Gene Therapy for Gynecologic Cancer
Authors
INTRODUCTION
Carcinogenesis can be considered an aberrancy of genetic function. As such, normal cells undergo initiation, promotion, clonal expansion, and progression in a stepwise process encompassing genetic alterations leading to a malignant phenotype. This pathobiology of genetic differentiation offers opportunities for innovative biologically targeted interventions. Gene therapy represents one such novel therapeutic strategy.
Gene therapy may be defined as the transfer of genetic material to a target cell to interfere with a gene's function, restore lost function, or initiate a new function.1 Gene therapy was initially envisioned as a treatment strategy for inherited genetic diseases. However, because most human malignancies are considered to be the result of acquired genetic alterations, gene therapy represents a rational investigational approach for the treatment of cancer.
Given that genetic intervention of gynecologic cancer is a rational investigational endeavor, this chapter seeks to provide a basic understanding of current vector technology, potential genetic targets relevant to gynecologic malignancies, and a summary of clinical trials in gynecologic cancer.
GENE THERAPY VECTOR STRATEGIES
Central to the realization of the potential of gene therapy approaches is the ability to accomplish efficient and specific gene delivery to cancer cells. To accomplish gene delivery, a variety of viral and nonviral systems have been developed. Initially, these systems were developed from unmodified liposomes, retroviral-based vectors, and adenoviral based vectors. A lesson of major importance in gene therapy trials to date is the realization that these vector systems are clearly inadequate to achieve a meaningful clinical effect. Moreover, complex host–vector interactions are now recognized that represent additional obstacles to be overcome by new generation vector systems.2 Current research in gene therapy vectors, therefore, is directed to surmount these recognized shortcomings.
ADENOVIRAL VECTORS
At present, the most promising vehicle for gene transfer to local–regional and disseminated cancer cells is the recombinant adenoviral vector, representing a family of DNA-based viruses. Recombinant adenoviral vectors can be produced at high titer, can directly transfect target tissue regardless of whether cells are dividing, and can package large amounts of DNA.1 Replication deficient adenoviruses can be constructed by deleting the E1, a viral gene, and replacing it with a transgene of interest. Given that a key limitation to the successful clinical translation of gene therapeutics is the efficient transfer of genetic material specifically to tumor cells, considerable effort has been devoted to the development of targetable adenoviral vectors. Immunologic targeting of adenoviral vectors has been accomplished by conjugation of ligands, such as fibroblast growth factor (FGF), to the adenoviral vectors.3 These vectors then demonstrate enhanced tumor targeting but retain functional viral mechanisms of cell entry and gene translation.4 Genetically engineered targeted adenoviral vectors have been developed based on knowledge of normal adenoviral entry into cells. It has been reported that coxsackie B virus and adenovirus share a common ubiquitous receptor, which has been designated coxsackie and adenovirus receptor (CAR).5 After knob-mediated attachment to the cell surface, the next step in infection is internalization of the virion by receptor mediated endocytosis potentiated by the interaction of arg-gly-asp (RGD) peptide sequences in the penton base with secondary host cell receptor integrins.6,7 Based on this understanding of adenoviral biology, Kraznykh and associates have endeavored to abolish native tropism by genetically manipulating the CAR receptor found on the HI loop of the adenoviral knob. Novel tropism is then introduced by retargeting the adenovirus to integrin receptors common to ovarian cancer cells by incorporating an RGD sequence into the HI loop of the adenoviral capsid structure.8 This genetically modified vector demonstrates enhanced gene transfer in both established cell lines and primary cell cultures, resulting in enhanced tumor cell death by encoded therapeutic genes.9,10
RETROVIRAL VECTORS
Retroviruses represent a class of vectors whose RNA genome is converted to DNA within the target cell. A key genetic element within the viral genome is the long terminal repeats (LTRs), which serve as promoter–enhancer regions and control expression of viral genes, including those genes required for viral integration (gag, pol, and env).11 By placing desired transgenes under control of the LTRs and eliminating critical proteins these viruses can be made replication incompetent. After delivery of the vector to the target cell by infection, the RNA enters the cell by fusion. The virally encoded enzyme, reverse transcriptase, converts the vector RNA into a RNA-DNA hybrid and then to double-stranded DNA. The DNA is integrated into the host genome of actively dividing cells where the host-cell machinery transcribes and translates the message resulting in a protein derived from the transgene of interest.11
A key limitation of the retroviral vectors is the requirement that cells be actively dividing for infection and subsequent gene translation to occur. As with adenoviral vectors, the possibility of random integration resulting in an undesirable phenotype remains a theoretical problem that has yet to be recognized in clinical trials.
REPLICATIVE ADENOVIRAL VECTORS
In a therapeutic strategy using replicative viral systems, target tumor killing by the viral agent is achieved as a direct consequence of viral replication.12 This concept of viral therapy was inspired early in this century by the observation of occasional tumor regressions in cancer patients with viral infections.13 Indeed, an early clinical trial in cervical carcinoma was performed after the isolation of the adenovirus in 1953.14 A lack of sustainable therapeutic efficacy has led investigators to dismiss viral therapy until more recently when molecular biologic techniques have allowed for greater manipulation of viral particles, resulting in new potential therapeutic agents.
With respect to candidate replicative viral agents, adenoviruses possess many of the relevant attributes recommending their employment in this context. In this regard, adenoviral vectors have been used extensively for a variety of gene therapy applications.1 Adenovirus has exhibited an unparalleled efficiency allowing effective infection of target cells in the context of in vivo gene delivery.1 Additionally, the cellular entry pathway of the virus has been extensively studied resulting in strategies to enhance viral tropism for target tumor cells.
EXPERIMENTAL VIRAL VECTORS
Lentiviruses are another class of retroviral vectors, which includes the best known virus, HIV. Lentiviruses have the advantage of retaining the ability to infect both dividing and nondividing cells. An attractive aspect of packaging transgenes within the lentiviral genome is the sustained expression (>6 months) that has been observed in experimental systems.15
Adeno-associated virus (AAV) is a DNA-based member of the human parvovirus family that depends on co-infection with a helper adenovirus for growth and replication. In contrast to alternative viral-based gene delivery systems, recombinant AAV lack all viral genes, are nonimmunogenic, and can infect dividing and quiescent cells.16 The most attractive aspect of AAV vectors is the stable and long-term expression of transgenes that has been observed in animal studies.16 Tropism-targeted AAV vectors have been developed for use in hematopoietic malignancies and may offer a means of enhancing therapeutic effect and therapeutic index.17,18
New developments in viral vector engineering have allowed the incorporation of tissue-specific promoter regions that drive the therapeutic genes of interest. In this manner, the transgene is only expressed in those tumor tissues constitutively expressing the site specific promoter.19,20 Another promising avenue of vector related research is the development of replication competent vectors to enhance local gene delivery. Conditionally replicative adenoviral vectors have been developed, which possess an IL-6 autocrine arc resulting in viral-mediated tumor cell oncolysis.21 ONYX has developed a vector that is an attenuated adenovirus that replicates in and causes selective lysis of cancer cells.22
NONVIRAL GENE DELIVERY
Safety concerns related to the use of viral particles as gene delivery vehicles in humans has led to research in nonviral delivery systems. In addition, the use of alternatives to viral particles often enables the investigator to package larger amounts of transgene DNA and avoids the immunogenicity inherent in most viral systems.
The simplest approach to nonviral delivery of genetic material is administration of naked plasmid DNA.23 Small amounts of naked DNA will be taken-up and translated by cells. This approach is probably limited in clinical use secondary to extremely poor gene transfer efficiency when delivered to systemic or regional tumors and the nonspecificity of the delivery system.
Cationic lipids have been the most extensively studied nonviral-based gene delivery system. Although these lipids do not appear to attain equivalent gene transfer efficiency as the adenovirus, lipid-based systems can accommodate large DNA sequences, can be manufactured in unlimited quantity, and have been safely applied in clinical trials to date.23
Alternative classes of synthetic cationic polymer vector systems have been investigated. Polyethylamines can mediate efficient gene transfer without the use of an endosome disruption component. In addition, biopolymers, such as gelatin and chitosan, have been used to develop gene delivery microspheres.24
The usefulness of a gene vector system is a function of both its gene transfer efficiency and its safety/toxicity in the proposed therapeutic scheme. Corroboration of the gene transfer efficiency is one of the goals of phase I human clinical gene therapy trials undergoing development. Whereas a number of vector approaches exist for ovarian cancer gene therapy, additional studies in humans will be required to refine the optimal vector system.
GENE THERAPY TREATMENT STRATEGIES FOR CANCER
Advances in molecular and tumor biology have contributed greatly to our understanding of the genetic alterations, oncogenes, and tumor-associated antigens associated with malignant transformation. Thus, gene therapy treatment strategies have been proposed that target alterations specific to tumor cells and tumor pathophysiology.
MOLECULAR CHEMOTHERAPY
Molecular chemotherapy is a strategy based on the delivery and selective expression of a gene-encoded toxin into cancer cells to achieve tumor eradication. With molecular chemotherapy, toxin expression can be accomplished by direct or indirect strategies. The direct approach uses a gene that encodes a toxin. In contrast, the indirect approach uses delivery of a prodrug that requires activation to be modified into a toxic metabolite that will ultimately lead to cell death (Fig. 1). The specificity of tumor eradication by these methods can be accomplished either by transductional targeting, whereby the toxin is specifically delivered to the tumor cells, or by transcriptional targeting, whereby tumor specific transcriptional activators are used to turn on the toxin gene.
|
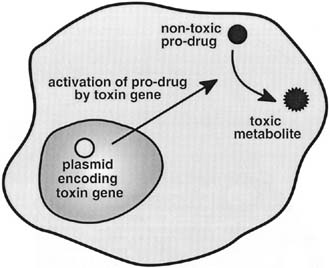
Indirect approaches to molecular chemotherapy have primarily focused on the delivery of the cytosine deaminase gene or the herpes simplex virus thymidine kinase (HSV-TK) gene. Preferential monophosphorylation of ganciclovir by HSV-TK is the mechanism that allows utilization of viral thymidine kinase for a cytotoxic effect.25 The mammalian thymidine kinase enzyme has a much lower affinity for the drug and, therefore, normal cells tend to be resistant to the toxic effects of ganciclovir when administered at low levels.26 With respect to HSV-TK-mediated cell killing, a serendipitous finding was that a greater number of tumor cells were actually killed than were initially transduced with the TK gene.27 This phenomenon is termed bystander effect. This bystander killing offers additional promise with regard to molecular chemotherapy strategies.
MUTATION COMPENSATION
Mutation compensation is a strategy used to achieve targeted ablation of a dominant oncogene, replacement of an altered tumor suppressor gene, or interference with the proper function of a growth factor or its receptor. Targeting may be directed at the level of DNA, mRNA, or the protein product.
Oligodeoxynucleotides are used to target a dominant oncogene at the DNA level by forming a triple helix. Such a structure can be formed by the association of a single strand of DNA with double-stranded DNA, which is purine-rich in one strand and pyrimidine-rich in the other. Thus, a triplex-forming oligonucleotide can be potentially designed to target polypurine–polypyrimidine double-stranded DNA in a sequence specific manner.28 Such regions are frequently found in the regulatory regions upstream of targeted genes.
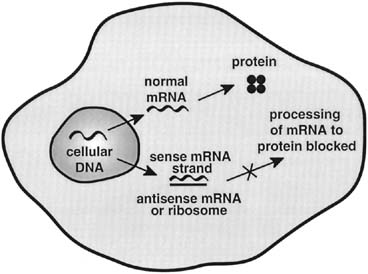
Antisense technology is being used to target dominant oncogenes at the level of mRNA. Antisense oligonucleotides are RNA sequences that are complimentary to the mRNA of the gene of interest.1 Antisense oligonucleotides function by binding to the target mRNA and blocking further processing of the genetic information by impaired transport, translational arrest, or initiating RNase degradation of mRNA (Fig. 2). Optimal targets for antisense ablation are those in which there is no normal counterpart mRNA with which to interact.
|
|
Ribozymes are small oligoribonucleotides that are capable of catalyzing RNA cleavage reactions at sequence-specific sites. The hammer-head class of ribozymes has the ability to fold into a characteristic secondary structure resembling the head of a hammer and cleaves the target RNA strand after GUA, GUC, or GUU triplet sequences.1
Recent advances in the understanding of tumorigenesis have led to the discovery of tumor suppressor genes. The intact gene appears to be critical in the repair of DNA damage that might otherwise result in the cell expressing a malignant phenotype. Additionally, intact tumor suppressor genes are thought to play a role in apoptosis or programmed cell death. These findings have led to a gene therapy strategy where the tumor suppressor genes are introduced into tumor cells.
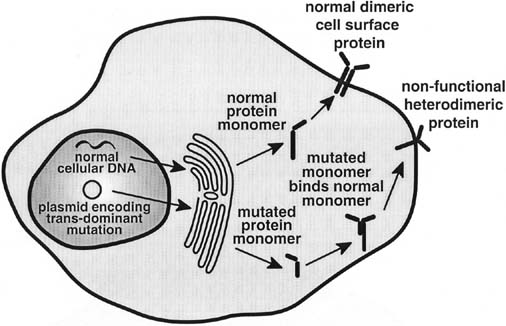
Dominant negative mutations are altered gene sequences that when translated produce a nonfunctional form of the protein of interest (Fig. 3). The use of dominant negative mutations to achieve functional inactivation of proteins involves the heterologous expression of a mutant protein that can inhibit the normal function of the native gene product produced by a cell. Dominant negative mutations have been most widely used to inhibit proteins that must associate into multimers to exert their function.
|
|
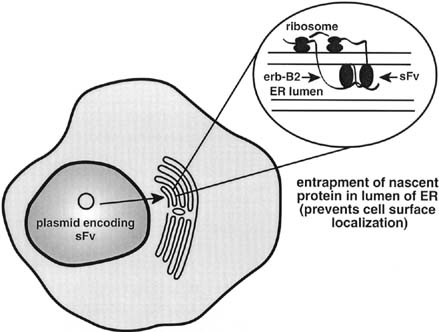
Single-chain antibodies represent an additional novel strategy to achieve targeted ablation of the protein product of a dominant oncogene. These molecules consist of the antigen binding variable light and heavy chains of an immunoglobulin molecule connected by a peptide spacer.1 The expression of a subcellular-directed (endoplasmic reticulum) form of the single-chain antibody targeted to the product of an oncogene can result in entrapment of the nascent protein (Fig. 4), thus preventing its subsequent translocation to the cell surface and abrogating the autocrine growth factor loop driving malignant transformation.
|
IMMUNOPOTENTIATION
The burgeoning knowledge of tumor immunology has led to the development of gene therapy strategies based on tumor-associated antigens and the ability of the immune system to recognize these molecules to achieve abrogation of a tumor cell population. This is termed immunopotentiation. Therapies that capitalize on immunopotentiation focus on two concepts, passive immunotherapy and active immunotherapy (Fig. 5).
|
|
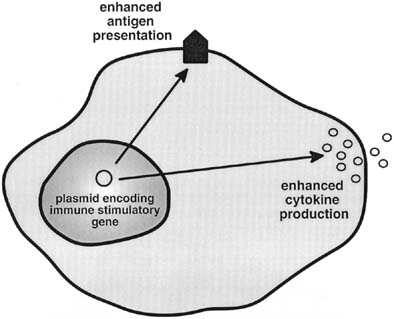
Passive immunotherapy refers to use of preformed immunologic elements to augment the immune response to tumor cells. To this end, tumor-infiltrating lymphocytes or tumor-associated lymphocytes can be isolated and expanded in vitro. After expansion, the lymphocytes are then readministered to the patient. Additionally, gene transfer technology can be used to modify the expanded cell population to augment production of cytokines or to influence traffic patterns of tumor-associated lymphocytes before reimplantation in the patient.
Active immunotherapy refers to the initiation or augmentation of an immune response directed against previously unrecognized or poorly immunogenic tumor antigens. Active immunotherapy involves techniques that increase expression of known tumor antigens or augment local concentrations of cytokines and costimulatory molecules to enhance the immune response against, as yet, unidentified tumor antigens.
VIROTHERAPY USING REPLICATIVE SYSTEMS
Initial attempts to derive conditionally replicative adenoviruses (CRADs) have focused on the achievement of tumor-selective replication. One such strategy has been the generation of type I conditionally replicative adenoviral vectors that are targeted to biologic factors modified in cancer cells. One such attenuated virus was developed to replicate only in cells lacking the cell cycle control protein p53.29 This virus, dl1520 (Onyx-015), has a deletion in the E1B-55kD gene product that is responsible for p53-binding and inactivation. Therefore, in theory, this deletion mutant would be unable to inactivate p53 in normal cells and would, consequently, be unable to replicate effectively. Alternatively, cancer cells lacking functional p53 would be sensitive to viral replication and subsequent oncolysis. Initial studies performed with this attenuated adenovirus showed sensitivity of p53 mutant ovarian cancer cell lines to cytopathic effect. In addition, tumor regression was achieved in murine xenograft models.30 Subsequent studies by Goodrum and associates and Turnell and associates, however, have determined that actual specificity of viral replication of dl1520 is not caused by the absence or presence of p53, but is based on the timing of viral replication in tumor cells.31,32 Thus, although the initial concept of targeting replication to the presence of a functional p53 gene was not realized with this virus, empiric efficacy in tumor treatment has been demonstrated.33
Another way to achieve tumor-specific adenoviral replication is to take advantage of altered cell cycle regulatory functions that occur in tumor versus normal tissue. In normal tissues, the G1-S phase checkpoint is intact; therefore, S-phase induction and viral replication are resisted. In almost all cancer cells, this checkpoint is lost as a result of any number of deletions or mutations; one such deletion is that of the RB1 gene.34 With this in mind, an adenovirus was created that carries a deletion of the retinoblastoma gene (Rb) binding site of E1A.35 Because of this deletion, the mutant adenovirus, designated Ad5-Δ24, cannot overcome the G1-S-phase checkpoint in quiescent cells as effectively as wild-type virus. Consequently, Ad5-Δ24 is unable to replicate and spread in normal tissues, thereby conferring a favorable therapeutic ratio to quiescent normal tissues. This virus has demonstrated efficacy in in vitro experiments of glioblastoma.36 In addition, a virus with the same functional deletion, designated dl922-947, replicates and lyses a broad range of cancer cells with irregularities in cell cycle checkpoints.37 Furthermore, intravenous administration reduces the incidence of metastases in a breast tumor xenograft model. Although this mutant demonstrates reduced S-phase induction and replication in nonproliferating normal cells, the virus is still capable of replicating in proliferating normal cells, which could prove problematic when translating this vector into the clinic; therefore, specific viral replication in tumor remains to be achieved with type I CRADs.
GENE THERAPY FOR GYNECOLOGIC MALIGNANCIES
Ovary
Because of the high mortality rate associated with ovarian carcinoma it is evident that research into novel therapeutic strategies, such as gene therapy, will be required if meaningful reductions in death caused by this disease are to be realized. Localization of ovarian cancer cells within the peritoneal cavity favors concentration of gene therapy vectors within this container to achieve an optimal therapeutic effect. Moreover, ovarian tumor cell lines and primary ovarian cell cultures have been shown to be transducible by gene therapy vectors in current use.
The toxin commonly used for ovarian carcinoma to accomplish molecular chemotherapy is the TK gene from the HSV. Experimental evidence has documented the ability of HSV-TK/GCV-based gene therapy to mediate tumor cell death and significantly improve survival in orthotopic murine models of intraperitoneal ovarian cancer.38
Mutation compensation or gene replacement strategies have also been extensively investigated in the context of ovarian carcinoma. Early examples of tumor suppressor gene replacement in ovarian carcinoma focused on the p53 gene. Janicek and associates, using antisense oligonucleotides targeted to p53 and c-myc, demonstrated growth inhibitory effects that functioned synergistically in some ovarian carcinoma cell lines.39 Wolf and associates then demonstrated that ovarian cancer cells are growth-inhibited by transfection with adenovirus-mediated p53 regardless of their endogenous p53 status.40 Additional utility of gene therapy may be found in its ability to enhance treatment of ovarian cancer cells with conventional chemotherapeutic agents. To this end, adenoviral mediated delivery of p53 has been demonstrated to sensitize ovarian cancer cells to paclitaxel and cisplatin in both in vitro and in vivo models of ovarian carcinoma.41,42PTEN and p16 are cell cycle regulatory genes that have been delivered by adenoviral-mediated gene transfer and resulted in growth inhibition in ovarian cancer cell lines.43,44Bax represents a member of the pro-apoptotic Bcl gene family. Delivery of adenovirus containing Bax gene constructs have been shown to induce ovarian tumor cell cytotoxicity and apoptosis in in vitro cell culture, as well as nude mouse models of ovarian carcinoma.45 Further utility of Bax-mediated gene therapy also may lie in its ability to sensitize ovarian cancer cells to the effects of ionizing radiation.46 A widely explored cell surface tyrosine kinase target is the erbB-2 protein product of the HER2-neu gene. This gene is frequently amplified in many human cancers, including ovarian carcinoma. Xing and associates have demonstrated prolonged survival in a mouse model of ovarian carcinoma using liposome-mediated gene transfer of the K1 gene, which produces the SV40 large T antigen known to suppress cell surface erbB-2 expression.47 Alternatively, Ueno and associates. have targeted down-regulation of the erbB-2 protein by liposome-mediated delivery of the E1A gene.48 These authors have noted significant ovarian tumor cell growth inhibition in vitro and prolonged survival in animal models of ovarian carcinoma. Moreover, these authors have demonstrated that E1, a mediated down-regulation of erbB-2, can sensitize cells to the toxic effects of paclitaxel chemotherapy.48 Single-chain antibodies delivered by adenoviral vectors and directed against cell surface erbB-2 have also resulted in tumor cell cytotoxicity and prolonged survival in animal models of ovarian cancer.49 Additionally, adenoviral-mediated gene transfer of anti-erbB-2 single-chain antibodies have been shown to sensitize erbB-2 overexpressing SKOV-3 ovarian cancer cells to the toxic effects of cisplatin.50
Gene-based strategies to enhance the immune response in ovarian carcinoma has focused on increasing local concentrations of cytokines, as well as up-regulating the presence of co-stimulatory molecules on the surface of ovarian cancer cells. In vitro studies of AAV-mediated IL-2 gene transfers have resulted in augmented local concentrations of IL-2 in ovarian cancer cell line cultures.51 Xu and associates have demonstrated prolonged survival with increased nitric oxide production after implantation of an ovarian cancer cell line transfected with the interferon beta gene into the peritoneal cavity of nude mice.52 Additionally, Son used liposome-mediated gene transfer of the interferon gamma gene in a murine model of metastatic ovarian carcinoma to prolong survival and inhibit tumor nodule growth.53 Finally, Gilligan and associates demonstrated enhanced immunogenicity of ovarian tumor cells adenovirally transduced to express the co-stimulatory molecule, B7-1.54
Cervix
Two HPV early genes, E6 and E7, play critical roles in the development and maintenance of a malignant phenotype and progression of disease. Several gene therapy strategies targeting E6 and E7 have been developed. Howley and others have shown that inactivation of p53 and Rb with the E6 and E7 oncoproteins, respectively, are key steps in the development of cervical cancer and immortalization of cervical epithelial cells.55
In light of these findings, Hamada and colleagues sought to explore the effects of a wild-type p53 recombinant adenoviral vector on cell growth in cervical carcinoma cells.56 In their study of HeLa cells, cell growth of infected HeLa cells was significantly suppressed in vitro, and the p53 protein was detected in these cells by both Western blot and immunohistochemistry. In a subsequent study, they were able to demonstrate that treatment of subcutaneous nodules in nude mice with this vector resulted in significant volume reduction.57 Von Knebel Doeberitz and others were able to demonstrate that expression of antisense HPV-18 E6 and E7 RNA could inhibit the growth of the cervical cancer cells.58
Further exploring the usefulness of directing strategies against HPV E6 and E7 oncogenes, Hamada and others studied the effects of introducing an antisense RNA transcript of E6 and E7 genes into cervical cancer cells.59 After construction of a recombinant adenoviral vector containing a cytomegalovirus promoter along with antisense RNA oligonucleotides of HPV16 E6 and E7 genes, they tested its effects on cell growth on the SiHa cervical cancer cell line, which is positive for HPV16 and contains wild-type p53. Their results indicated that these antisense transcripts suppressed growth of SiHa cells in a dramatic fashion. Similarly, in a study by Cho and associates, they were also able to demonstrate apoptotic changes in CaSki and SiHa cells after transfection with a plasmid containing HPV16 antisense nucleic acid.60 Moreover, molecular indications of apoptosis such as loss of mitochondrial transmembrane potential, release of mitochondrial cytochrome c into the cytoplasm, and activation of caspases 3 and 9 were seen.
P21, which is overexpressed in senescent fibroblasts, has been shown to bind and inhibit cyclin-dependent kinases, which are required for cell cycle activity.61,62 It has been recently suggested that p21 also can induce apoptotic cell death, as seen in vascular smooth muscle cells and retinoblastoma cells. In a recent study by Tsao and associates, multiple HPV-negative and positive human cervical cancer cell lines were infected with a recombinant adenovirus containing p21 cDNA.63 Massive cell death via apoptosis was observed in all cervical cell lines infected with this novel vector, which was most likely secondary to p21 transgene overexpression.
Bax, a member of the bcl-2 family, potently induces apoptosis via caspase-dependent and independent mechanisms and is a key downstream component of the p53 apoptotic pathway.64,65,66 Additionally, it has been shown that bax can function as a tumor suppressor.67 In work performed by Huh and others, multiple cervical cancer lines, including HeLa, C33A, and CaSki, and primary human cervical cancer cells lines were infected with a recombinant adenoviral vector containing the bax gene.68 Potent cytotoxicity via apoptosis and high transduction efficiency was observed in all three cervical cancer cell lines. Furthermore, primary human cervical cancer cells were isolated and infected with Ad/Bax and Ad/Cre; again, robust cytotoxicity via an apoptotic pathway was noted.
Wildner and others tested the efficacy of a replication competent adenovirus (Ad.TKRC) with the HSV-TK gene in nude mice with subcutaneous nodules of ME180 cervical carcinoma cells.69 Treatment of tumors with Ad.TKRC resulted in an antitumor response similar to that achieved by a replication-deficient Ad.TK with GCV. This finding indicated that Ad.TKRC alone without GCV has definitive oncolytic activity. There was no difference in antitumor effect between Ad.TK with GCV, Ad.TKRC alone, and Ad.TKRC followed by GCV 24 hours later. However, when GCV was administered 72 hours after administration of Ad.TKRC, survival was enhanced, with 60% of mice surviving more than 160 days. These data emphasize the importance of viral amplification and the conversion of transduced cells into adenoviral production cells. In a study by Bilsland and others, they evaluated a novel adenoviral suicide gene therapy vector expressing bacterial nitroreductase.70 By exploiting telomerase promoters in cervical cancer cells, they were able to use the bacterial nitroreductase gene that bioactivates the prodrug CB1954 into an active cytotoxic alkylating agent. Infection with adenoviral telomerase-NTR constructs resulted in an efficacious effect in telomerase-positive cervical cancer cell lines and cervical xenograft models.70,71
Some tumor cells fail to be immunogenic because of the absence of co-stimulatory molecules. Kaufmann and associates evaluated whether HPV16-positive cervical cancer cells transfected with the CD80 gene could elicit a HPV16 E7-specific cytotoxic T lymphocyte response in humans.72 Their work was the first to demonstrate that cervical cancer cells expressing CD80 were able to stimulate cytotoxic T lymphocytes using HPV16 E7 as tumor-associated antigens. In another work by Gilligan and associates, they were able to create a recombinant E1/E3-deleted adenovirus encoding the B7-1 co-stimulatory molecule.73 Cells expressing the B7-1 antigen were noted to have a higher level of T cell proliferation compared with tumor cells modified by a control virus containing the β-galactosidase gene. Thus, based on their evidence, adenoviral-mediated delivery of B7-1 to tumor cells may have some usefulness in the setting of immunopotentiation and, ultimately, the development of tumor vaccines by this route. Finally, Dall and others74 were able to construct a recombinant retroviral vector containing a single-chain antigen-binding fragment that recognizes specific epitopes that are frequently detected in cervical cancer but not in normal cervical epithelium.75 Using the aforementioned vector, they transferred the gene into a murine cytotoxic T cell line. They demonstrated that all recombinant clones were able to express the protein on the cell surface, and significant cytotoxicity was noted in cells expressing these epitopes in a MHC-independent manner.
In a recent study by Liu and associates, dendritic cells were pulsed with adeno-associated virus with the HPV16 E6 gene.76 This approach revealed transduced E6 gene mRNA expression along with chromosomal integration in infected dendritic cells. Rapid induction of a cytotoxic T cell response against primary cervical cancer cell lines was seen.
Uterus
At the present time, the usefulness of gene therapy in the setting of endometrial cancer is primarily based in in vitro studies. In a study by Kunishige and associates, the HSV-tk gene was transfected into the endometrial adenocarcinoma cell line, HHUA, via a nonviral vector.77 They were able to demonstrate a bystander effect and significant growth inhibition of HSV-tk-negative cells when the population of cells contained more than 3% HSV-tk-positive cells.
In another study by Niu and colleagues, they were able to transfer the thymidine kinase gene via a nonviral vector to cultured human and rat uterine leiomyoma cells.78 Again, a bystander effect was seen in leiomyoma cells transfected with the thymidine kinase gene and treated with ganciclovir. Forty-eight percent to 65% of cell death was witnessed when only 5% of leiomyoma cells were transfected with the TK gene. It also appeared that estradiol enhanced the bystander effect in rat leiomyoma cells. Similarly, in a study by Ural and colleagues, retroviral-mediated transfer of the HSV-tk gene was studied in the endometrial cancer cell line, EC4.79 With administration of ganciclovir, cell death and inhibition of proliferation was seen in vitro, along with a significant bystander effect.
Replacement of wild-type PTEN, a frequently altered gene in endometrial carcinomas, was evaluated by Sakurada and associates.80 Using a recombinant adenoviral vector, replacement of wild-type PTEN in endometrial cancer cell lines with inactivated PTEN resulted in decreased cell growth in vivo via apoptotic induction in addition to complete suppression of ex vivo tumor formation. Gene therapy approaches in uterine papillary serous carcinomas, an aggressive variant of endometrial carcinomas and histologically similar to papillary serous carcinomas of the ovary, has been studied as well. Ramondetta and associates studied the efficacy of adenoviral vectors containing p53 and p21 in a uterine papillary serous carcinoma cell line (SPEC-2) that contains a mutated form of p53.81 Both adenoviral vectors decreased cell proliferation; however, the effects of the adenoviral vector containing p21 were not as significant and were more transient when compared with the p53 vector. As such, the authors suggest that perhaps the p53 vector may be a more effective gene therapy agent in this subgroup of endometrial carcinomas.
CLINICAL TRIALS IN GENE THERAPY FOR GYNECOLOGIC MALIGNANCY
Encouraging results from laboratory investigations have resulted in the development of phase I clinical gene therapy trials for ovarian carcinoma. Clinical trials of molecular chemotherapy for ovarian cancer using the HSV-TK gene have been endeavored by several investigators, each using a unique gene delivery approach. In a trial reported by Robinson and colleagues, patients with recurrent ovarian cancer were injected in the peritoneal cavity with irradiated tumor cells from an established tumor cell line (PA-1) that had been retrovirally transduced to express the HSV-TK gene.82 The antitumor effect was intended to occur solely as the result of a bystander effect. Preliminary results of their phase I trial demonstrated acceptable toxicity. Link and associated used retroviral vector producer cells (VPC) (LTKOSN.2) that expressed HSV-TK to generate not only bystander-induced tumor cell killing but also direct tumor cell cytotoxicity when administered intraperitoneally to recurrent ovarian cancer patients.83 Preliminary results demonstrated acceptable toxicity. Four of nine patients were reported to have an antitumor response, although gene transfer was noted to be low. Alvarez and associates recently reported results of a phase I trial using unmodified adenoviral mediated delivery of the HSV-TK gene followed by infusion with ganciclovir in recurrent ovarian cancer patients.84 Transient and easily ameliorated vector-associated fever was experienced by 29% of treated patients. Reverse transcriptase polymerase chain reaction (RT-PCR) demonstrated the presence of mRNA in 13 of 13 evaluable patient ascites specimens. Hasenburg and colleagues reported their experience with intraperitoneal delivery of an adenoviral vector containing the HSV-TK gene in combination with an antiherpetic prodrug and topotecan.85 Ten patients underwent secondary debulking with delivery of gene therapy and five patients subsequently underwent second-look surgery. At second look, two of five patients were tumor-free and no tissue biopsy specimens demonstrated the presence of viral DNA. In a subsequent report, these same investigators noted that after a median follow-up period of 2.5 years, three of 10 patients experienced overall survival of 30, 30, and 31 months after treatment.86 While the authors suggest that these results appear to be longer than comparable results of second-line chemotherapy, the effectiveness of the gene therapy intervention is difficult to assess, because it is confounded by co-administration of interval debulking surgery and topotecan. Additionally, no data are presented to document actual in situ translation of the therapeutic gene.
Tait and associates, in an early phase I trial, examined the utility of intraperitoneal instillation of a retroviral vector encoding the BRCA1 gene in patients with recurrent of progressive ovarian cancer.87 Using Southern blot analysis on samples of tumor recovered from patients, these authors estimated that only 5% to 10% of tumor cells were transduced with the retroviral vector. Again, no significant toxicity was observed. Preliminary results from a second gene replacement phase I trial were reported by Buller and associates using an adenoviral vector encoding p53 in patients with recurrent ovarian carcinoma.88 Acceptable toxicity was observed and, interestingly, no significant adverse effects were observed from repetitive dosing of the adenovirus.
Two human clinical trials have been endeavored using vector strategies to administer therapeutic genes to patients with recurrent ovarian carcinoma. Hortobagyi and associates used cationic liposomes to deliver the E1A gene intrathoracic or intraperitoneal in 18 patients with recurrent ovarian cancer.89 The treatment was well tolerated and E1A gene expression was documented by immunohistochemistry accompanied by down-regulation of HER-2/neu expression in retrieved tumor cells. Alvarez and associates have also reported a phase I clinical trial using an adenoviral vector containing an anti-erbB-2 sFv encoding gene.90 Adenovirus was administered intraperitoneally in 15 patients with recurrent ovarian cancer. The treatment was well tolerated with no dose limiting toxicity observed, but no radiographic responses were noted. RT-PCR demonstrated expression of the anti-erbB-2 sFv encoding gene in peritoneal aspirates from 10 of 14 patients.
Finally, Vasey and associates have reported their initial experience with the ONYX-015 CRAD in ovarian cancer patients.91 Sixteen women received ONYX-015 intraperitoneally for a median of two cycles. While the treatment was well tolerated, no radiographic responses were demonstrated. Persistence of PCR evidence of adenovirus DNA for up to 10 days after administration was suggested as surrogate evidence of ongoing viral replication.
CONCLUSION
Advances in molecular biology, immunology, and virology have enabled the concept of genetic treatment of gynecologic cancer to become a reality. Research priorities include the development of new-generation vector systems, the further identification of gene therapeutic payloads, and the evaluation of these vector systems and therapeutic genes in clinical phase I trials. In view of the amount of work being performed in this rapidly moving field, there is reason to be optimistic about the development of gene therapy as a realistic clinical option for women afflicted with gynecologic malignancies.
REFERENCES
Barnes M, Deshane J, Rosenfeld M et al: Gene therapy and ovarian cancer. A review Obstet Gynecol 89:145, 1997 |
|
Elkas J, Baldwin R, Pegram M et al: Immunoglobulin in ovarian cancer ascites inhibits viral infection. Implications for adenoviral mediated gene therapy (No 52) Gynecol Oncol 72:456, 1999 |
|
Rogers B, Douglas J, Ahlem C et al: Use of a novel cross-linking method to modify adenovirus tropism. Gene Therapy 4:1387, 1997 |
|
Gu D, Gonzalez A, Printz M et al: Fibroblast growth factor 2 retargeted adenovirus has redirected cellular tropism. Evidence for reduced toxicity and enhanced anti-tumor activity in mice Cancer Res 59:2608, 1999 |
|
Bergelson J, Krithivas A, Celi L et al: The murine CAR homolog is a receptor for coxsackie B viruses and adenoviruses. J Virology 71:415-419, 1998 |
|
Bai M, Harfe B, Freimuth P: Mutations that alter an Arg-Gly-Asp (RGD) sequence in the adenovirus type 2 penton base abolish its cell rounding activity and delay virus reproduction in flat cells. J Virology 67:5198, 1993 |
|
Wickham T, Mathias P, Cheresh D et al: Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not attachment. Cell 73:309, 1993 |
|
Dmitriev I, Kraznykh V, Miller C et al: An adenovirus vector with genetically modified fibers demonstrates expanded tropism via utilization of a coxsackievirus and adenovirus receptor-independent cell entry mechanism. J Virology 72:9706, 1998 |
|
Vanderkwaak T, Wang M, Gomez-Navarro J et al: An advanced generation of adenoviral vectors selectively enhances gene transfer for ovarian cancer gene therapy approaches. Gynecol Oncol 74:227, 1999 |
|
Barnes M, Gomez-Navarro J, Strong T et al: A genetically targeted adenoviral vector for suicide gene therapy for ovarian carcinoma (No 111). Gynecol Oncol 76:259, 2000 |
|
Verma I, Somia N: Gene therapy. Promises, problems, and prospects Nature 389:239, 1997 |
|
Webb E, Smith C: Viruses in the treatment of cancer. Lancet 1:1206, 1970 |
|
Sinkovics J, Horvath J: New developments in the virus therapy of cancer: A historical review. Intervirology 36:193, 1993 |
|
Smith R, Huebner R, Rowe W et al: Studies on the use of viruses in the treatment of carcinoma of the cervix. Cancer 9:1211, 1956 |
|
Miyoshi H, Takahashi M, Gage F et al: Stable and efficient gene transfer into the retina using an HIV based lentiviral vector. Proc Natl Acad Sci USA 94:10319, 1997 |
|
Bueler H: Adeno-associated viral vectors for gene transfer and gene therapy. Biol Chem 380:613, 1999 |
|
Bartlett J, Kleinschmidt J, Boucher R et al: Targeted adeno-associated virus vector transduction of nonpermissive cells mediated by a bispecific F(ab' gamma)2 antibody. Nature Biotech 17:181, 1999 |
|
Yang Q, Mamounas M, Kennedy S et al: Development of novel cells surface CD34 targeted recombinant adeno-associated virus vectors for gene therapy. Hum Gene Ther 9:1929, 1998 |
|
Robertson M, Wang M, Siegal G et al: Use of a tissue specific promoter for targeted expression of the herpes simplex virus thymidine kinase gene in cervical carcinoma. Cancer Gene Ther 5:331, 1998 |
|
Chung I, Schwartz P, Crystal R et al: Use of L-plastin promoter to develop and adenoviral system that confers transgene expression in ovarian cancer cells but not in normal mesothelial cells. Cancer Gene Ther 6:99, 1999 |
|
Rancourt C, Piche A, Gomez-Navarro J et al: Interleukin-6 modulated conditionally replicative adenovirus as an antitumor/cytotoxic agent for cancer gene therapy. Clin Cancer Res 5:43, 1999 |
|
Heise C, Williams A, Xue S et al: Intravenous administration of ONYX-015, a selectively replicating adenovirus, induces antitumoral efficacy. Cancer Res 59:2623, 1999 |
|
Li S, Huang L: Nonviral gene therapy. Promises and challenges Gene Therapy 7:31, 2000 |
|
Roy K, Mao H, Huang S et al: Oral gene delivery with chitosan-DNA nanoparticles generates immunologic protection in a murine model of peanut allergy. Nat Med 5:387, 1999 |
|
Elion G, Furman P, Fyfe J et al: Selectivity of action of an antiherpetic agent, 9-(2-hydroxyethoxymethyl) guanine. Proc Natl Acad Sci USA 74:5716, 1977 |
|
Furman P, McGujirt P, Keller P et al: Inhibition by acyclovir of cell growth and DNA synthesis of cells biochemically transformed with herpes virus genetic information. Virology 102:420, 1980 |
|
Culver K, Ram Z, Walbridge S et al: In vivo gene transfer with retroviral vector-producing cells for the treatment of experimental brain tumors. Science 256:1550, 1992 |
|
Douglas J, Curiel D: Targeted gene therapy. Tumor Targeting 1:67, 1995 |
|
Bischoff J, Kirn D, Williams A et al: An adenovirus mutant that replicates selectively in p53 deficient human tumor cells. Science 274:373, 1996 |
|
Wildner O, Blaese R, Morris J: Therapy of colon cancer with oncolytic adenovirus is enhanced by the addition of herpes simplex virus-thymidine kinase. Cancer Res. 59:410, 1999 |
|
Goodrum F, Ornelles D: p53 status does not determine outcome of E1B 55 kilo-dalton mutant adenovirus lytic infection. J Virol 72:9479, 1998 |
|
Turnell A, Grand R, Gallimore P: The replicative capacities of large E1B-null group A and group C adenovirus are independent of host cell p53 status. J Virol 73:2074, 1999 |
|
Nemunaitis J, Khuri F, Ganly J et al: Phase II trial of intratumoral administration of ONYX-015, a replication selective adenovirus, in patients with refractory head and neck cancer. J Clin Oncol 19:289, 2001 |
|
Wang H, Moran E, Yaciuk P: E1A promotes association between p300 and pRB in multimeric complexes required for normal biologic activity. J Virol 69:7917, 1995 |
|
Fueyo J, Gomez-Manzano C, Alemany R et al: A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene 19:1, 1999 |
|
Suzuki K, Fueyo J, Krasnykh V et al: A conditionally replicative adenovirus with enhanced infectivity shows improved oncolytic potency. Clin Can Res 7:120, 2001 |
|
Heise C, Hermiston T, Johnson L et al: An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat Med 6:1134, 2000 |
|
Rosenfeld M, Wang M, Siegal G et al: Adenoviral-mediated delivery of herpes simplex virus thymidine kinase results in tumor reduction and prolonged survival in a SCID mouse model of human ovarian carcinoma. Clin Cancer Res 1:1571, 1995 |
|
Janicek M, Sevin B, Nguyen H et al: Combination anti-gene therapy targeting c-myc and p53 in ovarian cancer cell lines. Gynecol Oncol 59:87, 1995 |
|
Wolf J, Mills G, Bazzet L et al: Adenovirus mediated p53 growth inhibition of ovarian cancer cells is independent of endogenous p53 status. Gynecol Oncol 75:261, 1999 |
|
Song K, Cowan K, Sinha B: In vivo studies of adenovirus-mediated p53 gene therapy for cis-platin resistant human ovarian tumor xenografts. Oncology Res 11:153, 1999 |
|
Nielsen L, Lipari P, Dell J et al: Adenovirus mediated p53 gene therapy and paclitaxel have synergistic efficacy in models of human head and neck, ovarian, prostate, and breast cancer. Clin Cancer Res 4:835, 1998 |
|
Minaguchi T, Mori T, Kanamori Y et al: Growth suppression of human ovarian cancer cells by adenovirus mediated transfer of the PTEN gene. Cancer Res 59:6063, 1999 |
|
Wolf J, Kim T, Fightmaster D et al: Growth suppression of human ovarian cancer cell lines by the introduction of a p16 gene via a recombinant adenovirus. Gynecol Oncol 73:27, 1999 |
|
Tai Y, Strobel T, Kufe D et al: In vivo cytotoxicity of ovarian cancer cells through tumor-selective expression of the BAX gene. Cancer Res 59:2121, 1999 |
|
Barnes M, Arafat W, Gomez-Navarro J et al: Bax mediated radiosensitization of a human ovarian cancer cell line for cancer gene therapy (No. 35) Gynecol Oncol 72:452, 1999 |
|
Xing X, Matin A, Yu D et al: Mutant SV40 large T antigen as a therapeutic agent for HER-2/neu overexpressing ovarian cancer. Cancer Gene Ther 3:168, 1996 |
|
Ueno N, Bartholomeusz C, Herrmann J et al: E1A mediated paclitaxel sensitization in HER-2/neu overexpressing ovarian cancer SKOV-3.ip1 through apoptosis involving the caspase-3 pathway Clin Cancer Res 6:250, 2000 |
|
Deshane J, Grim J, Loechel S et al: Intracellular antibody against erbB-2 mediated targeted tumor cell eradication by apoptosis. Cancer Gene Ther 3:89, 1996 |
|
Barnes M, Deshane J, Siegal G et al: Novel gene therapy strategies to accomplish growth factor modulation induces enhanced tumor cell chemosensitivity. Clin Cancer Res 2:1089, 1996 |
|
Philip R, Clary B, Brunette E et al: Gene modification of primary tumor cells for active immunotherapy of human breast and ovarian cancer. Clin Cancer Res 2:59, 1996 |
|
Xu L, Xie K, Fidler I: Therapy of human ovarian cancer by transfection with the murine interferon beta gene. role of macrophage inducible nitric oxide synthase Human Gene Ther 9:2699, 1998 |
|
Son K: Cisplatin based interferon gamma gene therapy of murine ovarian carcinoma. Cancer Gene Ther 4:391, 1997 |
|
Gilligan M, Knox P, Weedon S et al: Adenoviral delivery of B7-1 (CD80) increases the immunogenicity of human ovarian and cervical carcinoma cells. Gene Ther 5:965, 1998 |
|
Howley P, Scheffner M, Munger K: Oncoproteins encoded by the cancer-associated human papillomavirus target the products of the retinoblastoma and p53 tumor suppressor genes. Cold Spring Harbor Symp Quant Biol 56:149, 1991 |
|
Hamada K, Zhang W, Alemany R, Wolf J et al: Growth inhibition of human cervical cancer cells with the recombinant adenovirus p53 in vitro. Gynecol Oncol 60:373, 1996 |
|
Hamada K, Alemany R, Zhang W et al: Adenovirus-mediated transfer of a wild-type p53 gene and induction of apoptosis in cervical cancer. Cancer Res 56:3047, 1996 |
|
von Knebel Doerberitz M, Oltersford T, Schwrz E et al: Correlation of modified human papillomavirus early gene expression with altered growth properties in C4-1 cervical carcinoma cells. Cancer Res 48:3780, 1988 |
|
Hamada K, Sakaue M, Alemany R et al: Adenovirus-mediated transfer of HPV 16 E6/E7 antisense RNA to human cervical cancer cells. Gynecol Oncol 63:219, 1996 |
|
Cho CW, Poo H, Cho YS et al: HPV E6 antisense induces apoptosis in CaSki cells via suppression of E6 splicing. Exp Mol Med 34:159, 2002 |
|
Noda A, Ning Y, Venable S et al: Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Exp Cell Res 211:90, 1994 |
|
Xiong Y, Hannon G, Zhang H et al: p21 is a universal inhibitor of cyclin kinases. Nature 366:701, 1993 |
|
Tsao Y, Huang S, Chang J et al: Adenovirus-mediated p21 gene transfer induces apoptosis of human cervical cancer cell lines. J Virol 73:4983, 1999 |
|
Xiang J, Chao DT, Korsmeyer SJ: BAX-induced cell death may not require interleukin 1 beta-converting enzyme-like proteases. Proc Natl Acad Sci USA 93:14559, 1996 |
|
Yin XM, Oltvai ZN, Koresmeyer SJ: Heterodimerzation with Bax is required for Bcl-2 to repress cell death. Curr Top Microbiol Immunol 1:4, 1995 |
|
Xiang H, Kinoshita Y, Knudson CM et al: Bax involvement in p53-mediated neuronal cell death. J Neurosci 18:1363, 1998 |
|
Yin C, Knudson CM, Korsmeyer SJ et al: Bax suppresses tumorigenesis and stimulates apoptosis in vivo. Nature 385:637, 1997 |
|
Huh WK, Gomez-Navarro J, Arafat WO et al: Bax-induced apoptosis as a novel gene therapy approach for carcinoma of the cervix. Gynecol Oncol 83:370, 2001 |
|
Wildner O, Morris J, Vahanian N et al: Adenoviral vectors capable of replication improve the efficacy of HSVtk/GCV suicide gene therapy of cancer. Gene Therapy 6:57, 1999 |
|
Bilsland AE, Anderson CJ, Fletcher-Monaghan AJ et al: Selective ablation of human cancer cells by telomerase-specific adenoviral suicide gene therapy vectors expressing bacterial nitroreductase. Oncogene 22:80, 2003 |
|
Plumb JA, Bilsland A, Kakani R et al: Telomerase-specific suicide gene therapy vectors expressing bacterial nitroreductase sensitize human cancer cells to the pro-drug CB1954. Oncogene 20:7797, 2001 |
|
Kaufmann A, Gissmann L, Schreckenberger C et al: Cervical carcinoma cells transfected with the CD80 gene elicit cytotoxic T lymphocyte response specific for HPV 16 E7 antigens. Cancer Gene Therapy 4:377, 1997 |
|
Gilligan M, Knox P, Weedon S et al: Adenoviral delivery of B7-1 (CD80) increases the immunogenicity of human ovarian and cervical carcinoma cells. Gene Therapy 5:965, 1998 |
|
Dall P, Hekele A, Beckmann M et al: Efficient lysis of CD44v7/8-presenting cells by genetically engineered cytotoxic T-lymphocytes—a model for immunogene therapy for cervical cancer. Gynecol Oncol 66:209, 1997 |
|
Dall H, Heider K-H, Hekele A et al: Surface protein expression and messenger RNA-splicing analysis of CD44 in uterine cervical cancer and normal cervical epithelium. Cancer Res 54:3337, 1994 |
|
Liu Y, Chiriva-Internati M, Grizzi F et al: Rapid induction of cytotoxic T-cell response against cervical cancer cells by human papillomavirus type 16 E6 antigen gene delivery into human dendritic cells by an adeno-associated virus vector. Cancer Gene Ther 8:948, 2001 |
|
Kunishige I, Samejima Y, Shiki Y et al: Suicide gene therapy for human uterine adenocarcinoma cells using herpes simplex virus thymidine kinase. Gynecol Oncol 72:16, 1999 |
|
Niu H, Simari R, Zimmermann E et al: Nonviral vector-mediated thymidine kinase gene transfer and ganciclovir treatment in leiomyoma cells. Obstet Gynecol 91:735, 1998 |
|
Ural AU, Takebe N, Adhikari D et al: Gene therapy for endometrial carcinoma with the herpes simplex thymidine kinase gene. Gynecol Oncol 76:305, 2000 |
|
Sakurada A, Hamada H, Fukushige S et al: Adenovirus-mediated delivery of the PTEN gene inhibits cell growth by induction of apoptosis in endometrial cancer. Int J Oncol 15:1069, 1999 |
|
Ramondetta L, Mills GB, Burke TW et al: Adenovirus-mediated expression of p53 or p21 in a papillary serous endometrial carcinoma cell line (SPEC-2) results in both growth inhibition and apoptotic cell death. potential application of gene therapy to endometrial cancer Clin Cancer Res 6:278, 2000 |
|
Robinson W, Adams J, Marrogi A et al: Vaccine therapy for ovarian cancer using herpes simplex virus thymidine kinase (HSV-TK) suicide gene transfer technique. A phase I study Gynecol Oncol 68:88, 1998 |
|
Link C, Eldeman M, Tennant L et al: LTKOSN.1 murine vector producer cell (VPC) for the in vivo delivery of the herpes simplex thymidine kinase (HSV-TK) gene for ovarian cancer Am Soc Gene Ther Abstract #915, 1999 |
|
Alvarez R, Gomez-Navarro J, Wang M et al: Adenoviral mediated suicide gene therapy for ovarian cancer (No. 107) Gynecol Oncol 76:258, 2000 |
|
Hasenburg A, Fischer D, Tong X et al: Histologic and immunohistochemical analysis of tissue response to adenovirus mediated herpes simplex thymidine kinase gene therapy of ovarian cancer. Int J Gyn Cancer 12:66, 2002 |
|
Hasenburg A, Tong X, Fischer D et al: Adenovirus mediated thymidine kinase gene therapy in combination with topotecan for patients with recurrent ovarian cancer. 2.5 year follow up Gynecol Oncol 83:549, 2001 |
|
Tait D, Obermiller P, Frazier S et al: A Phase I trial of retroviral BRCA1sv gene therapy in ovarian cancer. Clin Cancer Res 3:1959, 1997 |
|
Buller R, Pegram M, Runnebaum I: A Phase I/II trial of recombinant adenoviral human p53 intraperitoneal gene therapy in recurrent ovarian cancer (No. 37) Gynecol Oncol 72:452, 1999 |
|
Hortobagyi G, Ueno N, Xia W et al: Cationic liposome mediated E1A gene transfer to human breast and ovarian cancer cells and its biologic effects. a phase I clinical trial J Clin Oncol 19:3422, 2001 |
|
Alvarez R, Barnes M, Gomez-Navarro J et al: A cancer gene therapy approach using an anti-erbB2 single chain antibody encoding adenovirus (AD21). a phase I trial Clin Cancer Res 6:3081, 2000 |
|
Vasey P, Shulman L, Campos S et al: Phase I trial of intraperitoneal inkection of the E1B-5kd gene deleted adenovirus ONYX-015 (dll520) given on days 1 through 5 every 3 weeks in patients with recurrent/refractory epithelial ovarian cancer. J Clin Oncol 20:1562, 2002 |