed by the union of gametes), can be induced using dispermic fertilization (i.e. fertilized with two spermatozoa); however, this results in abnormal cellular development. Boveri noted that these abnormally developing eggs had incomplete chromosome sets compared to the parental cells. He was able to manipulate these dispermatically derived eggs such that some developed normally, and he found these to have a normal chromosome complement.14 From these experiments Boveri concluded that a definitive set of chromosomes is requisite for normal development, and therefore each chromosome must have individual characteristics. Furthermore, in 1903 Boveri related chromosomal behaviors to Mendel’s principles, stating:
"[the probability that] the characters dealt with in Mendelian experiments are truly connected to specific chromosomes is extraordinarily high"
Theodor Boveri
Walter Sutton was a young graduate working at the University of Kansas. Sutton characterized ‘reductive division’ in spermatids and oocytes of the Grasshopper Brachystola magna, and noticed that each received only one of each chromosome (as opposed to two copies in the parental cells). He then realized that when sperm and egg unite the resultant cell has a full complement for chromosomes derived equally from each parent. Sutton recognized that, in this way, chromosomes correspond to the physical basis of Mendel’s law of segregation.15
“The association of paternal and maternal chromosomes in pairs and their subsequent separation during the reduction division...may constitute the physical basis of the Mendelian law of heredity”
Walter Sutton
This theory became known as the Boveri-Sutton theory of chromosomal inheritance; however, it remained controversial for some years. Further studies, notably by Thomas Morgan Hunt working with Drosophila melanogaster validated this work. In 1915 Thomas Morgan Hunt published ‘the mechanisms of Mendelian heredity’, and linked genes to chromosomes:16
“The inheritance of all known cofactors can be sufficiently accounted for by the presence of genes in the chromosomes”
Thomas Morgan Hunt
Although the chromosome theory of inheritance became widely accepted, it remained unclear exactly what substance carries the factors which impart heredity. It was not until 1947 that the chemical components of chromosomes were delineated by Alfred Mirsky and Hans Ris.17 They isolated chromosomes from calf lymphocytes and were able to determine that chromosomes consist predominantly of nucleic acid and basic proteins, termed histones. At this time the prevailing opinion was that nucleic acids were chemically too simple to impart hereditary information. It was felt likely that nucleic acids provided the structural element of chromosomes, and proteins were likely to carry hereditary information. However, as subsequent experiments on micro-organisms were to demonstrate, it is deoxyribonucleic acid (DNA) that carries hereditary information.
In 1928 Frederick Griffith, working at Cambridge University, reported that mice simultaneously injected with an attenuated and non-encapsulated variant of the Type II pneumococcus bacteria (R strain), along a heat killed, encapsulated and virulent variant of Type III pneumococcus (S strain), frequently succumb to infection. Furthermore, the virulent encapsulated pneumococcus could then be cultured from the animals blood.18
“The inoculation into the subcutaneous tissues of mice of an attenuated R strain derived from one type, together with a large dose of virulent culture of another type killed by heating to 60°C, has resulted in the formation of a virulent S pneumococcus of the same type as that of the heated culture.”
Frederick Griffith
Although Griffith recognized that a heat stable element was responsible for his observations, its nature was not characterized until 1943. Oswald Avery, Colin MacLeod and Maclyn McCarthy working together at the Rockefeller Institute for Medical Research were able to isolate ‘transforming principle’ from large volumes of heat killed type III pneumococcus by alcohol extraction in pure ethanol for 8 hours. The precipitate was then deproteinized using a chloroform extraction, and the polysaccharide capsule was enzymatically degraded. The resultant viscous suspension demonstrated itself to be highly efficient at transforming Type II pneumococcus. Treatment with trypsin, chymotrypsin or ribonuclease did not affect its ability to transform pneumococcus; however, treatment with crude animal sources containing deoxyribonucleases resulted in ablation of its transforming ability.19
“The data obtained by chemical, enzymatic, and serological analysis together with the results of preliminary electrophoresis, and ultraviolet spectroscopy indicate that within the limits of methods, the active fraction contains no demonstrable protein, unbound lipid, or serologically active polysaccharide and consists principally, if not solely of a highly polymerized, viscous form of desoxyribonucleic acid… The evidence presented supports the belief that a nucleic acid of the desoxyribose type is the fundamental unit of the transforming principle.”
Oswald Avery
Avery’s results were initially received with some skepticism, as the view that proteins were likely to transmit hereditary information still prevailed.20 Indeed the Nobel Prize committee would later express regret for not having recognized this contribution. However, further studies, notably experiments on bacteriophage (viruses which infect bacteria), ultimately confirmed DNA as genetic material.
Alfred Hershey and Martha Chase worked together at the Carnegie Institute. They used radioactive isotopes of either phosphorous (32P) or sulphur (36S) to label bacteriophage particles. As DNA contains phosphorous and protein contains sulphur, the role of each of these macromolecules could be independently examined. Unlabeled bacterial cells were infected with labelled bacteriophages. Only the progeny of 32P-labelled bacteriophages were found to carry the radioactive marker after cell lysis.21
“The bulk of Phage sulfur remains at the cell surface, and takes no part in the multiplication of intracellular phage. The bulk of the phage DNA, on the other hand, enters the cell soon after adsorbtion of the phage to bacteria… We infer that sulfur-containing protein has no function in phage multiplication, and that DNA has some function”
Alfred Hershey
Although Hershey and Chase were somewhat circumspect as to the role of DNA in phage multiplication, and by extension as the hereditary material. However, that nucleic acid imparts hereditary information was over time accepted by the scientific community. Further investigations were to unlock the structure and function of this unique macromolecule.
THE DOUBLE HELIX
The individual component bases of nucleic acid were first reported in 1919, by Pheobius Levine.22 He treated nucleic acids derived from yeast samples with strong alkali and was able to isolate the individual nucleic acid bases (although his experiments in fact isolated the constituent bases of RNA, as DNA is alkali resistant). However, Levine theorized that the bases were constituent in equal proportions, and formed a generic repeating structure, and was too simple to impart hereditary information (the ‘tetranucleotide hypothesis’ of nucleic acids). A significant step in the understanding of the structure of DNA was the determination of the precise ratio of base composition of DNA. In a series of papers published in the early 1950s, Edwin Chargaff and colleagues from Columbia university were able to demonstrate that the ratio of ratios of purines and pyrimidines across different species is constant;23, 24, 25 Chargaff summarized these finding in his autobiography, Heraclitian Fire26 published in 1974;
“The regularities of the composition of DNAs – some friendly people later called them the ‘Chargaff rules’ – are as follows:
(a) the sum of the purines (adenine and guanine) equals that of the pyrimidines (cytosine and thymine);
(b) the molar ratio of adenine to thymine equals 1;
(c) the molar ratio of guanine to cytosine equals 1.”
Edwin Chargaff
Chargaff visited Cambridge in 1952, and was introduced to James Watson and Francis Crick. It is reported that they discussed the 1:1 ratio of adenine to thymine, and guanine to cytosine.27 It was shortly after this meeting, in April 1953, that Watson and Crick published their seminal paper in the journal Nature, on the structure of DNA – the double helix.28 Watson and Crick described two antiparallel helical coiled chains of DNA with the bases internally and the phosphate residues externally (Figure 2). They highlighted the importance of complimentary base paring for the first time;
“The novel feature of the structure is the manner in which the two chains are held together by the purine and pyrimidine bases… They are joined together in pairs, a single base from one chain being hydrogen bonded to a single base from the other chain… One of the pair must be a purine and the other a pyrimidine for bonding to occur… These pairs are adenine (purine) with thymine (pyrimidine) and guanine (purine) with cytosine (pyrimidine)… It has not escaped our notice that the specific pairing we have postulated immediately suggests a plausible copying mechanism for genetic material”
James Watson & Francis Crick
It should be noted that Rosalind Franklin, and Raymond Gosling also published X-ray diffraction data in the same issue of Nature on 25th April 1953. They also reported the helical structure the bases would be oriented inwards, with the phosphates positioned externally.29
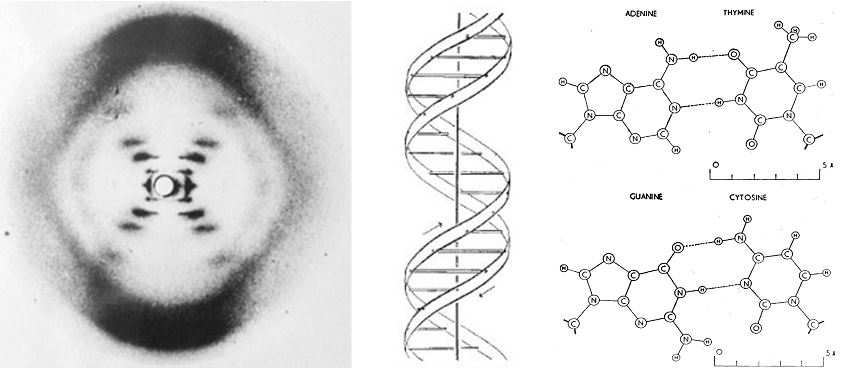 Figure 2 The X-ray crystallographic data, proposed antiparallel DNA helix, and hydrogen bonding between opposing nucleotides. Adapted from Watson and Cricks 1953 papers on the structure of DNA,28, 30 with permission.
Figure 2 The X-ray crystallographic data, proposed antiparallel DNA helix, and hydrogen bonding between opposing nucleotides. Adapted from Watson and Cricks 1953 papers on the structure of DNA,28, 30 with permission.
“We find that phosphate groups, or phosphorous atoms lie on a helix of diameter about 20Å., and the sugar and base groups must accordingly be turned inwards towards the helical axis’″
Rosalind Franklin
In a further paper published in Nature in May 1953, Watson and Crick speculated a mechanism for DNA duplication in which a single strand of DNA would act as a template for the formation of a second complimentary strand.31
“Our model for deoxyribonucleic acid is, in effect, a pair of templates, each of which is complementary to the other. We imagine that prior to duplication the hydrogen bonds are broken and the two chains unwind and separate. Each chain then acts as a template for the formation on to itself of a new companion chain, so that eventually we shall have two pairs of chains, where we had only one before’′
James Watson & Francis Crick
The hypothesis of semi-conservative DNA replication was confirmed in 1958 by Matthew Meselsohn and Franklin Stahl, working at the Californian Institute of Technology.30 They labelled Escherichia coli with heavy nitrogen (15N), by growth on heavy nitrogen supplemented media. The DNA from these bacteria could be distinguished by centrifugation owing to its increased density. They then switched bacteria raised on heavy nitrogen media to normal media (14N), and monitored the change in DNA density per replication cycle. It was found that after one cycle of replication an intermediate density band of DNA was identified. On further replication two bands, one corresponding to the intermediate band, and the other to DNA with light 14N only. Over successive duplications the concentration of the light band sequentially increased. From this they concluded that DNA duplicated semi-conservatively, as predicted by Watson and Crick:
“We find that the nitrogen of a DNA molecule is divided equally between two physically continuous subunits; that, following duplication, each daughter molecule receives one of these; and that the subunits are conserved through many duplications′’
Matthew Meselsohn and Franklin Stahl
In their Nature paper of May 1953 Watson and Crick hypothesized that the sequence of individual bases in the DNA molecule was likely to encode the genetic information:31
“It follows that in a long molecule many permutations are possible, and it therefore seems likely that the precise sequence of bases is the code which carries genetical information′’
James Watson & Francis Crick
The ‘genetic code’, however, would ultimately take some years to decipher.
GENETIC CODE AND THE CENTRAL DOGMA OF MOLECULAR BIOLOGY
In 1951 Frederick Sanger and Hans Tuppy, working at the University of Cambridge reported that phenyalanyl chain of insulin consisted of a linear chain of amino acids.32 They digested purified protein with trypsin, chymotrypsin and pepsin, and separated the resultant polypeptides by paper chromatography, and were then able to reconstruct the original protein sequence. This was the first demonstration that polypeptides consist of chain of amino acids linked together in sequence. In 1958 Vernon Ingram showed that a single amino acid change in human hemoglobin results sickle cell anemia.33 These findings, in conjunction with the advances in understanding of the structure of DNA led to the hypothesis that each gene consists of a linear sequence of nucleotides, which codes for a corresponding linear sequence of amino acids in a polypeptide. Two landmark papers published in 1961 would elucidate the nature of the genetic code.
Marshall Nirenberg and J. Heinrich Matthaei published the first report of an RNA coding for an amino acid in August 1961. They had extracted the cytoplasmic elements including ribosomes from Escherichia coli. Addition of a synthetic RNA (in this case a polyuracil RNA) resulted in the formation of a polypeptide consisting only of phenylalanine.34
“A stable, cell free system has been obtained from E.coli in which the amount of incorporation of amino acids into protein was dependent upon the addition of heat stable RNA preparations… Addition of synthetic polynucleotide, polyuridylic acid, specifically resulted in the incorporation of L-phenylalanine into a protein”
Marshall Nirenberg
Shortly after this publication, Francis Crick and colleagues published a paper speculating as to the nature of the genetic code for proteins.35 Using the T4 bacteriophage, they identified a strain which was unable to grow on a specific E. coli strain (K), and formed characteristic r plaque on a second strain (B). This bacteriophage strain was found to harbor an additional nucleotide in a gene (termed Cistron B). They found that proflavin, which mutates DNA by the removal of single nucleotides, restored the normal phenotype. Furthermore, removal of three nucleotides from the normal strain would gave a normal phenotype, however, removal of one or two nucleotides did not. From these results Crick and his colleagues were able to make strikingly accurate predictions about the nature of the genetic code;
“A group of three bases (or, less likely, a multiple of three bases) codes one-amino acid... The code is not of the overlapping type… The sequence of bases is read from a fixed starting point… the code is probably degenerate; that is, in general, one particular amino-acid can be coded by one of several triplets of bases”
James Crick
Between 1961 and 1966 Nirenberg and colleagues developed a technique to synthesize all 64 possible trinucleotides, and to test them against radioactive aminoacyl-tRNA preparations, to decipher the remaining genetic code. Nirenberg was awarded the Nobel prize in 1966 in recognition of this work.36 The ‘genetic code’ deciphered by this work is outlined in Figure 3.
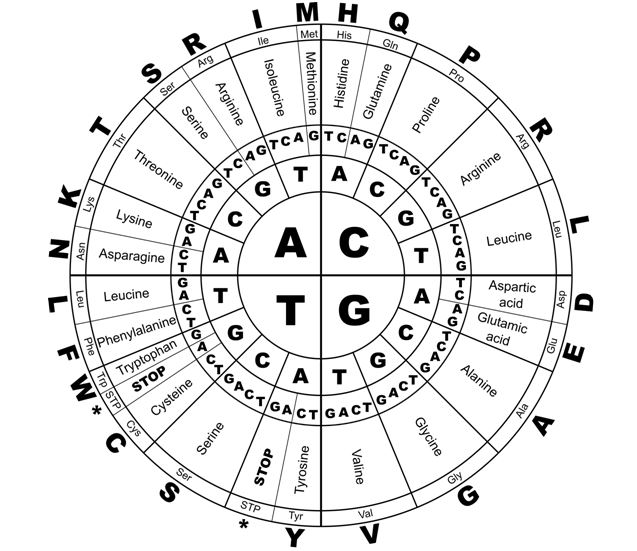 Figure 3 The genetic code for proteins
Figure 3 The genetic code for proteins
Although DNA was increasingly recognized as being the hereditary material from the publication of Avery’s data in 1943, the mechanics of how the information held in DNA becomes protein remained unclear. Initially it was proposed that protein were synthesized directly on the DNA itself, however, this was felt unlikely, as proteins were known to be synthesized in the cytoplasm rather than the nucleus.37 In 1947 Thomas Caspersson, working in Sweden reported a transient rise in RNA levels within the cytoplasm of cells actively synthesizing proteins.38 Further to this, in 1956 Elliot Volkin and Lazarus Astrachan used radioactive phosphorous to demonstrate that when E. coli are infected with the T4 bacteriophage, the radioactive phosphorous was taken up into the RNA fraction, and this RNA had a different base composition to that normally produced by E. coli.39 Two groups using different techniques published the discovery of transient ‘messenger’ RNAs, which transfer from the nucleus to the translational machinery, the ribosome in Nature in 1961.
Sidney Brenner, Francois Jacob and Matthew Meselsohn working at the University of Cambridge, demonstrated that on phage infection a transient, new viral RNA is generated. They grew E. coli on ‘heavy’ media, and then infected these cells with bacteriophage, and transferred them to light media. Using ultracentrifugation they were able to demonstrate that no new ribosomes were synthesized on phage infection, however, a new RNA with relatively rapid turnover, which had a base composition corresponding to phage DNA was attached to the pre-existing ribosomes;40
“a special type of RNA molecule, or ‘messenger RNA’, exists which brings genetic information from genes to non-specialized ribosomes’
Sidney Brenner
A second group led by James Watson in Harvard was also able to demonstrate that E. coli generate transient RNAs which localize to the ribosome, outside of the context of phage infection.41 They ‘pulsed‘ E. coli cells with 32P-labelled uracil for 20 seconds, then lysed the cells, and examined the ribosomes, and found that the new radioactively labelled RNA co-sediments with ribosomes:
“Here we present evidence that RNA molecules physically similar to phage-specific RNA exist in normal E.coli and that, under suitable ionic conditions, they are associated with ribosomal particles… A messenger role for pulse-RNA fits nicely with its specific attachment to 70S ribosomes, the site of protein synthesis”
James Watson
It was these discoveries which led Francis Crick to propose what he termed the ‘central dogma of molecular biology’:42
“The Central Dogma. This states that once ‘information’ has passed into protein it cannot get out again. In more detail, the transfer of information from nucleic acid to nucleic acid, or from nucleic acid to protein may be possible, but transfer from protein to protein, or from protein to nucleic acid is impossible. Information means here the precise determination of sequence, either of bases in the nucleic acid or of amino acid residues in the protein”
Francis Crick
The central dogma is sometimes abbreviated to ‘DNA makes RNA makes protein’, however, this is an over simplification, and Crick reiterated this in 1970, after the discovery of retroviral reverse transcriptase (which converts RNA to DNA):43
“The central dogma of molecular biology deals with the detailed residue-by-residue transfer of sequential information. It states that such information cannot be transferred from protein to either protein or nucleic acid”
Francis Crick
That DNA was the hereditary material, and encoded proteins through a triplet code, which is translated into a messenger RNA and transcribed to protein in the cytoplasm by ribosomes was established. Efforts were subsequently focused on developing techniques to sequence nucleic acids.
DNA SEQUENCING AND THE HUMAN GENOME PROJECT
The first report of a technique for sequencing DNA was by Raymond Wu of Cornell University in 1970.44
However, this technique was limited to short sequences, from the 5’ end of DNA. Two further techniques were reported in the mid 1970s which were labor intensive,44, 45 but used with some success, notably the bacteriophage Φ174 genome of 5375 bases was sequenced by Frederick Sanger and colleagues in 1977.46 However, in 1978 Frederick Sanger reported a new rapid and simple sequencing technique, which would ultimately transform the discipline of genetics.47
Sanger and colleagues took advantage of the recent discovery of the Thermococcus aquaticus heat stable DNA polymerase48 could be used to amplify DNA sequences using a complimentary primer to the DNA template (the ‘polymerase chain reaction’). Furthermore, it was discovered that 2,3, dideoxythymidine (ddTTP) inhibits the activity of DNA polymerase when incorporated into a growing DNA chain. Sanger was able to synthesize 2,3,dideoxy-derivatives for each nucleotide (ddATP/ddTTP/ddCTP/ddGTP). These ‘chain terminating’ nucleotides were then added to a standard polymerase chain reaction mix individually (i.e. four reactions undertaken in parallel). As the chain lengthens it is terminated when the dideoxynucleotide is incorporated; however, as the normal base is in great excess in the reaction mix this occurs randomly, and with sufficient cycles a termination event will occur at every nucleotide of that type in the DNA sequence which is being amplified. This results in a mix of DNA fragments of different lengths. The four reactions are the separated on the basis of size, in parallel by electrophoresis, and the DNA sequence determined by ‘reading’ the bands in order, as each band represented the nucleotide at which a termination event had occurred. This technique was to become known as Sanger sequencing, and Frederick Sanger was jointly awarded the Nobel Prize for chemistry in 1980 for “contributions concerning the determination of base sequences in nucleic acids”.
Once established Sanger sequencing was rapidly employed. The entire 172,282 base pair sequence of the Epstein-Barr virus was determined using Sanger sequencing, and was published in 1984.49 In 1995 the 1.8 Mb genome of the pathogenic bacteria Haemophilus influenzae organism was published, using the modified ‘shotgun’ sequencing technique for the first time.50 ‘Shotgun’ sequencing uses the Sanger technique to generate reads of randomly fragmented target DNA. The short sequences then compared and aligned to regenerate the whole template sequence. This technique requires considerable computing power, and it was advances in computing that allowed the most ambitious sequencing project – the Human Genome Project.
The human genome project was undertaken simultaneously by a publically funded international consortium, and a private company, Celera Genomics. It is estimated that the total cost of this project was over £3 billion, and took 13 years. In 2003 the two groups published their initial data and analysis simultaneously in Science and Nature.51, 52
“The Human Genome Project is but the latest increment in a remarkable scientific program whose origins stretch back a hundred years to the rediscovery of Mendel's laws and whose end is nowhere in sight. In a sense, it provides a capstone for efforts in the past century to discover genetic information and a foundation for efforts in the coming century to understand it”
The Human Genome Project Consortium
The latest analysis of the human genome has identified approximately 19,000 genes, along with large areas of intergenic DNA, initially labelled as ‘junk DNA’. However, projects such as ENCODE53 are now elucidating complex regulatory roles of non-coding DNA.54
Sanger sequencing has been superseded by a number of ‘next generation sequencing’ (NGS, also referred to as ‘high throughput’) technologies. In total over 180 organisms have had their genomes fully sequenced including micro-organisms, archaea, plants, fungi, insects and animals. It is now possible using NGS technologies to sequence genome, transcriptome (by RNA-seq), DNA:protein interactions (CHIP-seq), and epigenetic changes (bisulphite sequencing), even to the single cell level.55 Projects such as the International Genome Sample Resource, and the ‘100,000 Genomes Project’ in the United Kingdom are generating and sharing vast quantities of genomic data, which not only gives insight into the genetic causes of human disease, but also as to the natural degree of variation present and tolerated in the human genome.
EPIGENETICS
Epigenetics (epi = ‘upon’) refers to chemical alterations to chromatin which regulate its function. Specifically epigenetic changes include DNA methylation, histone modifications, chromatin structure and ultimately regulation of gene expression.56, 57
DNA methylation occurs at the 5’ carbon of cytosine residues when in the sequence 5’-CpG-3’, with approximately 75% of CpG dinucleotides methylated in human somatic cells.58 The distribution of CpG dinucleotides in the genome is non-random, with the majority of these motifs clustered in short CpG dense regions, often associated with the 5’ promoter sequences of open reading frames, termed CpG Islands (CpGIs).59 The remainder of the genome is CpG deplete, with genomic, non-CpGI motifs essentially universally methylated.58 Analysis of the human genome predicts approximately 29,000 CpGIs, with an estimated 60% of promoter sequences associated with CpGIs.51
It has long been recognised that CpGI methylation state predicts transcriptional activity, with hypermethylation associated with a repressive transcriptional state and hypomethylation associated with an active transcriptional state.60
DNA methylation is not the sole epigenetic event responsible for the transcriptional state of a DNA sequence. Indeed DNA methylation alone may not automatically impose a repressive transcriptional state.61 Modifications to the histone proteins which make up the nucleosome complex, also regulate DNA function and gene expression. Specific histone modifications are considered markers of chromatin structure, with acetylation of histone tails at lysine residues, and methylation of lysine 4 in histone 3 (H3K4me3) associated with a permissive chromatin structure, and methylation at lysine 9 in histone 3 (H3K9me2) and trimethylation of histone 3 lysine 27 (H3K27Me3) associated with a repressive chromatin structure, and silencing of gene expression.62
Epigenetic regulation is thought to exert regulatory control over the generic genome, with differential global epigenetic changes driving cellular differentiation.63 Different epigenetic markers are found in different tissues of the same individual, making interpretation complex. Currently an international consortium is undertaking epigenetic analysis of different tissue types in the Human Epigenome Project, to give a reference for the normal epigenetic variation seen in the human epigenome.64
The role of epigenetics in human disease is becoming increasingly recognised. Disorders of imprinting represent a protypical example of epigenetic aberrations causing human disease.65
Imprinted genes demonstrate parent of origin specific differential CpGI methylation. The developmental disorder Angelman syndrome, presents in childhood with learning difficulties, ataxia, seizures, and characteristic affect. In Angelman syndrome there is disruption of imprinting at the UBE3 locus.66 This locus is typically paternally imprinted (i.e. the allele inherited from the father is silenced), with the maternal allele expressed. However, in Angelman syndrome, individuals fail to inherit a normally expressed maternal copy of UBE3.
Epigenetic aberrations are also increasingly recognised as important in the development of cancer.67 Changes in DNA methylation patterns are commonly observed in the cancer cells. Generalized hypomethylation of non-CpGI motifs has been demonstrated in numerous cancers.68 Focal CpGI hypermethylation is a common event in the cancer cell, possibly affecting hundreds of CpGI containing genes. Furthermore, these aberrant methylation patterns are non-random and tumor specific.69 Genes which regulate the cell cycle, differentiation, apoptosis, angiogenesis and invasive behaviors are all predicted to be susceptible to hypermethylation in the cancer cell.70 Specifically hypermethylation of the promotor region of the mismatch repair tumor suppressor gene MLH1 has been found in >20% colorectal cancers.71
Historically it was thought that CpGI hypermethylation was a focal event, resulting in the silencing of individual transcripts. However, large-scale epigenetic silencing events, analogous to gross genetic changes such as loss of heterozygosity (LOH), or large scale deletions, termed Long-Range Epigenetic Silencing (LRES), have been demonstrated in multiple cancer types. 72, 73, 74
The complexities of gene regulation by multiple overlapping mechanisms are only beginning to be elucidated, however, it is clear that epigenetic mechanisms play a critical role in both health and disease.
CURRENT APPLICATIONS
There are a great number of individual genetic tests in use in modern clinical genetics. Here we present a brief overview of important techniques currently used in clinical practice. It is important to briefly consider the possible sources of DNA from a patient. Typically DNA is derived from white blood cells after standard phlebotomy; however, in some cases other sources of DNA are required. If a genetic event occurs at a point of development after fertilization, an individual will be mosaic for that change (i.e. the genetic change will only be present in some tissues). This can sometimes be seen with mosaic skin changes, and DNA can be derived from fibroblasts derived from a skin biopsy to identify mosaic alterations. In pregnancy small amounts of fetal DNA cross the placental barrier and can be isolated from maternal blood. This has been exploited in Non-Invasive Pre-Natal (NIPT) testing for gender and aneuploidies. Fetal DNA can also be derived from chorionic villus or amniotic fluid sampling.
Cytogenetics
Cytogenetics refers to the study of the number, structure and function of chromosomes. Standard karyotyping involves isolating the chromosomes from a cell in metaphase and staining the chromosomes to reveal the characteristic banding pattern. Gross changes such as deletions or duplications can then be identified. Karyotyping is a commonly used technique, and is indicated in the context of developmental delay, multiple congenital abnormalities, still birth, a clinical suspicion of a cytogenetic disorder such a trisomy 21, and some in malignancies, notably leukemias.75 The limitation of karyotyping is that only large chromosomal changes are identifiable by light microscopy, with the lower limit of a deletion or duplication detectable by G-banded karyotyping between 5 and 10 Mb.76
Fluorescence in situ hybridization (FISH) is further cytogenetic technique which can be used to identify specific chromosomal rearrangements. A fluorescent DNA probe is hybridized to the chromosome in metaphase, which identifies a specific region of the chromosome. Again this can be used to target specific duplications or deletions, and can be particularly important for some cancers, chromosomal rearrangements. The Philadelphia chromosome, a translocation between chromosome 9 and 22 (t(9;22)(q34.1;q11.2)), can be identified using this technique, and is used to direct the use of specific tyrosine kinase inhibitors in the treatment of chronic myeloid leukemia.77
Due to the limitations of traditional karyotyping with respect to small cytogenetic changes, the technique has been largely superseded by the much more sensitive comparative genome hybridization Array (CGH Array) technology.78 This technique is now a front-line investigation undertaken to investigate children with learning difficulties or dysmorphic facial features and is often requested by pediatricians before referral to genetic services. In this technique, a patient’s genomic DNA sample is fragmented, and labelled with a fluorescent tag. This DNA is then mixed in equal quantities with a control sample, known to have a full complement of chromosomal DNA, also fragmented and labelled with a different fluorescent tag. The DNA fragments are then competitively hybridized to a DNA chip which has representative probes from across the genome. Fluorescence analysis indicates for each probe the relative quantities of control versus patient DNA, and will identify small deletions or duplications. It is important to note that the probes are distributed across the entire genome, however, they are short sequences and the array does not give full coverage, so the exact size and location of duplications or deletions cannot be identified by this technique. Furthermore balanced translocations will not be identified using this technique, as although there are structural changes to the chromosomes, the whole genome complement will remain. This technique is extremely sensitive, and has identified a number of new syndromes which cannot be identified by traditional karyotyping. A protypical example of a microdeletion syndrome is Koolen de Vries syndrome, in which a 17q21.31 microdeletion of between 500 and 650 kb . This condition is characterized by hypotonia, orofacial dyspraxia, learning difficulties, characteristic dysmorphic facial features and epilepsy.79 It is important to note that not all microdeletions/duplications are pathogenic, and indeed there are a great many individuals who carry benign copy number variants. As such the results of CGH array can be difficult to interpret, and referral to a clinical genetic service is often indicated. The British charity Unique (www.rarechromo.co.uk) has collated an up to date library of information leaflets for known microdeletion and microduplication syndromes.
Targeted gene testing
In recent years there has been a rapid expansion in the availability of affordable sequencing technologies. Targeted, or single gene testing is used when a clinician has a high index of suspicion from the clinical phenotype that a particular gene is responsible for their patient’s condition. A good example would be the sequencing of the BRCA genes in a women diagnosed bilateral, or unilateral triple negative breast cancer under the age of 50.80 The advantages of targeted gene testing are that it is generally more rapid, cheaper, and as only an individual or a limited number of genes are sequenced it is less likely that a variant of unknown significance (VUS), that is to say a sequence change for which the pathogenicity is not known, will be identified. It is difficult to counsel patients about VUS as uncertainty remains, and decisions regarding cascade testing for family members are particularly challenging.
Gene panels
Increasingly it is recognized that different genetic changes may result in overlapping phenotypes which are difficult to distinguish even for experienced clinicians. Furthermore, there are conditions such as retinitis pigmentosa, or the aortopathies, that have significant genetic heterogeneity.81 Historically clinicians would individually target individual genes in order, according to clinical probability, however, this strategy can be time consuming and expensive. Using current high throughput sequencing technologies, panels of genes related to specific clinical phenotypes can now be sequenced in parallel. The advantage of this is that there is less reliance on subjective interpretation of fine phenotypic detail. Furthermore this allows rapid diagnostics as the tests are run simultaneously. Often this is approach is more economical, as although an individual gene sequencing test may be cheaper, multiple tests are often required to come to a diagnosis. The corollary to sequencing more genes is the increased possibility of detecting VUS, which are often difficult to interpret, and therefore difficult to counsel patients about. There are now many gene panels for diverse conditions including cancer, cardiac, immunological, metabolic, neurological and developmental disorders.82
Whole exome sequencing
Whole exome sequencing (WES) is a technique which uses high throughout NGS technologies to sequence all the exons of known coding sequences in the human genome. The most recent WES platforms also sequence additional biologically relevant non-coding elements such as microRNAs and long non-coding RNA sequences This typically represents between 1 and 2% of the genome.83 The obvious advantage of this is the technique does not rely on detailed phenotypic assessment of the patient, as it will identify changes in any coding sequence. The technique was first reported as being used to successfully diagnose a patient in 2011, with severe inflammatory bowel disease in which conventional approaches had failed to give a diagnosis.84 WES remains a comparatively expensive technology, and currently its use in the United Kingdom National Health Service is assessed on a case-by-case basis, and is typically limited to patients in whom a diagnosis has not been reached through conventional testing, or when there is a particular time pressure to achieve a diagnosis, or as part of research initiatives such as the ‘Deciphering Developmental Disorders’ study.85 However, as the cost of sequencing continues to decline it is likely that the role of WES will increase, and indeed WES may in time supersede targeted or panel testing as a front line test. As the amount of the genome sequenced increases the likelihood identifying VUS or incidental clinically significant genetic changes also increase. Although this can be reduced by only interrogating the exome data for particular genes of interest, effectively using the exome as a panel, this still represents a significant challenge when counselling patients for this test.
Whole genome sequencing
As the name suggests, whole genome sequencing (WGS) provides comprehensive sequencing coverage of the entire genome. WGS is now a commercially available technology. When it is considered that the first human genome took some 13 years, 200 scientists and over £3 billion to complete, the rate at which this technology has developed can be appreciated.51, 52 Currently WGS remains in the research setting within the United Kingdom, with the 100,000 Genomes Project continuing to recruit patients.86 In the coming years projects such as this will give insight into the both the causes of disease, and the natural variation tolerated by the human genome. It will also be interesting to observe to what degree there is an increased return in diagnosis over and above exome sequencing coupled with CGH Array. When compared to WGS, WES is rapid, more economical, and requires approximately one tenth of the computing power.83 However, the role of non-coding DNA sequences is increasingly recognized as important, and WES does not interrogate these sequences. It should be highlighted that WGS generates a large number of variants, particularly in non-coding sequences, as these sequences are typically less conserved. Variation in non-coding sequence is currently less well characterized, and consequently the clinical relevance of these changes is more difficult to interpret.83
Epigenetic testing
Currently targeted epigenetic investigations are available in specific circumstances, such as the testing for MLH1 promoter hypermethylation in colorectal cancer. Efforts are continuing to collate a library of tissue specific epiginomic data, and in the future more tissue specific targeted epigenetic tests may become available, however clinical epigenetic testing remains in its infancy.
FUTURE PERSPECTIVES
With the advent of affordable, rapid next generation sequencing technology the potential for individuals to have access to their genomic data is rapidly increasing. Technologies, such as CRISPR/Cas9 gene editing, hold the potential to revolutionize medicine, and have recently been successfully used to treat a child with leukemia in the United Kingdom.87 The first human trial of gene editing in the manipulation of immune cells to target lung cancer is currently underway in China.88 How these new technologies will shape future clinical practice is hard to predict, however, as our understanding of genomic information, and novel genetic technologies increases, the possibility of truly individualized medicine becomes conceivable.
REFERENCES
Balter M. 2007. Seeking Agriculture’s Ancient Roots. Science; 316, p1830-1835. |
|
Brown TA, Jones MK, Powell W, Allaby RG. 2008. The Complex Origin of Domesticated Crops in the Fertile Crescent. Trends in Ecology and Evolution, 24, p103-109. |
|
MacHugh DE, Larson G, Orlando L. 2016. Taming the Past: Ancient DNA and the Study of Animal Domestication. Annual Reviews in Animal Biosciences; 5 6.1-6.23. |
|
Darwin C. 1859. On the Origin of Special by Means of Natural Selection. John Murray, Albermarle Street; London. |
|
Griffiths AJF, Miller JH, Suzuki DT, Lewontin RC, Gelbart WM. 2000. An Introduction to Genetic Analysis; Seventh Edition. New York, W.H.Freeman. |
|
Mendel G. 1866. Versuche über Plflanzenhybriden. Verhandlungen des naturforschenden Vereines in Brünn, Bd. IV für das Jahr 1865, Abhandlungen, 3–47. (Translated by William Bateson, 1902) |
|
Miescher. 1871. Uber Die Chemische Zusammensetzung der Eiterzellen. Medicinisch-Chemische Untersuchungen; 4, p441-460. |
|
Dahn R. 2008. Discovering DNA: Freidrich Miescher and the Early Years of Nucleic Acid Research. Human Genetics; 122, p565-581. |
|
Altmann R. 1889. Ueber Nucleinsäuren. Archiv Für Anatomie and Physiologie; p524-536. |
|
Paweletz N. 2001. Perspectives: Walther Flemming: Pioneer of Mitosis Research. Nature Reviews: Molecular and Cellular Biology; 2, p72-75. |
|
Flemming W. 1882. Zellsubstanz, Kurn und Zellteilung. F.C.Vogel, Leipzig. |
|
Waldeyer HW. 1888. Über Karyokinese und ihre Beziehungen zu den Befreuchtungsvorgä. Archiv für Mikroskopische Anatomie; 32, p1-22. |
|
13. Martins LACP. 1999. Did Sutton and Boveri Propose the So-Called Sutton-Boveri Chromosome Hypothesis? Genetics and Molecular Biology; 22, p261-271. |
|
Boveri T. 1902. Über Mehrpolige Mitosen als Mitten zur Analyse des Zellkerns. Verh. Phys.-med. Ges; 35, p67-90. |
|
Sutton WS. 1902. On the Morphology of the Chromosome Group in Brachystola magna. Biol. Bull.; 4m p24-39. |
|
Morgan TYH, Sturtevant AH, Muller HJ. Bridges CB. 1915. The Mechanisms of Mendelian Hereditary. New York, Henry Holt. |
|
Mirsky AE, Ris H. 1947. The Chemical Composition of Isolated Chromosomes. The Journal of General Physiology; 31, p7-18. |
|
Griffith F. 1928. The Significance of Pneumococcal Types. Journal of Hygeine; 27, p113-159. |
|
Avery OT, MacLeod CM, McCarthy M. 1944. Studies on the Chemical Nature of the Substance Inducing Transformation of Pneumococcal Types: Induction of Transformation by a Desoxyribonucleic Acid Fraction Isolated from Pneumococcus Type III. Journal of Experimental Medicine; 72, p137-158. |
|
Deichmann U. 2004. Early Responses to Avery et al.’s Paper on DNA as Hereditary Material. Historical Studies in the Natural Sciences; 34, p207-232. |
|
Hershey AD, Chase M. 1952. Independent Functions of Viral Protein and Nucleic Acid in Growth of Bacteriophage. Journal of General Physiology; 36, p39-56. |
|
Levine PA. 1919. The Structure of Yeast Nucleic Acid: IV. Ammonia Hydrolysis. Journal of Biological Chemistry; 40, p 415–424. |
|
Chargaff, Vischer E, Doniger R, Green C, Misani F.1949. The Composition of Desoxypentose Nucleic Acids of Thymus and Spleen. Journal of Biological Chemistry; 177, p429-438. |
|
Chargaff E, Zamenhof S, Green C. 1950.Human Desoxypentose Nucleic Acid: Composition of Human Desoxypentose Nucleic Acid. Nature; 165, p756-757. |
|
Chargaff E, Lipshitz R. 1953. Composition of Mammalian Desoxyribonucleic Acids. Journal of the American Chemical Society; 75, p3658-3661. |
|
Chargaff E. 1974. Heraclitian Fire: Sketches from a Life before Nature. Rockefeller University Press. ISBN 9780874700886. |
|
Manchester KL. 2008. Historical Opinion: Erwin Chargaff and his ‘Rules’ for the Base Composition of DNA: Why did he Fail to See the Possibility of Complementarity? Trends in Biochemical Sciences; 33, p65-70. |
|
Watson JD, and Crick FHC. 1953 A Structure for Deoxyribose Nucleic acid. Nature; 171, p737–738. |
|
Franklin RE, Gosling RG. 1953. Molecular Configuration of Sodium Thymonucleate. Nature 171, p740-741. |
|
Watson J, Crick FHC. 1953. Genetical Implications of the Structure of Deoxyribonucleic Acid. Nature, 171, p964-967. |
|
Meselsohn M, Stahl F. 1958. The Replication of DNA in Escherichia coli. Proceedings of the National Academy of Sciences; 44, p671-682. |
|
Sanger F, Tuppy H. 1951. The Amino-acid Sequence in the Phenylalanyl Chain of Insulin. 2. The Investigation of Peptides from Enzymatic Hydrolysates. The Biochemical Journal; 49, p481-490. |
|
Hunt JA, Ingram VM. 1958. Abnormal Human Haemoglobins. II. The Chymotrypyic Digestion of the Trypsin-Resistant Core of Haemoglobin A and S. Biochimica et Biophysica Acta; 28, p546-549. |
|
Nirenberg MW, Matthaei JH. 1961. The Dependence of Cell-free Protein Synthesis in E.coli upon Naturally Occurring or Synthetic Polyribonucleotides. Proceedings of the National Academy of Sciences; 47, p1588-1602. |
|
Crick FHC, Barnett L, Brenner S, Watts-Tobin RJ. 1961. General Nature of the Genetic Code for Proteins. Nature; 192, p1227-1232. |
|
Nirenberg M. 2004. Historical Review: Deciphering the Genetic Code – a Personal Account. Trends in Biochemical Sciences; 29, p 46-54. |
|
Cobb M. 2015. Who Discovered Messenger RNA? Current Biology; 25, R523-548. |
|
Caspersson T. 1947. The Relationship between Nucleic Acid and Protein Synthesis. Symposium for the Society of Experimental Biology; 1, 9127-151. |
|
Volkin E, Astrachan L. 1956. Phosphorous Incorporation in Escherichia coli Ribonucleic Acid after Infection with Bacteriophage T2. Virology, 2, p146-161. |
|
Brenner S, Jacob F, Meselsohn M. 1961. An Unstable Intermediate Carrying Information from Genes to Ribosomes for Protein Synthesis. Nature; 190, p 576-581. |
|
Gros F, Hiatt H, Gilbert W, Kurkland CG, Risebrough RW, Watson JD. 1961. Unstable Ribonucleic Acid Revealed by Pulse Labelling of Escherichia coli. Nature; 190, p581-585. |
|
Crick FHC. 1958. The Biological Replication of Macromolecules. Symposium of the Society of Experimental Biology; 12, p138-163. |
|
Crick FHC. 1970. Central Dogma of Molecular Biology. Nature; 227, p561-563. |
|
Wu R. 1972. Nucleotide Sequence Analysis of DNA. Nature; 236, p198-200. |
|
Sanger F, Coulson AR. 1975. A Rapid Method for Determining Sequences in DNA by Primed Synthesis with DNA Polymerase. Journal of Molecular Biology; 94, p441-448. |
|
Maxam AM, Gilbert W. 1977. A New Method for Sequencing DNA. Proceedings of the National Academy of Sciences; 74, p560-564. |
|
Sanger F, Air GM, Barrell BG, Brown NL, Coulson AR, Fiddes JC, Hutchison III CA, Slocombe PM, Smith M. 1977. Nucleotide Sequence of Bacteriophage Phi X-174 DNA. Nature; 265, p687-695. |
|
Sanger F, Nicklen S, Coulson AR. 1977. DNA Sequencing with Chain-terminating Inhibitors. Proceedings of the National Academy of Sciences; 74, p5463-5467. |
|
Baer R, Bankier AT, Biggin MD, Deininger PL, Farrell PJ, Gibson TJ, Hatfull G, Hudson GS, Satchwell SC, Séquin C. 1984. DNA Sequence and Expression of the B95-8 Epstein - Barr virus Genome. Nature, 310, p207-211. |
|
Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM. 1995. Whole Genome Random Sequencing and Assembly of Haemophilus influenza Rd. Science; 269, p496-512. |
|
International Human Genome Sequencing Consortium, et al. 2003. Initial Sequence and Analysis of the Human Genome. Nature; 409, p860-891. |
|
Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, et al. The Sequence of the Human Genome. Science; 291, p1304-1351. |
|
The ENCODE Project Consortium. 2012. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature; 489, p57-74. |
|
Chi KR. 2016. The Dark Side of the Human Genome. Nature; 538, p275-277. |
|
Gawad C, Koh W, Quake SR. 2016. Single-cell Genome Sequencing: Current State of the Science. Nature Reviews Genetics; 17, p175-188. |
|
Bird A. 2002. DNA Methylation and Epigenetic Memory. Genes and Development, 16, p6-21. |
|
Turner BM. 2002. Cellular Memory and the Histone Code. Cell, 111, p285-291. |
|
Erhlich M, Gama-Sosa MA, Huang LH, Midgett RM, Kuo KC, McCune RA, Gehrke C. 1982. Amount and Distribution of 5-Methylcytosine in Human DNA from Different Types of Tissues of Cells. Nucleic Acid Research, 10, p2709-2721. |
|
Gardiner-Garden M, Frommer M. 1987. CpG Islands in Vertebrate Genomes. Journal of Molecular Biology, 196, p261-282. |
|
Holliday R, Pugh JE. 1975. DNA Modification Mechanisms and Gene Activity during Development. Science, 187, p226-232. |
|
Jones PA, Laird PW. 1999. Cancer Epigenetics Comes of Age. Nature Genetics, 21, p163-167. |
|
Jenuwein T, Allis CD. 2001. Translating the Histone Code. Science 293, p1074-1080. |
|
Wu H, Sun YI. 2006. Epigenetic Regulation of Stem Cell Differentiation. Paediatric Research; 59, 21R-25R. |
|
Roadmap Epigenomics Consortium, et al. Integrative Analysis of 111 Reference Human Epigenomes. Nature; 518, p317-330. |
|
Reik W, Walter J. 2001. Genomic Imprinting: Parental Influence on the Genome. Nature Reviews Genetics; 2, p21-32. |
|
Lalande M, Calciano. 2007. Molecular Epigenetics of Angelman Syndrome. Cellular and Molecular Life Sciences; 64, p947-960. |
|
Jones PA, Baylin SB. 2002. The Fundamental Role of Epigenetic Changes in Cancer. Nature Reviews: Genetics, 3, p415-428. |
|
Feinberg AP, Vogelstein B. 1983. Hypomethylation Distinguishes Genes of Some Human Cancer From Their Normal Counterparts. Nature, 301, p89-92. |
|
Costello JF, Fruhwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, Wright FA, Feramisco JD, Peltomaki P, Lang JC, Schuller DE, Yu L, Bloomfireld CD, Caligiuri LA, Yates A, Nishikawa R, Su Huang HJ, Petrelli NJ, Zhang X, O’Dorisio MS, Held WA, Cavenee WK, Plass C. 2000. Aberrant CpG-Island Methylation has Non-Random and Tumour-Specific Patterns. Nature Genetics, 24, p132-138. |
|
Esteler M. 2007. Epigenetic Gene Silencing in Cancer: the DNA Hypermethylome. Human Molecular Genetics, 16, R11, R50-59. |
|
Li X, Yao X, Wang Y, Hu F, Wang F, Jiang Y, Liu Y, Wang D, Sun G, Zhao Y. 2013. MLH1 Promoter Methylation Frequency in Colorectal Cancer Patients and Related Clinopathological and Molecular Features. PLoS One; 8, e59064. |
|
Frigola J, Song J, Stirzaker C, Hinshelwood RA, Pienado MA, Clark SJ. 2006. Epigenetic Remodeling in Colorectal Cancer Results in Coordinate Gene Suppression across an Entire Chromosomal Band. Nature Genetics, 38, p540-549. |
|
Dallosso AR, Hancock AL, Szemes M, Tsai HH, Laxmi C, Malik S, Moorwood K, Ward A, Sarkar A, Barasch J, Vuononvirta R, Jones C, Pritchard-Jones K, Royer-Pokora B, Lee SB, Brown KW, Malik K. 2009. Frequent Long-Range Epigenetic Silencing of Protocadherin Gene Cluster on Chromosome 5q31 in Wilms’ Tumour. PLoS: Genetics; 5, e1000745. |
|
Dallosso AR, Øster B, Greenhough A, Thorsen K, Curry TJ, Owen C, Hancock AL, Szemes M, Paraskeva C, Frank M, Anderson CL, Malik K. 2012. Long-Range Epigenetic Silencing of Chromosome 5q31 Protocadherins is involved in Early and Late Stages of Colorectal Tumorigenesis through Modulation of Oncogenic Pathways. Oncogene; 31, p4409-4419. |
|
Riegel M. 2014. Human Molecular Cytogenetics: From Cells to Nucleotides. Genetics and Molecular Biology; 37, p194-209. |
|
Martin CL, Warburton D. 2015. Detection of Chromosomal Aberrations in Clinical Practice: From Karyotype to Genome Sequence. Annual Reviews of Genomics and Human Genetics; 16, p309-326. |
|
Kuzrock R, Kantarjian H, Drunker BJ, Talpaz M. 2003. Philadelphia Chromosome-Positive Leukemias: From Basic Mechanisms to Molecular Therapeutics. Annals of Internal Medicine; 138, p819-831. |
|
Bejjani BA, Shaffer LG. 2006. Application of Array-Based Comparative Genomic Hybridization to Clinical Diagnostics. Journal of Molecular Diagnostics; 8, p528-533. |
|
Koolen DA, Sharp AJ, Hurst JA, Firth HV, Knight SJ, Goldenberg A, Saugier-Veber P, Pfundt R, Vissers LE, Destrée A, Grisart B, Rooms L, Van der Aa N, Field M, Hackett A, Bell K, Nowaczyk MJ, Mancini GM, Poddighe PJ, Schwartz CE, Rossi E, De Gregori M, Antonacci-Fulton LL, McLellan MD, Garrett JM, Wiechert MA, Miner TL, Crosby S, Ciccone R, Willatt L, Rauch A, Zenker M, Aradhya S, Manning MA, Strom TM, Wagenstaller J, Krepischi-Santos AC, Vianna-Morgante AM, Rosenberg C, Price SM, Stewart H, Shaw-Smith C, Brunner HG, Wilkie AO, Veltman JA, Zuffardi O, Eichler EE, de Vries BB. 2008. Clinical and Molecular Delineation of the 17q21.31 microdeletion syndrome. Journal of Medical Genetics; 45, p710-720. |
|
National Institute for Health and Care Excellence. 2015. Familial Breast Cancer: Classification, Care and Managing Breast Cancer and Related Risks in People with a Family History of Breast Cancer. Clinical Guidance [CG164]. |
|
Perez-Carro R, Corton M, Sánchez-Navarro I, Zurita O, Sanches-Bolivar N, , Sánchez-Alcudia R, Lelieveld SH, Aller E, Lopez-Martines MA, López-Molina MI, Fernandez-San Jose P, Blanco-Kelly F, Riviero-Alvarez R, Gilissen C, Millan JM, Avila-Fernandez A, Ayuso C. 2016. Panel-Based NGS Reveals Novel Pathogenic Mutations in Autosomal Recessive Retinitis Pigmentosa. Scientific Reports; 6, article no. 19531. |
|
Rehm HL. 2013. Disease-Targeted Sequencing: A Cornerstone in the Clinic. Nature Reviews: Genetics; 14, p295-300. |
|
Warr A, Robert C, Hume D, Archibold A, Deeb N, Watson M. 2016. Exome Sequencing: Current and Future Perspectives. Genes, Genomes, Genetics; 5, p1543-1550. |
|
Worthey EA, Mayer AN, Syverson GD, Helbling D, Bonacci BB, Decker B, Serpe JM, Tscannen MR, Veith RL, Basehore MJ, Broeckel U, Tomita-Mitchell A, Arca MJ, Casper JT, Margolis DA, Bick DP, Hessner MJ, Routes JM, Verbsky JW, Jacob HJ, Dimmock DP. 2011. Making a Definitive Diagnosis: Successful Clinical Application of Whole Exome Sequencing in a Child with Intractable Inflammatory Bowel Disease. Genetics in Medicine; 13, p255-262. |
|
Deciphering Developmental Disorders Consortium. 2015. Large Scale Discovery of Novel Genetic Causes of Developmental Disorders. Nature; 519, p223-228. |
|
Marx V. 2015. The DNA of a Nation. Nature; 524, p503-505. |
|
Reardon S. 2015. Leukaemia Success Heralds Wave of Gene-Editing Therapies. Nature, 527, p146-147. |
|
Cyranowski D. 2016. Chinese Scientists to Pioneer First Human CRISPR trail. Nature; 535, p476-477. |