Genetics of Sexual Differentiation
Authors
INTRODUCTION
Sexual determination in mammals is genetically and hormonally controlled.1, 2 The role of hormones in gonadal differentiation was identified initially through organ transplantation or ablation of endogenous structures in mammalian embryos and subsequently through analysis of humans with hormone biosynthetic or receptor defects.3 The role of genes has been identified through genetic analysis of individuals with aberrant gonadal differentiation.4 These genes have been cloned and analyzed for their functions, interactions, and temporal expression in sex determination.5, 6 In this chapter, the conceptual framework for sexual differentiation and the major bits of experimental evidence that underlie this framework are reviewed.
GONADAL PHYSIOLOGY AND ANATOMY
Human gonads serve several functions. Gonads are the repository of germ cells and influence the maturation of these cells into oocytes and spermatocytes. Gonads produce the hormones that control the development of the internal and external genitalia and that regulate pubertal development and the reproductive cycle, including sperm maturation in the male, and ovulation and menstruation in the female. These functions are mediated by the specialized cells of the gonad that include germ cells, support cells, and hormone-producing cells.
The germ cells of the differentiated gonads reside in compartments that place them in functional contact with support cells. Through these functional contacts, support cells provide for the maintenance and maturation of the germ cells. In the seminiferous tubules of the testes, Sertoli cells form functional contacts with sperm cell precursors. Tight junctions between Sertoli cells provide a barrier from many substances that circulate in the blood, which must be transported through the Sertoli cells to the germ cells. Multiple support cells in the follicles of the ovary form contacts with the oocytes. The nonvascular, epithelial granulosa cells form cell-to-cell communication with the granulosa cells via gap junctions. The vascular, stromal, thecal cells also play an important role in hormone production.
The different gonadal cells produce functionally important hormones. The Leydig cells in the testes produce testosterone and the Sertoli cells produce inhibin. The theca cells in the ovary produce testosterone, which is converted to estradiol by the enzyme aromatase in the granulosa cells. After ovulation, the granulosa and theca cells are converted into a corpus luteum that secretes estradiol and progesterone.
GONADAL EMBRYOLOGY
The human, early embryonic gonad has the potential to differentiate either as a testis or an ovary (Fig. 1). This potential is intrinsic to the gonad and is unaffected by transplantation into an embryo of the opposite sex. The early mammalian gonad differentiates along the distal margin of the mesonephros. The cells in the gonadal ridge originate in the mesonephros and the coelomic epithelium. By 40–50 days of gestation, the gonadal ridge is committed to form a testis.7, 8 The tight packing of germ cells and somatic (early Sertoli) cells to form seminiferous cords is the first morphologic sign of testis determination.
|
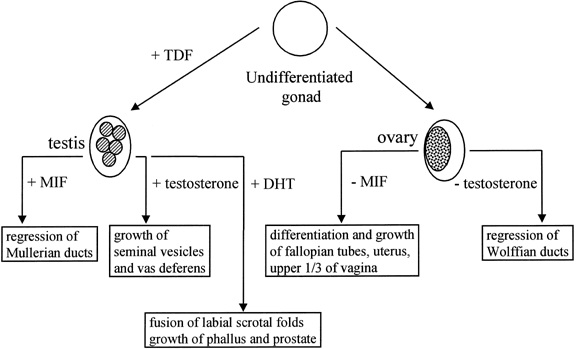
The gonadal ridge in female embryos remains undifferentiated until germ cells have invaded. The invasion occurs at 6–9 weeks of development, although ovarian follicles are not seen until 13 weeks.9, 10 The first sign of ovarian development is the appearance of germ cells entering meiosis, thus initiating oogenesis. The oogonia are enclosed by granulosa cells and a basal lamina, forming a primitive follicle. If a follicle is not formed, the oogonia degenerate. The absence of germ cell migration in male embryos leads to the formation of seminiferous tubules without spermatogonia, the “Sertoli-only syndrome”.
Around 5 weeks of development, human germ cells originate in the primitive streak and then migrate along the posterior mesentery into the gonad.11, 12 The cells are bipotential for their development regardless of their genetic constitution. The gonads of chimeric mice formed by the aggregation of early XX and XY embryos have spermatogonia with either an XX or an XY chromosomal constitution.13 Through their contact with Sertoli cells, the germ cells differentiate into spermatogonia. In the absence of contact with Sertoli cells, the bipotential germ cells differentiate into oogonia. Undifferentiated male germ cells that migrate or are transplanted into ectopic locations, such as under the capsule of the kidney, differentiate as oogonia.14, 15 A follicle is not formed in these ectopic locations and the oogonium regresses.16
The state of differentiation of the germ cells varies between newborn males and females. The spermatogonia in the primitive testis undergo a series of mitotic cell divisions and are then finally arrested in a premeiotic state. At puberty, the process of sperm maturation includes the series of meiotic divisions. The oogonia in the early ovary undergo a series of mitotic divisions and then enter meiosis, where they are arrested in the dictyotene state before the formation of the first polar body. At puberty, oogenesis resumes, going through the series of meiotic divisions and ovulation.
Up to four million germ cells are present in the ovary during embryonic life. The majority regress and are lost; thus, only 20,000 germ cells are present in the gonad at the time of birth.9 This process of germ cell loss continues through the life of the female. Only 400–500 oocytes are ovulated and the remainder are lost through atresia.16
The differentiation of support cells is influenced by the genetic constitution of these cells and the environment in which the differentiation takes place. In XX/XY chimeras, the majority of cells that differentiate into Sertoli cells contains a Y chromosome.13, 17 These studies suggest that the precursors act in response to an internal genetic signal to become Sertoli cells. Leydig cell differentiation is dependent on the formation of the Sertoli cells and seminiferous tubules, although the specific cellular signal is not known. In the ovary, the differentiation of granulosa and theca cells is dependent on the presence of oogonia.
GENETICS OF TESTIS DETERMINATION
The observation of dimorphic sex chromosomes in 1905 led to the idea that a gene on the Y chromosome was testis determining.18 In humans, this was not confirmed until methods for cytogenetic analysis improved in the 1950s. In that era, the presence of a Y chromosome was shown to be male sex-determining, regardless of the number of X chromosomes present, and the absence of a Y chromosome was shown to be female sex-determining.19, 20 Amid these observations, a number of individuals were observed who had genetic disorders of sexual development (DSD), genetic sex of one type and a phenotypic sex of the opposite type. These individuals were classified either as 46,XX testicular DSD (males) or 46,XY complete gonadal dysgenesis (females); the latter occurred because their gonads regressed after an initial period of differentiation. A model for explaining genetic sex reversal was proposed in 1965 in which nonhomologous recombination between X and Y chromosomes translocated the testis-determining factor from the Y onto the X chromosome.21 This hypothesis was proved correct for many cases of 46,XX testicular DSD and was instrumental for the cloning of the Y-linked testis-determining factor (Fig. 2A). Still, not all cases of sex reversal could be explained by this observation, and the study of individuals with other forms of genetic sex reversal has been important for identifying autosomal- and X-linked sex-determining genes (see Fig. 2B).
|
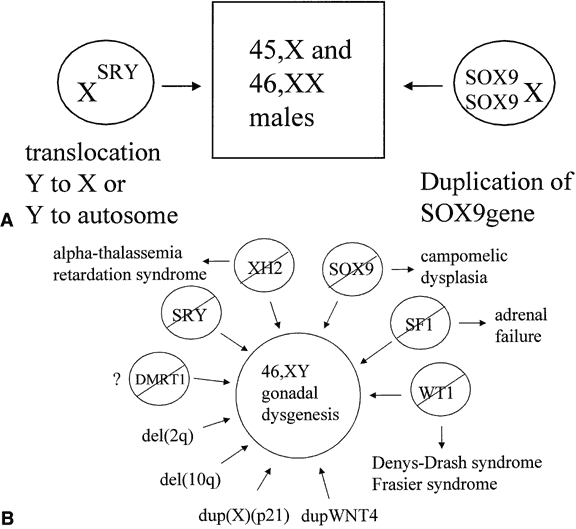
From the 1970s on, a number of candidate genes for the testis-determining factor, including HY antigen, bkm, and ZFY, were proposed and then subsequently excluded.22, 23, 24 In 1990, the gene called sex-determining region Y (SRY in humans, Sry in mice) was shown to be the testis-determining factor on the Y chromosome.5, 6 This gene was found on the Y chromosomes of most other eutherian mammals in which it was tested (e.g. chimp, rabbit, pig, horse, cow, and tiger). It was also found in all human and mouse XX males who have this phenotype on the basis of nonhomologous recombination between the X and Y chromosomes. Deletion of the SRY/Sry gene prevents testes differentiation from occurring.5, 25 The timing of SRY/Sry expression in the gonadal ridges of the humans and mice was as expected (i.e. at the induction of testicular differentiation).26, 27 The human and mouse SRY/Sry genes are not functionally equivalent. XX mice transgenic for an intact Sry gene have normal testis differentiation, whereas XX mice transgenic for an intact human SRY gene do not.28
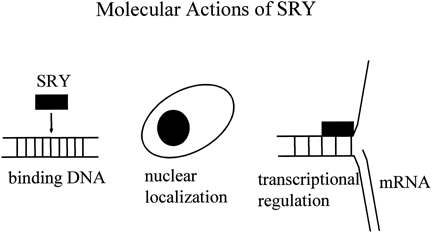
The SRY proteins from all mammalian species contain an HMG box with a nuclear localization signal, suggesting that the proteins might act as nuclear transcription factors.29 The HMG boxes of the SRY proteins from various species all bind DNA at a similar consensus sequence (AATAAC) and bend DNA to a varying degree (Fig. 3).30 The structures of the SRY proteins, even among closely related species of mice and primates, are very different outside the HMG box.31, 32 The mouse (M. musculus) Sry gene encodes glutamine/histidine-rich repeat 223 amino acid residues in length at the N-terminus of the protein. This is highly polymorphic in different mouse species and shortened in nonmusculus species. The human SRY gene does not contain a glutamine/histidine-rich region. The mouse Sry protein with the long glutamine-rich region functions as a transcriptional activator.33 Shortening of the glutamine-rich domain prevents transcriptional activation from occurring both in vitro and in vivo in transgenic mice.34, 35 There is no evidence that SRY functions as a transcriptional activator in other species. Rather, it might function as an architectural factor that binds other transcriptional regulators. The identification of SIP1, a human protein with two PDZ-protein-binding domains that can interact with the C-terminal seven amino acids of SRY, suggests a mechanism by which this can happen.36
|

A number of autosomal genes have been cloned that play a role in testis determination (see Fig. 2B). The first to be identified was the Wilms' tumor suppressor, WT1.37, 38 This gene encodes a transcription factor with zinc-finger domains that bind to specific DNA sequences. The gene is expressed both in the undifferentiated gonadal ridge and in the sex cords after testicular determination in human embryos, suggesting that it has roles both in early gonadal development and auxiliary effects in the testis pathway.39 Many different isoforms are produced through alternate splicing of the WT1 RNA. Deletion of WT1 from the germline is associated with Wilms' and other tumors, especially gonadoblastoma, and with renal and gonadal anomalies.40, 41 Germline mutation varies the efficiency of splicing WT1 RNA, thus yielding different ratios of the WT1 isoform that lead to sex reversal rather than normal gonadal differentiation.42, 43
SOX9 (SRY-like HMG-box protein 9) was the second autosomal, sex-determining gene to be identified.44 Mutations in this gene are associated with campomelic dysplasia, a skeletal malformation syndrome in which the 46,XY individuals commonly have sex reversal.45 SOX9 encodes a protein with two activation domains, suggesting that it acts by upregulation of other genes.46 The expression of SOX9 in the gonads of 46,XY human embryos follows a pattern similar to that of SRY.26 The expression commences with testicular induction and increases over the next several days with maximal detection observed over the sex cords, most likely in Sertoli cells. No expression in the gonads of early 46,XX female embryos, is consistent with a primary role for SOX9 in testicular determination; however, low levels of SOX9 transcripts in the embryonic rete ovarii are detected at a later time. Several bits of genetic evidence suggest that all of the effects of SRY in causing male sexual differentiation may channel through SOX9. An individual with 46,XX testicular DSD with a chromosomal duplication encompassing the SOX9 gene suggests that enhanced expression of SOX9, even in the absence of SRY, is sufficient to cause testis determination (see Fig. 2A).47 Similarly, derepression of SOX9 expression in XX gonads leads to male development in Odsex mice.48 Ordinarily, this derepression might be mediated by Sry.
Steroidogenic factor-1 (SF1), a transcription factor that regulates the expression of a number of genes involved in steroid hormone production and male sexual differentiation, falls into the class of so-called orphan nuclear receptors.49, 50 In addition to a zinc-finger DNA binding domain, the gene encodes a receptor domain for an as-yet-unidentified ligand. Human SF1 is expressed in the adrenal gland and the gonadal ridge before testicular differentiation. The expression in both tissues continues after the period of testicular differentiation.39 Testicular expression is observed both in Leydig and Sertoli cells, suggesting a role both in testis differentiation and in hormone production. Mutations in SF1 have been identified in 13% of cases of 46,XY pure gonadal dysgenesis.51 Mutations in the SF1 gene have also been associated with adrenal hypoplasia congenita.52
Homozygous mutations in DHH, a signalling molecule that plays a role in morphogenesis, have been identified in up to 20% of cases of 46,XY (partial) DSD and heterozygous mutations have been identified in up to 50% of cases of 46,XY complete gonadal dysgenesis.53
Homozygous disruption of the human R-spondin1 (RSPO1) gene causes 46,XX testicular DSD in the absence of the SRY gene, as well as palmoplantar hyperkeratosis and predisposition to squamous cell carcinoma of the skin. The R-spondins are a family of small growth factors. This finding demonstrates that the SRY gene is not necessary for testicular development.54
At least two X chromosomal loci have been implicated to play a role in gonadal development. When mutated, XH2 results in the alpha-thalassemia, mental retardation syndrome.55, 56 Some 46,XY individuals have gonadal dysgenesis as part of their phenotype, suggesting that XH2 plays a role in testicular development. This gene encodes a helicase that unwinds DNA, making it accessible to transcription factors. The dosage-sensitive site has apparent anti-testis properties, when overexpressed, but is not essential for testis determination. This region was identified in individuals with 46,XY gonadal dysgenesis who had duplications of the X chromosome in the region of band Xp21 and is not subject to X chromosome inactivation in these individuals with partial duplication.57, 58 Deletion of a gene within this region has been associated with adrenal hypoplasia congenita. As a result, the gene has been called DAX1 (dosage-sensitive sex reversal, adrenal hypoplasia congenita, and X chromosome).59 DAX1 encodes an orphan nuclear receptor with zinc fingers for DNA binding and a domain for binding an unknown ligand. Low levels of DAX1 expression are observed before and after testicular induction in humans, most notably over the sex cords after the Sertoli cells have differentiated.39 As a result, the anti-testis properties of DAX1 overexpression might act before or after the expression of SRY.
WNT4 may also function as a dosage-sensitive, sex-determining gene. Targeted, homozygous deletion of this gene results in masculinization of XX mice. These mice have a marked reduction in the number of oocytes in their ovaries at birth,60 with the remaining oocytes being observed in the process of degeneration. A few sex-cordlike structures have been observed in these ovaries and the supporting cells in these sex cords express the Sertoli cell markers, Mis and Dhh. In addition, Leydig cell differentiation is not suppressed and testosterone biosynthesis occurs. Inactivation of Wnt4 and Foxl2 causes testis differentiation in XX mice, resulting in the formation of testis tubules and spermatogonia.61 Mutations in Wnt4 have extragonadal effects on sexual differentiation. Homozygous mutant mice have regress of their Mullerian ducts and masculinization of their Wolffian ducts. Overexpression of WNT4 was observed in the lymphocytes of a 46,XY female with gonadal dysgenesis whose chromosomal constitution included a 1p31-p35 duplication.62 This duplicated segment included the WNT4 gene. In transfected cells, WNT4 upregulated DAX1, suggesting a common mechanism for the two forms of dosage-sensitive sex reversal.
Several testis-determining genes have been identified in 46,XY knockout mice with gonadal dysgenesis. These include fibroblast growth factor 9 (Fgf9) and Dmrt1. Homozygous knockout of the Fgf9 gene results in lung hypoplasia and, in XY embryos, a variety of gonadal phenotypes, ranging from gonadal dysgenesis to testicular hypoplasia.63 To date, mutations in the Fgf9 gene have not been described in humans with sex reversal.
The Dmrt family of genes contains a zinc finger-like DNA binding domain homologous to the sex-determining genes in C. elegans (Mab-3) and Drosophila (dsx). Both invertebrate proteins are functionally related because they are required for differentiation of male-specific sense organs, regulation of yolk protein transcription, and normal male mating behavior.64 They show expression exclusively in gonadal ridge and early ridge that is enhanced in males.65, 66 Homozygous knockout of the mouse Dmrt1 gene causes severe testicular hypoplasia in XY mice, a phenotype similar to that observed in humans with a deletion of the short arm of chromosome 9.67, 68 However, chromosome 9 contains three DMRT genes. Deletion mapping and mutation analysis have been unable to distinguish which of these genes is essential for the testicular development.69
The products of several testis-determining genes interact to regulate MIS, a downstream target of sexual differentiation (Fig. 4). SOX9 binds to a high-affinity site in the MIS promoter and initiates transcription.70 If the high-affinity site is eliminated, no transcription is initiated. Other factors binding to one another and to the MIS promoter act as quantitative regulators of MIS transcription. SF1 binds to the promoter and to SOX9 to increase transcription.71 Elimination of the SF1 binding site reduces the levels of Mis transcripts in mice, but to a level of expression that is still sufficient to cause complete Mullerian duct regression.70 An isoform of WT1 associates with SF1 to form a heterodimer that increases MIS expression.72 WT1 missense mutations, associated with 46,XY gonadal dysgenesis, do not synergize with SF1. DAX1 antagonizes the synergy between SF1 and WT1, most likely through a direct interaction with SF1.72, 73, 74, 75 These findings suggest that WT1 and DAX1 may also functionally oppose each other during testis development by modulating SF1-mediated transactivation.
|
WT1 also plays a role in regulating the expression of SRY.76 WT1 is known to be temporally expressed in the gonadal ridge before as the expression of SRY—as might be expected for a transcriptional regulator. This induction of SRY expression occurs through a proximal EGR1-like DNA-binding sequence in the core promoter of the SRY gene.
GENETICS OF OVARIAN DEVELOPMENT
The identification of 45,X monosomy in the 1950s as the genetic basis for Turner syndrome provided a beginning for understanding the genetic control of ovarian development.20 In Turner syndrome, pure gonadal dysgenesis occurs because differentiation along the ovarian pathway is initiated but not completed.7 During normal oocyte maturation, X chromosome reactivation occurs and genes from both X chromosomes are expressed, but in 45,X monosomy, the level of expression of these genes is reduced.77, 78 The locations of these X-linked genes are suggested by nonmosaic structural variants of the X chromosome that have been observed in individuals with gonadal dysgenesis.79 The most common structural variant associated with this phenotype is an isodicentric chromosome of the long arm of X.80 Here, the long arm, the centromere, and part of the short arm are duplicated and the remainder of the short arm is deleted. Because genes on the short arm are necessary for viability, the isodicentric chromosome is preferentially inactivated in somatic cells, but it can be reactivated in oocytes. When X chromosome reactivation occurs in oocytes, suboptimal dosage for genes on the short arm of the X chromosome may lead to oocyte regression and gonadal dysgenesis.
Studies of individuals with deletions of either the short arm or the long arm of the X chromosome provide a further indication of where these dosage-sensitive genes reside.79, 81, 82 The phenotypes associated with these deletions fall into two categories: gonadal dysgenesis and secondary amenorrhea. Deletions at the same site on the X chromosome in different individuals may be associated with either gonadal dysgenesis or with secondary amenorrhea. These differences in phenotype reflect whether regression of all of the ovarian follicles occurred before puberty or after. This seeming paradox of identical genetic events being associated with phenotypes of varying severity can be explained either by the presence of different genes at these locations (hence the deletions are not identical), by the presence of different alleles at these sites, or by differences in modifiers of these genes.
EVIDENCE FOR OTHER GENES IN THE SEX-DETERMINING PATHWAYS
Research to date has provided a conceptual framework for understanding normal and abnormal human sexual development and its abnormalities; however, many other genes are likely to play a role. Clues for the identity of these genes have emerged from a variety of sources. In some familial cases of 46,XY gonadal dysgenesis, mutations in the SRY genes of the transmitting fathers suggest that factors that interact with the SRY protein may modulate the phenotype to be either normal or abnormal.83 Cases of familial sex reversal do not show linkage to known sex-determining genes and are likely to be linked to novel genes.4 One of these genes has been mapped via linkage analysis to the long arm of chromosome 5.84 The existence of other genes in the sex-determining pathway has been inferred from the occurrence of multiple sporadic cases of 46,XY mixed or pure gonadal dysgenesis associated with chromosomal deletions at 2q31 and 10q25.85, 86
Based on the gonadal phenotypes observed in mice with homozygous knockout mutations, several genes have been identified that play a role in early gonadal development. Emx2 is a homeobox gene homologous to the Drosophila head gap gene, ems. Emx2 appears to be necessary for development of the epithelial components of the developing urogenital system.33 Mice homozygous for an Emx2 knockout mutation have disruption of the dorsal telencephalon and loss of the kidneys, ureters, gonads, and genital tracts. The M33 gene is a homolog of the Drosophila polycomb genes that maintains the repressed state of homeotic and other developmentally regulated genes by mediating changes in higher order structure. In M33 knockout mice, half of the embryos die. The formation of gonadal ridge is retarded in surviving embryos and the XY mice demonstrated sex reversal.87
Another means for looking for male-female differences in gene expression in the developing gonad became a possibility with the advent of high-density microarrays of cDNA or oligonucleotides on silicon chips. Using this approach, the Nexin-1 and Vanin-1 genes showed sexually dimorphic expression in the gonads before testicular differentiation. The sexually dimorphic expression was confirmed using in situ hybridization. None of these findings have been linked to genetic studies to learn whether expression of either gene is necessary for testicular differentiation.
SUMMARY
This chapter has presented an overview of the current understanding about male and female sex determination in humans. Compared to our understanding about gonadal development in the experimental animals, Caenorhabditis elegans and Drosophila, this knowledge is still very rudimentary, but the suggestion of developmental pathways is starting to emerge.
Several features of the male sex-determining pathway are apparent. In males, testicular differentiation is switched on by expression of the Y-linked SRY gene. This gene is neither necessary nor sufficient, and its effects may be substituted by genes downstream. Among these genes is SOX9, which has structural similarity to SRY and, when overexpressed, may cause testicular differentiation to occur. Overexpression of other genes, including DAX1 and WNT4, may override the normal male sex-determining pathway, producing gonadal dysgenesis. The products of many of these genes may interact to regulate genes involved in the development of extragonadal phenotypes, both reproductive and other. Mutation in these genes is associated with sex reversal, frequently with extragonadal phenotypes.
The timing and events of ovarian development differ markedly from those involved in testicular development. Many of the same genes are expressed in both developing testis and ovaries. Among these, correct expression of WNT4 is known to be critical to ovarian development. Unlike testicular development, germ cell atresia is an important theme in the developing ovary, with the tempo of germ cell atresia being critical for determining the ovarian phenotype.
REFERENCES
Ostrer H: Alterations of sex differentiation in males: from candidate genes to diagnosisand treatments. Curr Pharm Des. 2004;10(5):501-11. |
|
Capel B: The battle of the sexes. Mech Dev 92: 89–103, 2000 |
|
Jost A, Vigier B, Prepin J et al: Studies of sex determination in mammals. Recent Prog Horm Res 29: 1–41, 1973 |
|
Ostrer H: Sex determination: Lessons from families and embryos. Clin Genet 59: 207–215, 2001 |
|
Gubbay J, Collignon J, Koopman P et al: A gene mapping to the sex-determining region of the mouse Y chromosome is a member of a novel family of embryonically expressed genes. Nature 346: 245–250, 1990 |
|
Sinclair AH, Berta P, Palmer MS et al: A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 346: 240–244, 1990 |
|
Singh RP, Carr DH: The anatomy and histology of XO human embryos and fetuses. Anat Rec 155: 369–383, 1966 |
|
Jirasek JE: Morphogenesis of the genital system in the human. Birth Defects 13: 13–39, 1977 |
|
Baker TG: A quantitative and cytological study of germ cells in human primordial oocytes. Proc R Soc Lond [Biol] 158: 417–433, 1963 |
|
Baker TG, Franchi LL: The fine structure of oogonia and oocytes in human ovaries. J Cell Sci 2: 213–224, 1967 |
|
Ginsburg M, Snow MHL, McLaren A: Primordial germ cells in the mouse embryo during gastrulation. Development 110: 521–528, 1990 |
|
Witschi E: Migration of the germ cells of human embryos from the yolk sac to the primitive gonadal folds. Contribution Embryology, Carnegie Institute, Washington 32: 67, 1948 |
|
Burgoyne PS, Buehr M, Koopman P et al: Cell-autonomous action of the testis-determining gene: Sertoli cells are exclusively XY in XX-XY chimaeric mouse testes. Development 102: 443–450, 1988 |
|
Francavilla S, Zamboni L: Differentiation of mouse ectopic germinal cells in intra- and perigonadal locations. J Exp Zool 233: 101–109, 1985 |
|
Upadhyay S, Zamboni L: Ectopic germ cells: Natural model for the study of germ cell sexual differentiation. Proc Natl Acad Sci 79: 6584–6588, 1982 |
|
Motta PM, Makabe S: Elimination of germ cells during differentiation of the human ovary: an electron microscope study. Eur J Obstet Gynecol Reprod Biol 22: 271–286, 1986 |
|
Palmer SJ, Burgoyne PS: In situ analysis of fetal, prepubertal and adult XX-XY chimeric mouse testes: Sertoli cells are predominantly, but not exclusively, XY. Development 112: 265–268, 1991 |
|
Wilson EB: THE CHROMOSOMES IN RELATION TO THE DETERMINATION OF SEX IN INSECTS. Science. 1905 Oct 20;22(564):500-502. |
|
FORD CE, JONES KW, POLANI PE et al: A sex-chromosome anomaly in a case of gonadal dysgenesis (Turner's syndrome). Lancet. 1959 Apr 4;1(7075):711-3. |
|
JACOBS PA, STRONG JA: A case of human intersexuality having a possible XXY sex-determining mechanism. Nature. 1959 Jan 31;183(4657):302-3. |
|
Ferguson-Smith MA: X-Y chromosomal interchange in the aetiology of true hermaphroditism and of XXKlinefelter's syndrome. Lancet. 1966 Aug 27;2(7461):475-6. |
|
Wachtel SS, Ono S, Koo GC et al: Possible role for H--Y antigen in the primary determination of sex. Nature. 1975 Sep 18;257(5523):235-6. |
|
Singh L, Phillips C, Jones KW: The conserved nucleotide sequences of Bkm, which define Sxr in the mouse, aretranscribed. Cell. 1984 Jan;36(1):111-20. |
|
Page DC, Mosher R, Simpson EM et al: The sex-determining region of the human Y chromosome encodes a finger protein. Cell. 1987 Dec 24;51(6):1091-104. |
|
Berta P, Hawkins JR, Sinclair AH et al: Genetic evidence equating SRY and the testis-determining factor. Nature. 1990 Nov 29;348(6300):448-50. |
|
Hanley NA, Hagan DM, Clement-Jones M et al: SRY, SOX9, and DAX1 expression patterns during human sex determination andgonadal development. Mech Dev. 2000 Mar 1;91(1-2):403-7. |
|
Koopman P, Munsterberg A, Capel B et al: Expression of a candidate sex-determining gene during mouse testisdifferentiation. Nature. 1990 Nov 29;348(6300):450-2. |
|
Koopman P, Gubbay J, Vivian N et al: Male development of chromosomally female mice transgenic for Sry. Nature. 1991 May 9;351(6322):117-21. |
|
Dubin RA, Ostrer H: Sry is a transcriptional activator. Mol Endocrinol. 1994 Sep;8(9):1182-92. |
|
Giese K, Pagel J, Grosschedl R: Distinct DNA-binding properties of the high mobility group domain of murine and Proc Natl Acad Sci U S A. 1994 Apr 12;91(8):3368-72. |
|
Whitfield LS, Lovell-Badge R, Goodfellow PN: Rapid sequence evolution of the mammalian sex-determining gene SRY. Nature. 1993 Aug 19;364(6439):713-5. |
|
Tucker PK, Lundrigan BL: Rapid evolution of the sex determining locus in Old World mice and rats. Nature. 1993 Aug 19;364(6439):715-7. |
|
Miyamoto N, Yoshida M, Kuratani S et al: Defects of urogenital development in mice lacking Emx2. Development 124: 1653–1664, 1997 |
|
Dubin RA, Coward P, Lau YF et al: Functional comparison of the Mus musculus molossinus and Mus musculus domesticusSry genes. Mol Endocrinol. 1995 Dec;9(12):1645-54. |
|
Bowles J, Cooper L, Berkman J et al: Sry requires a CAG repeat domain for male sex determination in Mus musculus. Nat Genet. 1999 Aug;22(4):405-8. |
|
Poulat F, de Santa Barbara P, Desclozeaux M et al: The human testis determining factor SRY binds a nuclear factor containing PDZprotein interaction domains. J Biol Chem. 1997 Mar 14;272(11):7167-72. |
|
Gessler M, Poustka A, Cavenee W et al: Homozygous deletion in Wilms tumours of a zinc-finger gene identified bychromosome jumping. Nature. 1990 Feb 22;343(6260):774-8. |
|
Call KM, Glaser T, Ito CY et al: Isolation and characterization of a zinc finger polypeptide gene at the humanchromosome 11 Wilms' tumor locus. Cell. 1990 Feb 9;60(3):509-20. |
|
Hanley NA, Ball SG, Clement-Jones M et al: Expression of steroidogenic factor 1 and Wilms' tumour 1 during early humangonadal development and sex determination. Mech Dev. 1999 Sep;87(1-2):175-80. |
|
Riccardi VM, Sujansky E, Smith AC et al: Chromosomal imbalance in the Aniridia-Wilms' tumor association: 11p interstitialdeletion. Pediatrics. 1978 Apr;61(4):604-10. |
|
Yunis JJ, Ramsay NK: Familial occurrence of the aniridia-Wilms tumor syndrome with deletion J Pediatr. 1980 Jun;96(6):1027-30. |
|
Schedl A, Hastie N: Multiple roles for the Wilms' tumour suppressor gene, WT1 in genitourinarydevelopment. Mol Cell Endocrinol. 1998 May 25;140(1-2):65-9. |
|
Hastie ND: Dominant negative mutations in the Wilms tumour (WT1) gene cause Denys-Drashgenitourinary development. Hum Mol Genet. 1992 Aug;1(5):293-5. |
|
Foster JW, Dominguez-Steglich MA, Guioli S et al: Campomelic dysplasia and autosomal sex reversal caused by mutations in an Nature. 1994 Dec 8;372(6506):525-30. |
|
Hovmoller ML, Osuna A, Eklof O et al: Camptomelic dwarfism. A genetically determined mesenchymal disorder combined withsex reversal. Hereditas. 1977;86(1):51-62. |
|
McDowall S, Argentaro A, Ranganathan S et al: Functional and structural studies of wild type SOX9 and mutations causingcampomelic dysplasia. J Biol Chem. 1999 Aug 20;274(34):24023-30. |
|
Huang B, Wang S, Ning Y et al: Autosomal XX sex reversal caused by duplication of SOX9. Am J Med Genet. 1999 Dec 3;87(4):349-53. |
|
Bishop CE, Whitworth DJ, Qin Y et al: A transgenic insertion upstream of sox9 is associated with dominant XX sexreversal in the mouse. Nat Genet. 2000 Dec;26(4):490-4. |
|
Ikeda Y, Shen WH, Ingraham HA et al: Developmental expression of mouse steroidogenic factor-1, an essential regulatorof the steroid hydroxylases. Mol Endocrinol. 1994 May;8(5):654-62. |
|
Lala DS, Rice DA, Parker KL: Steroidogenic factor I, a key regulator of steroidogenic enzyme expression, is Mol Endocrinol. 1992 Aug;6(8):1249-58. |
|
Lin L, Philibert P, Ferraz-de-Souza B et al: Heterozygous missense mutations in steroidogenic factor 1 (SF1/Ad4BP, NR5A1) areassociated with 46,XY disorders of sex development with normal adrenal function. J Clin Endocrinol Metab. 2007 Mar;92(3):991-9. Epub 2007 Jan 2. |
|
Achermann JC, Ito M, Ito M et al: A mutation in the gene encoding steroidogenic factor-1 causes XY sex reversal andadrenal failure in humans. Nat Genet. 1999 Jun;22(2):125-6. |
|
Canto P, Soderlund D, Reyes E et al: Mutations in the desert hedgehog (DHH) gene in patients with 46,XY complete puregonadal dysgenesis. J Clin Endocrinol Metab. 2004 Sep;89(9):4480-3. |
|
Parma P, Radi O, Vidal V et al: R-spondin1 is essential in sex determination, skin differentiation andmalignancy. Nat Genet. 2006 Nov;38(11):1304-9. Epub 2006 Oct 15. |
|
McPherson EW, Clemens MM, Gibbons RJ et al: X-linked alpha-thalassemia/mental retardation (ATR-X) syndrome: a new kindredwith severe genital anomalies and mild hematologic expression. Am J Med Genet. 1995 Jan 30;55(3):302-6. |
|
Reardon W, Gibbons RJ, Winter RM et al: Male pseudohermaphroditism in sibs with the alpha-thalassemia/mental retardation Am J Med Genet. 1995 Jan 30;55(3):285-7. |
|
Bernstein R, Koo GC, Wachtel SS: Abnormality of the X chromosome in human 46,XY female siblings with dysgeneticovaries. Science. 1980 Feb 15;207(4432):768-9. |
|
Bardoni B, Zanaria E, Guioli S et al: A dosage sensitive locus at chromosome Xp21 is involved in male to female sexreversal. Nat Genet. 1994 Aug;7(4):497-501. |
|
Zanaria E, Muscatelli F, Bardoni B et al: An unusual member of the nuclear hormone receptor superfamily responsible for Nature. 1994 Dec 15;372(6507):635-41. |
|
Vainio S, Heikkila M, Kispert A et al: Female development in mammals is regulated by Wnt-4 signalling. Nature. 1999 Feb 4;397(6718):405-9. |
|
Ottolenghi C, Pelosi E, Tran J et al: Loss of Wnt4 and Foxl2 leads to female-to-male sex reversal extending to germcells. Hum Mol Genet. 2007 Dec 1;16(23):2795-804. Epub 2007 Aug 29. |
|
Jordan BK, Mohammed M, Ching ST et al: Up-regulation of WNT-4 signaling and dosage-sensitive sex reversal in humans. Am J Hum Genet. 2001 May;68(5):1102-9. Epub 2001 Mar 29. |
|
Colvin JS, Green RP, Schmahl J et al: Male-to-female sex reversal in mice lacking fibroblast growth factor 9. Cell. 2001 Mar 23;104(6):875-89. |
|
Raymond CS, Shamu CE, Shen MM et al: Evidence for evolutionary conservation of sex-determining genes. Nature. 1998 Feb 12;391(6668):691-5. |
|
Moniot B, Berta P, Scherer G et al: Male specific expression suggests role of DMRT1 in human sex determination. Mech Dev. 2000 Mar 1;91(1-2):323-5. |
|
Raymond CS, Kettlewell JR, Hirsch B et al: Expression of Dmrt1 in the genital ridge of mouse and chicken embryos suggests arole in vertebrate sexual development. Dev Biol. 1999 Nov 15;215(2):208-20. |
|
Ion R, Telvi L, Chaussain JL et al: Failure of testicular development associated with a rearrangement of 9p24.1proximal to the SNF2 gene. Hum Genet. 1998 Feb;102(2):151-6. |
|
Ottolenghi C, Veitia R, Quintana-Murci L et al: The region on 9p associated with 46,XY sex reversal contains several transcripts Genomics. 2000 Mar 1;64(2):170-8. |
|
Veitia R, Nunes M, Brauner R et al: Deletions of distal 9p associated with 46,XY male to female sex reversal: Genomics. 1997 Apr 15;41(2):271-4. |
|
Arango NA, Lovell-Badge R, Behringer RR: Targeted mutagenesis of the endogenous mouse Mis gene promoter: in vivodefinition of genetic pathways of vertebrate sexual development. Cell. 1999 Nov 12;99(4):409-19. |
|
De Santa Barbara P, Bonneaud N, Boizet B et al: Direct interaction of SRY-related protein SOX9 and steroidogenic factor 1 Mol Cell Biol. 1998 Nov;18(11):6653-65. |
|
Nachtigal MW, Hirokawa Y, Enyeart-VanHouten DL et al: Wilms' tumor 1 and Dax-1 modulate the orphan nuclear receptor SF-1 in Cell. 1998 May 1;93(3):445-54. |
|
Crawford PA, Dorn C, Sadovsky Y et al: Nuclear receptor DAX-1 recruits nuclear receptor corepressor N-CoR tosteroidogenic factor 1. Mol Cell Biol. 1998 May;18(5):2949-56. |
|
Ito M, Yu R, Jameson JL: DAX-1 inhibits SF-1-mediated transactivation via a carboxy-terminal domain thatis deleted in adrenal hypoplasia congenita. Mol Cell Biol. 1997 Mar;17(3):1476-83. |
|
Lalli E, Bardoni B, Zazopoulos E et al: A transcriptional silencing domain in DAX-1 whose mutation causes adrenalhypoplasia congenita. Mol Endocrinol. 1997 Dec;11(13):1950-60. |
|
Hossain A, Saunders GF: The human sex-determining gene SRY is a direct target of WT1. J Biol Chem. 2001 May 18;276(20):16817-23. Epub 2001 Feb 20. |
|
Kratzer PG, Chapman VM: X chromosome reactivation in oocytes of Mus caroli. Proc Natl Acad Sci 78: 3093–3097, 1981 |
|
Monk M, McLaren A: X-chromosome inactivation in foetal germ cells of the mouse. J Embryol Experiment Morph 63: 75–84, 1981 |
|
Simpson JL: Abnormal sexual differentiation in humans. Ann Rev Genet 16: 193–224, 1982 |
|
Therman E, Patau K: Abnormal X chromosomes in man: origin, behavior and effects. Humangenetik 25: 1–16, 1974 |
|
Simpson JL: Genes, chromosomes, and reproductive failure. Fertil Steril 33: 107–116, 1980 |
|
Krauss CM, Turksoy N, Atkins L et al: Familial premature ovarian failure due to an interstitial deletion of the long arm of the X chromosome. New Engl J Med 317: 125–131, 1987 |
|
Sarafoglou K, Ostrer H: Familial sex reversal: A review. J Clin Endocrinol Metab 85: 483–493, 2000 |
|
Jawaheer D, Juo SH, Le Caignec C et al: Mapping a gene for 46,XY gonadal dysgenesis by linkage analysis. Clin Genet. 2003 Jun;63(6):530-5. |
|
Chung YP, Hwa HL, Tseng LH et al: Prenatal diagnosis of monosomy 10q25 associated with single umbilical artery and sex reversal: Report of a case. Prenat Diagn 18: 73–77, 1998 |
|
Slavotinek A, Schwarz C, Getty JF et al: Two cases with interstitial deletions of chromosome 2 and sex reversal in one. Am J Med Genet 86: 75–81, 1999 |
|
Katoh-Fukui Y, Tsuchiya R, Shiroishi T et al: Male-to-female sex reversal in M33 mutant mice. Nature 393: 688–692, 1998 |