Genes Causing Azoospermia and Oligozoospermia
Authors
INTRODUCTION
It is estimated that infertility affects approximately 10% of the population. Although historically a significant percentage of male-factor infertility was diagnosed as idiopathic, recent studies have clearly demonstrated a genetic etiology. This chapter offers a brief review of the criteria used in the diagnosis of azoospermia/severe oligozoospermia, followed by a discussion of the role of Y chromosomal and autosomal genes in the etiology of this condition.
CRITERIA FOR THE DIAGNOSIS OF AZOOSPERMIA OR SEVERE OLIGOZOOSPERMIA
The production of mature spermatozoa is a highly complex, though poorly understood, sequence of coordinated mitotic, meiotic, and differentiation events. Because of the complex nature of this process, elucidation of the molecular mechanisms that regulate spermatogenesis has proved difficult. It is apparent, however, that many genes with intricately interwoven pathways are required for the production of spermatozoa in appropriate numbers and of sufficient quality to achieve successful fertilization.
The purpose of spermatogenesis is to provide a haploid vehicle through which the paternal genetic information may be represented in the offspring. All of the mitotic, meiotic, and differentiative steps that are required for the production of haploid spermatozoa occur in the seminiferous tubules of the testes. In the human, spermatogenesis requires approximately 72 days from stem cell to terminally differentiated spermatozoa.1 The normal male will produce, on average, 150–250 million sperm during this time. Azoospermia is characterized by the absence of sperm in the semen; severe oligospermia is defined as the presence of fewer than 5 million sperm/mL of ejaculate.2 In the absence of clinical intervention, successful fertilization is rare in both cases.
The diagnosis of azoospermia or severe oligozoospermia is arrived at after a thorough reproductive, developmental, medical, surgical, and exposure history is obtained. Many laboratory tests are available to evaluate different aspects of male fertility, but semen analysis is a primary source of informative data about the number and quality of spermatozoa in the ejaculate. If persistent azoospermia or severe oligozoospermia is evident after analysis of a number of semen samples, evaluation of the endocrine status is warranted.3 This information can be used to determine whether infertility is due to any of the following:
- Pretesticular factors resulting from inadequate hormonal stimulation of spermatogenesis
- Testicular causes due to impaired testicular function, which generally result in elevated gonadotropin levels
- Post-testicular abnormalities resulting from obstruction or reproductive tract dysfunction and characterized by normal gonadotropin levels.
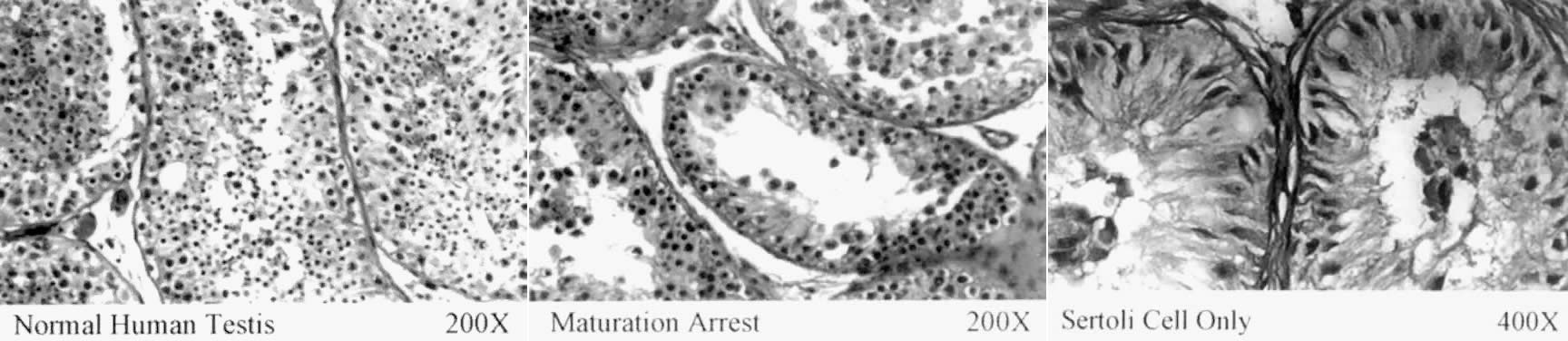
Testicular biopsy is an informative tool for distinguishing between testicular and post-testicular causes of infertility. Primary testicular failures, such as Sertoli-cell-only syndrome (SCOS) and maturation arrest (Fig. 1), can be differentiated with this procedure.4, 5 Discussion of diagnosis and treatment options for obstructive causes of azoospermia is beyond the scope of this chapter and has been discussed in detail by other authors.6, 7, 8 No treatment options are currently available in patients with severe germ cell failure; however, assisted reproductive techniques, such as intracytoplasmic sperm injection, may be used if isolated spermatozoa are present.9 Elucidation of the molecular mechanisms responsible for the control of spermatogenesis is an essential first step in the development of therapeutic options for this group of patients.
|
ROLE OF THE Y CHROMOSOME IN TESTIS DETERMINATION AND SPERMATOGENESIS
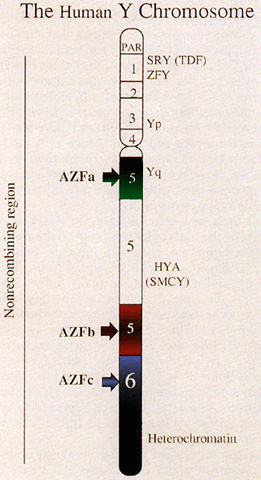
Unlike autosomes, the Y chromosome is unique in that it has no homologue. Recombination in this chromosome is restricted to a small telomeric pseudoautosomal region, which homologously pairs with the X chromosome during male meiosis. The remainder of the Y is thus nonrecombining and represents approximately 95% of its length (Fig. 2).
|
All aspects of the mammalian male phenotype, including spermatogenesis, are either directly or indirectly due to the activity of the Y chromosome. The central role played by the mammalian Y chromosome in sex determination was first described in 1959, with the report that XO patients were female and XXY patients were male.10, 11 This indicated that in mammals (unlike in Drosophila, whose sex is determined by X:autosome ratio), a dominant Y chromosome-located gene(s) was the primary testis determinant, termed TDY (testis determining Y). TDY initiates primary sex determination by inducing an indifferent gonad to form a testis. This testicular environment, in turn, initiates secondary sexual differentiation, causing the early germ cells to follow the male pathway by forming prospermatogonia, rather than meiotic oocytes. Thus, a functional TDY is an absolute requirement for male fertility.12
The localization and eventual cloning of TDY was made possible by deletion mapping of the Y using XX male and XY female patients.13, 14, 15 These individuals result from rare, illegitimate pairing of the pseuodoautosomal region of the Y with the X chromosome which leads to translocation of a small, variable portion of the Y chromosome onto the X and a corresponding loss of a portion from the Y. If this portion involves TDY then XX(Y+) sex-reversed males and XY(del) infertile females are produced. XX(Y+) persons are phenotypically male but sterile because of the presence of two X chromosomes, which invariably leads to profound defects in the mitotic stages of spermatogenesis. Identification of the specific gene responsible for testis determination proved difficult, with a number of candidates proposed and subsequently rejected before the true gene was found. The first gene associated with the Y chromosome was the male-specific minor histocompatibility antigen gene H-Y.16, 17, 18, 19 Because it was the only locus known to map to the Y, it became an excellent candidate for TDY until deletion mapping unequivocally assigned it to an area outside of that responsible for testis determination.20, 21, 22 The first convincing TDY candidate gene was ZFY (Zinc finger protein on the Y), identified by Page and colleagues23 in 1987. As additional XX(Y+) males were identified with very small Y translocations that did not include ZFY, it became apparent that this gene could not be the testis-determining factor gene (TDF).24 In a collaborative European study, a careful examination of XX male patients with critical Y translocations led Goodfellow and Fellous to isolate a gene they named SRY and to show that it was the elusive TDF gene (see Fig. 2).25, 26 Prima facie evidence that SRY was necessary and sufficient for primary testis determination was provided by constructing an XX Sry transgenic mouse. These mice developed as phenotypically sterile males, reiterating the human XX male syndrome.27
Deletion mapping of the Y chromosome has also provided valuable evidence about other biological functions of the Y chromosome, namely those involved in the control of spermatogenesis. In 1976, Tiepolo and Zuffardi28 identified four azoospermic patients with large cytologically visible de novo deletions of the distal half of Yq. They postulated that this region of the Y chromosome contained genes essential for spermatogenesis and designated this locus the azoospermic factor (AZF) region. Specific regions responsible for spermatogenesis have been ascribed to the Y chromosome of other organisms, suggesting that this is an evolutionarily conserved function.29, 30
More refined mapping of AZF (and other regions of the Y chromosome) has been greatly facilitated by the generation of a large number of Y-specific sequence-tagged sites (STS). An STS is a short stretch of known genomic sequence defined by the polymerase chain reaction (PCR) with specific primers. STSs were used to map specific AZF loci on the Y chromosome based on deletions observed in infertile men.31, 32 Extensive analysis for the presence of 76 Yq-specific STSs in males with idiopathic azoospermia or severe oligospermia has suggested that there are three distinct regions in Yq, termed AZFa (proximal), AZFb (central), and AZFc (distal) associated with this phenotype (see Fig. 2).33
Much of the Y chromosome is occupied by repeated sequences of unknown function. This has led many to believe that the Y was a repository of degenerating sequences with little biological importance and few real genes. Systematic examination of the Y chromosome, however, has revealed that there are more genes than had previously been appreciated and that these genes may play critical roles in the regulation of normal testicular function. Recent efforts have focused on the isolation of such specific genes from all three AZF regions.
An AZF candidate was first proposed in 1993 with the description of a novel multicopy gene family designated YRRM (Y chromosome RNA recognition motif).34 YRRM (since renamed RBM) is a highly conserved family of Y-specific genes that belong to a superfamily of RNA-binding proteins. RBM is expressed in the nuclei of male germ cells; its germ cell-specific expression suggests that regulation of RNA metabolism may be important for the proper control of spermatogenesis. A member of this gene family mapping to interval 6 was deleted in several infertile patients. However, because RBM is a multigene family, specific roles for individual family members must be correlated with the azoospermic phenotype; as yet, no definitive proof for its being AZF has been reported.
In a study by Reijo and associates,35 13% of subjects with nonobstructed azoospermia carried de novo deletions in the 6D-6E interval of the Y chromosome; deletions in this area were also seen in severely oligospermic men. No members of the RBM family were detected in this area, but a new AZF candidate, designated DAZ (deleted in azoospermia) was proposed. Initially thought to be a single-copy gene, DAZ was deleted in a high percentage of the azoospermic population tested by the authors. It, too, bears an RNA recognition motif, but it is expressed in the cytoplasm of premeiotic testicular germ cells, making it an excellent candidate for a factor that might be important in the maintenance of germ stem cell populations. Additional support for the role of DAZ in spermatogenesis is provided by an examination of the Drosophila homologue boule, in which a loss of function mutation results in azoospermia.36 Recent data indicate that multiple copies of DAZ are present in the AZFc region and that a functional homologue (DAZLA) exists on human chromosome 3.37, 38 Thus, a direct association between deletions in DAZ and azoospermia is complicated by multiple copies of the gene and by the abundant expression of its autosomal homologue in the testis. In addition, as with RBM, this deletion is present in only a fraction of the patients studied, suggesting that other Y loci and likely other chromosomes are involved in the complicated phenotype of azoospermia or severe oligozoospermia. To date, there has been no formal proof that DAZ is essential for human spermatogenesis, because no intragenic mutations have been found.39 A recent report by Ruggiu and co-workers40 on gene targeting in the mouse demonstrated that the autosomal mouse homologue of DAZ (Dazla) is essential for the development and survival of germ cells in the ovary and testis. Much remains to be resolved about the role of DAZ in spermatogenesis; it is clear, however, that DAZ remains a candidate azoospermia gene with a likely role in the etiology of some types of infertility.
In 1996 a new gene mapping to AZFa was described on the basis of its homology to the Drosophila developmental gene fat facets.41 This gene, DFFRY, has a homologue mapping to the X chromosome and is expressed in many human tissues, including the testis. It encodes a protein whose sequence suggests a function in the regulation of protein stability via the ubiquitin pathway. In a recent report, DFFRY was deleted in three azoospermic patients:42 two had a testicular phenotype resembling Sertoli-cell-only syndrome, and the third exhibited diminished spermatogenesis. This is the first gene from AZFa that has been reported to be deleted in azoospermic persons; its further characterization is ongoing.
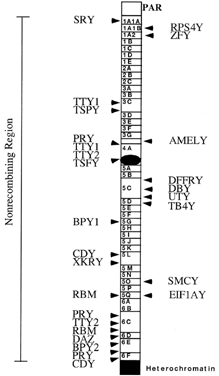
The transcriptional map of the Y chromosome was recently expanded by Lahn and Page,43 who reported isolation of a number of new genes mapping to various regions. Fig. 3 shows the latest transcriptional map of the Y chromosome; included are these new sequences. The newly isolated genes fall into two classes: single-copy genes with a wide range of expression and homology on the X, and multicopy genes with no X homologues that appear to be expressed specifically in the testis. Of particular interest are those new genes located in the AZFa-c regions, DBY and TB4Y (AZFa), E1FAY (AZFb), PRY, and CDY (AZFc). Although one may speculate about the function of these genes based on their homology to other known genes, much more information is required before specific biological functions may be assigned. It is quite clear, however, that there are a number of genes in those areas of the Y chromosome known to be associated with infertility, and that there are likely more to be found.
|
Identification of Y-specific STSs and genes has provided the clinician with an opportunity to assess the relative contribution of deletions of certain Y sequences to the infertile phenotype. Although a direct cause-and-effect relationship cannot yet be unequivocally assigned to particular deletions, it is possible to rule out large deletions in certain areas of the Y chromosome based on STS analysis. In addition, as intracytoplasmic sperm injection is being increasingly used to assist fertilization in azoospermic or severely oligozoospermic persons, and because the Y chromosome of these persons will be passed on to their sons, assessment of the status of the Y chromosome in these patients is of considerable value. As mentioned, PCR-based analysis of Yq-deleted persons has been extensively used to map AZF loci. Multiplex PCR (PCR using more than one STS per reaction) is a rapid, cost-effective, and highly reliable method of analyzing the presence (or absence) of large portions of the Y chromosome (Fig. 4). It must be recognized that small deletions in areas not covered by these STSs will not be detected, and that this method is only suitable for assessment of relatively large deletions.
|
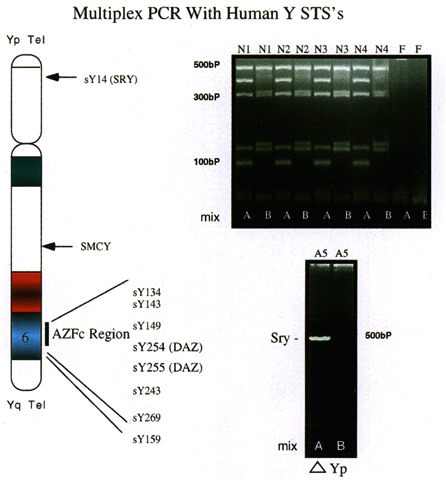
In a recent study by Foresta and associates,44 using samples from subjects with well-defined forms of idiopathic testicular damage (azoospermia with SCOS and oligozoospermia with hypospermatogenesis), 37.5% of patients with azoospermia had one or more Yq STSs missing. Twenty-two percent of patients with severe oligozoospermia were also missing one or more of these STSs. Fertile controls and the fathers or brothers of these patients did not show any abnormality, indicating that these were de novo deletions. Of the deletions observed in this highly selected group of patients, 72.7% overlapped the DAZ gene and 36.4% overlapped the RBM gene; 18.2% of patients had deletions outside of both DAZ and RBM, confirming other reports suggesting that other genes in interval 6 may be associated with infertility.39, 41 In addition, the severity of the phenotype was not correlated with the severity of the deletion, indicating that other genes outside of AZF may modulate the effects of AZF deletions. In this regard, it is noteworthy that even in this highly selected population, the vast majority (62.5%) of patients have no detectable Yq deletions. Thus, either small deletions unamenable to PCR analysis in as yet unidentified genes are responsible for these phenotypes, or there are other genes not on the Y chromosome that participate in the regulation of spermatogenesis. The latter hypothesis will be considered in the next section.
AUTOSOMAL GENES AND THE REGULATION OF SPERMATOGENESIS
Little is known about autosomal defects and infertility in humans, so much of what is known about the contribution of non-Y-chromosome genes in the control of spermatogenesis has been derived from animal models. To date, mouse mutations affecting nearly every stage of spermatogenesis have been described; this section examines a few spontaneously occurring and induced mutations that are associated with the severe impairment of fertility. Because most infertility in humans presents as an apparently isolated condition, those animal models that have infertility as their only sequela will be discussed in the most detail.
Table 1 lists a number of autosomal loci and genes that, when deleted, cause infertility. These mutations have arisen naturally (e.g. juvenile spermatogonial depletion [jsd]) or have been induced by homologous recombination ('knock outs'); the order in which they are discussed corresponds to the stage(s) of spermatogenesis affected. Thus, the jsd mutation, which effects stem-cell renewal, is considered first. From a clinical point of view, because these genes are autosomal, fertility problems are likely only in the homozygous state. However, as infertility in these animals arises in the context of only one genetic mutation in the presence of an intact Y chromosome, it is clear that these autosomal genes play a role in spermatogenesis for which Y genes cannot substitute.
Table 1. Autosomal loci and genes that cause infertility when deleted
Gene/Locus | Mutant Phenotype | Reference |
jsd Juvenile spermatogonial depletion locus | One round of spermatogenesis followed by failure of stem cells to repopulate testis | Beamer et al. 198845
|
Bmp8b Bone morphogenetic protein | Failure/reduction in germ-cell proliferation and increased spermatocyte apoptosis | Zhao et al. 199646
|
Dazla Mouse homologue of human DAZ | Decreased development and survival of germ cells
| Ruggiu et al. 199740
|
Rxr β Retinoid X receptor β | Oligo-astheno-teratozoospermia
| Kastner et al. 199647
|
Pms2 DNA mismatch repair | Abnormal chromosomal synapsis and production of small numbers of spermatozoa with abnormal morphology | Baker et al. 199548
|
Mlh1 DNA mismatch repair | Pachytene arrest
| Edelman et al. 199549
|
Crem Cyclic AMP-responsive element modulator | Spermiogenesis arrest and germ-cell apoptosis
| Nantel et al. 199650
|
mHR6B Mouse homologue of human RAD6 | Impairment of spermatid elongation and condensation/morphologic abnormalities of spermatozoa | Roest et al. 199651
|
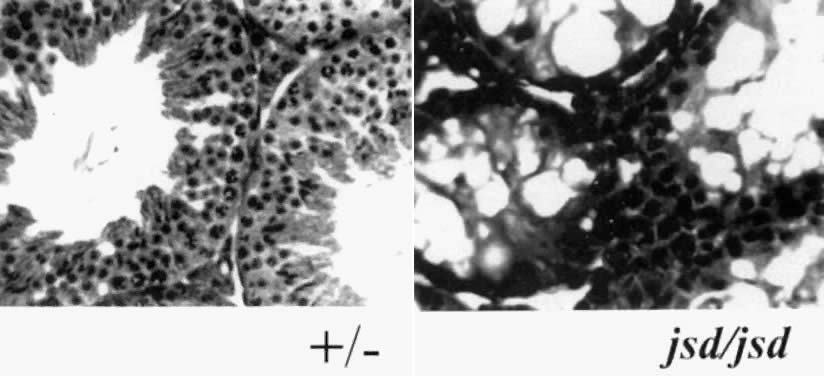
Spontaneously occurring mutations that result in infertility are, by definition, selected against during evolution. If, however, the heterozygote condition has no effect on fertility and if only one sex is affected by the homozygote condition, this would be an ideal model for the study of an autosomal gene involved in spermatogenesis. Some spontaneously occurring mouse mutations fulfill the aforementioned criteria and, significantly, show no other phenotypic effects in other organ systems. Of these, jsd is of particular interest because the phenotype of homozygous mutant animals is reminiscent of human Sertoli-cell-only conditions. The jsd mutation, reported in 1988 by Beamer and colleagues,45 defines a locus affecting postnatal proliferation of spermatogonial cells. The phenotype in these animals was apparent only after puberty, when a single round of apparently normal spermatogenesis occurred; thereafter, mature sperm were not produced. Histologic analysis of the seminiferous tubules in postpubertal 9-week-old animals indicated seminiferous tubules devoid of spermatogenesis; these tubules were small in diameter and populated by Sertoli cells and the occasional type A spermatogonia. The authors thus proposed that the defect was due to a failure of spermatogonial stem-cell renewal, resulting in insufficient numbers of stem cells for normal spermatogenesis to occur. Reminiscent of many types of testicular failure in humans, serum FSH levels in these animals were significantly elevated (in comparison to their heterozygote littermates). Fig. 5 presents the typical histology of a normal, heterozygous mouse testis (+/-) versus that of a homozygous mouse testis (jsd/jsd). The latter histologic sample resembles the histologic sample of a Sertoli-cell-only human testis (see Fig. 1C) suggesting that this animal model may provide some insight into the control of spermatogonial stem-cell renewal in humans. Beamer and colleagues45 mapped the jsd locus to mouse chromosome 1; Rohozinski and Bishop52 and Bradley et al.53 identified the mutated gene responsible for this phenotype as mUtp14b. The mUtp14b gene was retrotransposed from a ubiquitously expressed gene located on the X chromosome, apparently acquiring a germ cell specific function. Additional studies by Rohozinski et al.54 suggest that some azoospermic/severely oligospermic individuals have deletions in a related retroposed gene, UTP14c, found on chromosome 13 in humans.
|
In 1996, Zhao and co-workers46 reported that the generation of mice with a targeted mutation in bone morphogenetic protein 8B (Bmp8b) resulted in male germ-cell deficiency and infertility. BMPs belong to the transforming growth factor-B superfamily of secreted signaling molecules. A number of BMPs have been identified, with diverse functions centered around the regulation of embryonic development. Bmp8b is expressed in the placenta during embryogenesis and in male germ cells during postnatal development; early postnatal expression of Bmp8b is seen in spermatogonia and primary spermatocytes.55 After 3.5 weeks of age, it is expressed specifically in round spermatids. In the Bmp8b homozygous mutant mouse, two testis-specific abnormalities are apparent: the first influences germ-cell proliferation, and the second affects pachytene spermatocyte survival.
In the study by Zhao and Hogan,55 homozygous mutant males had much smaller testicles and partial to nearly complete germ-cell depletion. Sertoli and Leydig cells were apparently normal. Although two of the mutant males sired litters, they later became sterile. Thus, the authors suggested that Bmp8b is required for both the normal mitotic proliferation and differentiation of germ cells and for the survival of pachytene spermatocytes. The contribution of the human homologue of this gene to the control of spermatogenesis has yet to be established.
In the mouse, the homologue of the human Y chromosome gene DAZ is located on an autosome and has been named Dazla. As mentioned, DAZ and Dazla encode proteins containing RNA-binding motifs; in mice, Dazla is expressed in both male and female germ cells. Targeted disruption of Dazla results in male and female infertility due to an early loss of germ cells.40 Interestingly, heterozygotes exhibited reduced numbers of sperm with a high percentage of morphologic abnormalities, suggesting a quantitative requirement for the Dazla protein. Fertility impairment caused by Dazla mutation in both sexes is in contrast to the male-specific infertility phenotype resulting from mutation of the Drosophila homologue boule. These data support the notion that DAZ participates in spermatogenesis but indicates that, in the mouse, an autosomal DAZ is sufficient for the maintenance of fertility. Recent studies in human populations indicates that a single nucleotide polymorphism (SNP) in exon 3 of the autosomal DAZL gene was more prevalent in a group of non-obstructed azoospermic/oligospermic individuals than in normal fertile controls, suggesting a role for DAZL in human spermatogenesis.56
It has long been known that vitamin A is required for normal testicular function.57 The identification of nuclear receptors for metabolites of this vitamin, namely all-trans (and nine-cis) retinoic acid, has allowed examination of the molecular mechanisms of retinoid action to begin. A report by Kastner and associates,47 indicating a male-sterility phenotype in mice with a mutation of retinoid X receptor b (Rxr b), provides new insight into the role of retinoids in spermatogenesis. It must be noted, however, that there is significant (approximately 50%) in utero or perinatal lethality associated with the homozygous phenotype; thus the biological role of Rxr b seems more pleiotropic than that evidenced by other genes discussed in this chapter. However, it is of interest for two reasons:
- The authors suggested that the defect is in the Sertoli cells, based on the normal expression pattern of Rxr b (unique to Sertoli cells) and the histology of the mutant testis. Homozygotes were characterized by early and progressive accumulation of lipids in the Sertoli cells, which preceded a failure of spermatid maturation and release. It is of note that accumulation of lipids has been seen in the Sertoli cells of some infertile men.58
- Although the redundancy of the retinoid receptor pathway has been well established,59 Rxr b seems uniquely qualified to mediate the normal functioning of spermiogenesis. Taken together, the data from this mutant population of mice suggest that Rxr b may play a role in the pathology of some types of male infertility.
The integrity of meiosis is maintained, in part, by the action of genes that correct replicative mismatches and that ensure appropriate repair and recombination of homologous chromosomes. The relative contributions of the mouse homologues of DNA mismatch repair genes PMS-2 and MLH-1 to the maintenance of normal spermatogenesis have been assessed by the use of targeted mutations in each of these genes.48, 49 Male mice defective in Pms2 exhibit a marked decrease in sperm production, with grossly abnormal morphology. Examination of seminiferous tubules revealed a disruption in spermatogenesis beginning at the primary spermatocyte stage, coincident with meiosis I. Ultrastructural examination of mutant germ cells revealed abnormalities in chromosome synapsis. No meiotic abnormalities were observed in female mice, but both male and female homozygotes displayed an increased propensity for tumors in a number of somatic cell types. The data suggest a specific role for Pms2 in maintenance of the integrity of male meiosis; however, because of the increased incidence of tumorigenesis in these animals, the relative contribution of this type of mutation to human infertility is unknown.
A similar phenotype of sterility is seen in Mlh1-deficient mice, although the apparent cause of this failure is different. Examination of these males revealed an absence of spermatozoa in the epididymides, with testes that were half the normal size compared to those of normal littermates. Normal spermatogonia as well as Sertoli and Leydig cells were apparent, with no spermatocytes beyond the pachytene stage. Further examination revealed meiotic arrest after synapsis, but before chiasma formation. Thus, these mice failed to produce spermatids and spermatozoa and had an increased incidence of spermatocyte apoptosis. Unlike the Pms2 knockout, both male and female Mlh1-deficient mice are sterile. Because Mlh1 is not specifically required for spermatogenesis, but rather is required for the maintenance of normal meiosis, its role in human male infertility may be restricted to a small percentage of the population.
An autosomal gene whose involvement in the process of spermiogenesis has been postulated based on mutant mice generated by homologous recombination is cyclic AMP-responsive element modulator (Crem). Crem is a transcriptional activator expressed in postmeiotic germ cells. Nantel and colleagues50 reported that mice heterozygous for the disrupted Crem exhibited reduced fertility, with decreased testicular weight, a 46% reduction in the number of spermatozoa, and a twofold increase in the number of spermatozoa with aberrant structures. Thus, mutation in one copy of this gene is deleterious to spermatogenesis in the mouse. Homozygous mutant males lacked spermatozoa in their seminal fluid; histologic analysis of their seminiferous tubules indicated that spermatogenesis is interrupted at the very early spermatid stage, with apparently normal Sertoli and Leydig cells. A tenfold increase in apoptosis in Crem-deficient mice was observed, suggesting that lack of Crem causes germ cells to stop differentiating and to undergo apoptosis. Homozygous females were completely normal, and no other defect in male Crem-deficient mice was reported. An activator of CREM in the testis, the gene ACT, also appears important for normal fertility; disruption of this gene in mice results in severely reduced sperm counts and abnormalities in sperm morphology.60 Subsequent studies analyzing ACT gene structure in infertile males has suggested that different ACT haplotypes interacting with CREM may influence human male subfertility.61 Thus CREM and the proteins that modulate its function may participate in the regulation of human spermatogenesis.
The ubiquitin system is believed to play an important role in the stabilization, refolding, and translocation of proteins; it participates in many cellular processes, including stress response, cell cycle regulation, DNA repair, and gene expression.62 RAD6 is a yeast ubiquitin conjugating DNA repair enzyme believed to act by influencing chromatin structure. Human (HR6 A and B) and mouse (mHR6 A and B) homologues of RAD6 have been identified, and the function of one of these homologues has been assayed by the generation of mice mutant for mHR6B.51 The phenotype of these mutants is that of male sterility, with no other obvious defects in male or female mice. The defect is due to failure of progression through the elongation and condensation steps of spermatid development, with secondary defects in Sertoli cell morphology. A wide range of gross morphologic irregularities were observed in tubules that had a few spermatozoa, reminiscent of the histology evident in some types of human infertility.63
It is apparent that in animal models of infertility, autosomal genes play an important role in the regulation of spermatogenesis. A number of autosomal genes acting at many different stages of spermatogenesis have been identified; as more autosomal genes are eliminated by targeted mutation, it is likely that more will be found that are indispensable to male fertility. What, then, is the relative contribution of these genes to human infertility? Most of the above phenotypes are apparent only in the homozygous condition, suggesting that they will be responsible for fewer cases of infertility compared to the Y chromosome, which is present in the essentially heterozygous condition. However, because many females homozygous for these genes are fertile, they can act as carriers for these mutations, transmitting the defect to their offspring. It is clear that much more information must be obtained before the relative contribution of mutations in autosomal genes to human male infertility can be assessed.
SUMMARY
Recent advances in our understanding of the molecular mechanisms that regulate spermatogenesis may provide new insight into cases of idiopathic infertility. Although deletions in the Y chromosome are likely responsible for a significant percentage of azoospermia and severe oligospermia, a direct relationship between deletions in specific Y chromosome genes and the presenting phenotype has yet to be determined. In addition, much remains to be uncovered about the contribution of autosomal genes to this disease. The current technological ability to achieve pregnancy in azoospermic and severely oligospermic patients mandates an understanding of the defects involved so that appropriate information may be made available as to the likelihood of transmission of these defects to the offspring.
REFERENCES
Clermont Y: The cycle of the seminiferous epithelium in man. Am J Anat 112: 35, 1986 |
|
World Health Organization: Laboratory Manual for the Examination of Human Semen and Semen-Cervical Mucus Interactions, 2nd ed. New York, Cambridge University Press, 1992 |
|
Sigman M, Lipshultz L, Howards S: Evaluation of the subfertile male. In Lipshultz L, Howards S (eds): Infertility in the Male, pp 173–193. St. Louis, Mosby, 1997 |
|
Levin HS: Testicular biopsy in the study of male infertilty: Its current usefulness, histologic techniques and prospects for the future. Hum Pathol 10: 569, 1979 |
|
Meinhardt E, McRae CU, Chisholm GD: Testicular biopsy in the evaluation of male infertility. Br Med J 3: 577, 1973 |
|
Silber S: Surgical management of male-factor infertility. In Seibel M (ed): Infertility: A Comprehensive Text, pp 371–386. Stamford, CT, Appleton and Lange, 1997 |
|
Thomas A, Howards S: Microsurgical treatment of male infertility. In Lipshultz L, Howards S (eds): Infertility in the Male. St. Louis, Mosby, 1977 |
|
Southwick GJ, Temple-Smith PD: Epididymal microsurgery: Current techniques and new horizons. Microsurgery 9: 266, 1988 |
|
Palermo G, Joris H, Devroey P et al: Pregnancies after intracytoplasmic injection of single spermatozoon into an oocyte. Lancet 340: 17, 1992 |
|
Jacobs PA, Strong JA: A case of human intersexuality having a possible XXY sex-determining mechanism. Nature 183: 302, 1959 |
|
Ford CE, Jones KW, Polani PE et al: A sex-chromosome anomaly in a case of gonadal dysgenesis. Lancet 711, 1959 |
|
McLaren A: The making of male mice. Nature 351: 96, 1991 |
|
Vergnaud G, Page DC, Simmler MC et al: A deletion map of the human Y chromosome based on DNA hybridization. Am J Hum Genet 38: 109, 1986 |
|
Muller U, Doulon T, Schmid M et al: Deletion mapping of the testis determining locus with DNA proves in 46,XX males and 46,XY and 46,X,dic(Y) females. Nucleic Acids Res 14: 6489, 1986 |
|
Disteche CM, Casanova M, Saal H et al: Small deletions of the short arm of the Y chromosome in 46, XY females. Proc Natl Acad Sci USA 83: 7841, 1986 |
|
Rary JM, Cummings DK, Jones HW Jr, Rock JA: Assignment of the H-Y antigen gene to the short arm of chromosome Y. J Heredity 70: 78, 1979 |
|
Agulnik A, Mitchell M, Lerner JL et al: A mouse Y chromosome gene encoded by a region essential for spermatogenesis and expression of male-specific minor histocompatibility antigens. Hum Mol Gen 3: 873, 1994 |
|
Scott DM, Ehrmann IE, Ellis PS et al: Identification of a mouse male specific transplantation antigen H-Y. Nature 376: 695, 1995 |
|
Wang W, Meadows L, denHaan JM et al: Human H-Y: A male specific histocompatibility antigen derived from the SMCY protein. Science 269: 1558, 1995 |
|
Goodfellow PJ, Darling SM, Thomas NS, Goodfellow PN: A pseudoautosomal gene in man. Science 234: 740, 1986 |
|
O'Reilly AJ, Affara NA, Simpson E et al: A molecular deletion map of the Y chromosome long arm defining X and autosomal homologous regions and the localisation of the HYA locus to the proximal region of the Yq euchromatin. Hum Mol Genet 1: 379, 1992 |
|
Cantrell A, Bogan JS, Simpson E et al: Deletion mapping of H-Y antigen to the long arm of the human Y chromosome. Genomics 13: 1255, 1992 |
|
Page DC, Mosher R, Simpson E et al: The sex determining region of the human Y chromosome encodes a zinc finger protein. Cell 51: 1091, 1987 |
|
Palmer MS, Sinclair AH, Berta P: Genetic evidence that ZFY in not the testis determining factor. Nature 342: 937, 1989 |
|
Sinclair AH, Berta P, Palmer MS et al: A gene from the human sex determining region encodes a protein with homology to a conserved DNA binding motif. Nature 346: 240, 1990 |
|
Berta P, Hawkins JR, Sinclair AH et al: Genetic evidence equating SRY as the testis determining factor. Nature 348: 448, 1990 |
|
Koopman T, Gubbay J, Vivian N et al: Male development of chromosomally female mice transgenic for SRY. Nature 351: 117, 1991 |
|
Tiepolo L, Zuffardi O: Localization of factors controlling spermatogenesis in the nonfluorescent portion of the human Y chromosome long arm. Hum Genet 34: 119, 1976 |
|
Hennig W: Y chromosome function and spermatogenesis in Drosophila hydei. Adv Genet 23: 179, 1985 |
|
Burgoyne PS, Levy ER, McLean A: Spermatogenic failure in male mice lacking HY antigen. Nature 320: 170, 1986 |
|
Ma K, Sharkey A, Kirsch S et al: Towards the molecular localisation of the AZF locus: Mapping of microdeletions in azoospermic men within 14 subintervals of interval 6 of the human Y chromosome. Hum Mol Genet 1: 29, 1992 |
|
Vogt PH, Chandley AC, Hargreave TB et al: Microdeletions in interval 6 of the Y chromosome of males with idiopathic sterility point to disruption of AZF, a human spermatogenesis gene. Hum Genet 89: 491, 1992 |
|
Vogt PH, Edelmann A, Kirsch S et al: Human Y chromosome azoospermia factors (AZF) mapped to different subregions in YQ11. Hum Mol Genet 5: 933, 1996 |
|
Ma K, Inglis JD, Sharkey A et al: A Y chromosome gene family with RNA-binding protein homology: Candidates for the azoospermia factor AZF controlling human spermatogenesis. Cell 75: 1287, 1993 |
|
Reijo R, Lee TY, Salo P et al: Diverse spermatogenic defects in humans caused by Y chromosome deletions encompassing a novel DNA-binding protein gene. Nature 10: 383, 1995 |
|
Eberhart CG, Maines JZ, Wasserman SA: Meiotic cell cycle requirement for a fly homologue of human deleted in azoospermia. Nature 381: 783, 1996 |
|
Saxena R, Brown LG, Hawkins T et al: The DAZ gene cluster on the human Y chromosome arose from an autosomal gene that was transposed repeatedly amplified and pruned. Nature Genet 14: 292, 1996 |
|
Seboun E, Barbaux S, Bourgeon T et al: Gene sequence, localization and evolutionary conservation of DAZLA, a candidate male sterility gene. Genomics 41: 227, 1997 |
|
Vereb M, Agulnik AI, Houston JT et al: Absence of DAZ gene mutations in cases of non-obstructed azoospermia. Mol Hum Reprod 3: 55, 1997 |
|
Ruggiu M, Speed R, Taggart M et al: The mouse Dazla gene encodes a cytoplasmic protein essential for spermatogenesis. Nature 389: 73, 1997 |
|
Jones MH, Furlong RA, Burkin H et al: The Drosophila developmental gene fat facets has a human homologue in Xp11. 4 which escapes X inactivation and has related sequences on Yq11.2. Hum Mol Genet 5: 1695, 1996 |
|
Brown GM, Furlong RA, Sargent CA et al: Characterisation of the coding sequence and fine mapping of the human DFFRY gene and comparative expression analysis and mapping to the Sxrb interval of the mouse Y chromosome of the Dffry gene. Hum Mol Genet 7: 97, 1998 |
|
Lahn BT, Page D: Functional coherence of the human Y chromosome. Science 278: 1997 |
|
Foresta C, Ferlin A, Garolla A et al: Y chromosome deletions in idiopathic severe testiculopathies. J Clin Endocrinol Metab 82: 1075, 1997 |
|
Beamer W, Cunliffe-Beamer TL, Shultz KL et al: Juvenile spermatogonial depletion (jsd): A genetic defect of germ cell proliferation of male mice. Biol Reprod 38: 899, 1988 |
|
Zhao GQ, Deng K, Labosky P et al: The gene encoding bone morphogenetic protein 8B is required for the initiation and maintenance of spermatogenesis in the mouse. Genes Devel 10: 1657, 1996 |
|
Kastner P, Mark M, Gansmuller A et al: Abnormal spermatogenesis in RXR b mutant mice. Genes Devel 10: 80, 1996 |
|
Baker SM, Bronner E, Zhang L et al: Male mice defective in the DNA mismatch repair gene PMS2 exhibit abnormal chromosome synapsis in meiosis. Cell 82: 309, 1995 |
|
Edelman W, Cohen PE, Kane M et al: Meiotic pachytene arrest in MLH1-deficient mice. Cell 85: 1125, 1995 |
|
Nantel F, Monaco L, Foulkes NS, Masquiller D et al: Spermiogenesis deficiency and germ-cell apoptosis in CREM mutant mice. Nature 380: 159, 1996 |
|
Roest HP, van Klaveren J, deWit J et al: Inactivation of the HR6B ubiquitin-conjugating DNA repair enzyme in mice causes male sterility associated with chromatin modification. Cell 86: 799, 1996 |
|
Rohozinski J, Bishop CE: The mouse juvenile spermatogonial depletion (jsd) phenotype is due to a mutation in the X-derived retrogene, mUtp14b. Proc Natl Acad Sci U S A. 2004 Aug 10;101(32):11695-700. Epub 2004 Aug 2. |
|
Bradley J, Baltus A, Skaletsky H et al: An X-to-autosome retrogene is required for spermatogenesis in mice. Nat Genet. 2004 Aug;36(8):872-6. Epub 2004 Jul 18. |
|
Rohozinski J, Lamb DJ, Bishop CE: UTP14c is a recently acquired retrogene associated with spermatogenesis andfertility in man. Biol Reprod. 2006 Apr;74(4):644-51. Epub 2005 Dec 14. |
|
Zhao GQ, Hogan BL. Evidence that mouse Bmp8a (Op2) and Bmp8b are duplicated genes that play a role in spermatogenesis and placental development. Mech Dev. 1996 Jul;57(2):159-68. |
|
Teng YN, Lin YM, Lin YH et al: Association of a single-nucleotide polymorphism of the J Clin Endocrinol Metab. 2002 Nov;87(11):5258-64. |
|
Eskild W, Hansson V: Vitamin A functions in the reproductive organs. In Blomoff R (ed): Vitamin A in Health and Disease. New York, Marcel Dekker, 1994 |
|
Johnson AD: Testicular lipids. In Johnson AD, Gomes WR, Vandemark NL (eds): The Testis. New York, Academic Press, 1970 |
|
Leid M, Kastner P, Chambon P: Multiplicity generates diversity in the retinoic acid signalling pathways. Trends Biochem Sci 17: 427, 1992 |
|
Kotaja N, De Cesare D, Macho B et al: Abnormal sperm in mice with targeted deletion of the act (activator of cAMP-responsive element modulator in testis) gene. Proc Natl Acad Sci U S A. 2004 Jul 20;101(29):10620-5. Epub 2004 Jul 9. |
|
Christensen GL, Wooding SP, Ivanov IP et al: Sequencing and haplotype analysis of the activator of CREM in the testis (ACT) gene in populations of fertile and infertile males. Mol Hum Reprod. 2006 Apr;12(4):257-62. |
|
Hoechstrasser M: Ubiquitin, proteasomes and the regulation of intracellular protein degradation. Curr Opin Cell Biol 7: 215, 1995 |
|
Aitken RJ, Baker HWG, Irvine DS: On the nature of semen quality and infertility. Hum Reprod 10: 248, 1995 |