
Hypogonadotropic Hypogonadism: A Genetics Based Approach
Authors
INTRODUCTION
Hypogonadotropic hypogonadism is defined as low levels of the sex steroids estrogen and progesterone (hypogonadism) in patients who are hypogonadotropic (low) or have inappropriately normal levels of gonadotropins FSH and LH. The vast majority of cases of HH are diagnosed in men. In women, this disorder generally presents as primary amenorrhea, which is discussed elsewhere in this text and has a multitude of other causes, including but not limited to CNS lesions, trauma, and malnutrition. Several areas on the genome have been implicated in HH. A large number of patients with HH will be found to have a known mutation; those without a known mutation can provide an opportunity to investigate for mutations in candidate genes that have not yet been proven to be associated with HH. Continuing research has led to the discovery of multiple genes that are essential for both the development and proper function of gonadotropin releasing hormone (GnRH) neurons in the hypothalamus which when dysfunctional lead to both normosmic hypogonadotropic hypogonadism (nHH) and Kallmann syndrome (anosmic HH). Individual mutations appear to have variable penetrance and expressivity. This chapter focuses on the genetic defects identified as causing hypogonadotropic hypogonadism. While this text is focused on female patients, male phenotypes are discussed as a complete reproductive family history of both males and females is an essential part of the work-up of a patient with suspected HH.
There are two biological pathways that can lead to HH. The first has its roots in embryology. GnRH secreting neurons migrate from the olfactory bulb to the hypothalamus during embryologic development (Figure 1). Mutations that interfere with this migration lead to anosmic HH, also known as Kallmann syndrome.1 The second pathway involves loss of function mutations that result in abnormal function of GnRH secreting neurons that underwent normal embryonic migration. These mutations cause normosmic hypogonadotropic hypogonadism (nHH). While some mutations have been shown to cause both Kallmann syndrome and nHH, the distinction between the two phenotypes is useful from a clinical and didactic perspective and is used in this chapter.
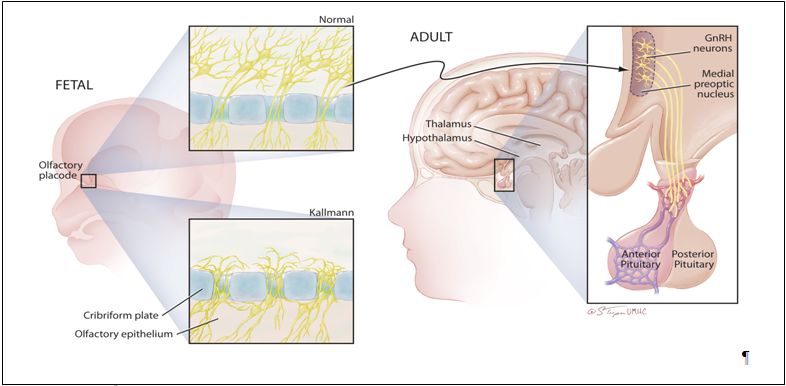 Fig. 1. The developmental history of the GnRH neuron. The clinical association of anosmia with delayed puberty emphasizes the developmental history of hypothalamic GnRH neurons. GnRH neurons arise in the epithelial layer of the olfactory placode during fetal development, and then migrate through the cribiform plate and along the vomeronasal axons to arrive in the hypothalamus. Migration in patients with Kallman syndrome often stops just dorsal to the cribiform plate. After normal migration, a few thousand GnRH neurons are found scattered over several hypothalamic nuclei in the human adult. The majority of GnRH nuclei can be found in the arcuate and preoptic areas of the medial basal hypothalamus. These hypothalamic GnRH cell bodies project axons along the median eminence to terminate near the hypophyseal portal capillary system where they secrete minute amounts of GnRH in a pulsatile fashion
Fig. 1. The developmental history of the GnRH neuron. The clinical association of anosmia with delayed puberty emphasizes the developmental history of hypothalamic GnRH neurons. GnRH neurons arise in the epithelial layer of the olfactory placode during fetal development, and then migrate through the cribiform plate and along the vomeronasal axons to arrive in the hypothalamus. Migration in patients with Kallman syndrome often stops just dorsal to the cribiform plate. After normal migration, a few thousand GnRH neurons are found scattered over several hypothalamic nuclei in the human adult. The majority of GnRH nuclei can be found in the arcuate and preoptic areas of the medial basal hypothalamus. These hypothalamic GnRH cell bodies project axons along the median eminence to terminate near the hypophyseal portal capillary system where they secrete minute amounts of GnRH in a pulsatile fashion
KALLMANN SYNDROME
Kallmann syndrome has a prevalence of 1/8000 to 1/60,000 and is more commonly found in males than females.1, 2 There are multiple genetic defects that can cause Kallmann syndrome (Table 1). Not surprisingly, multiple inheritance patterns have been reported, including X-linked recessive, autosomal dominant and autosomal recessive. Frequently inheritance is sporadic and does not correlate with any specific pattern.
The KAL1 gene is located on the X chromosome at the Xp22.3 locus and encodes an extracellular glycoprotein called anosmin 1. This protein is involved in cellular adhesion and cell migration.2 A mutation in the KAL1 gene leads to failure of GnRH neuronal migration during embryologic development. Adults therefore present with anosmia and HH. Patients with mutations in the KAL1 gene typically have a severe phenotype. Males tend to have cryptorchidism, microphallus, and small testes, while females present with classic anosmic HH.3 All reported families with KAL1 mutations and hypogonadotropic hypogonadism have exclusively anosmic hypogonadotropic hypogonadism. In the case of a patient with Kallmann syndrome, a family history of normosmic HH makes a KAL1 mutation unlikely. Patients with the KAL1 mutation frequently have additional abnormalities, including renal agenesis, synkinesia.4 Thus, evaluation of the genitourinary system is an important step in the evaluation of the patient with hypogonadotropic hypogonadism, particularly those with known KAL1 mutations.
Table 1: Gene defects and clinical characteristics in patients with hypothalamic hypogonadism. Adapted from Beate, et al., Int J Endocrinol, 20105
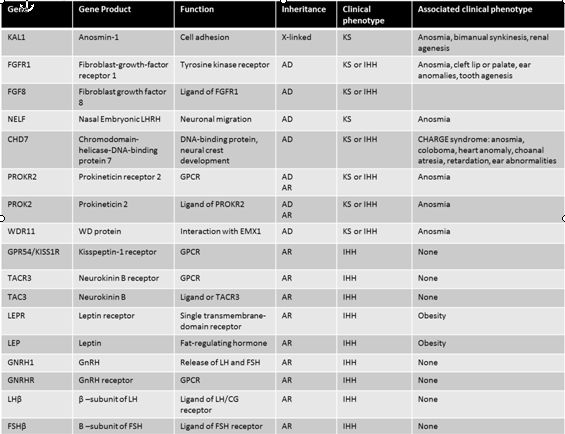
Fibroblast growth factor receptor 1 (FGFR1) is located on chromosome 8. FGFR1 is also implicated in cell migration and requires the same cofactor, heparin sulfate, as anosmin.1, 6 Thus, FGFR1 and KAL1 are likely to be two separate genes that are involved in the migration of GnRH neurons to the hypothalamus. This is supported by the finding that patients with a mutation in FGFR1 and KAL1 can present with very similar phenotypes, although the presentation is more variable with FGFR1.
The phenotype reported with FGFR1 mutation is incredibly variable.7 In a series of 80 patients with FGFR1 mutations individuals were described with normosmic or anosmic HH. In addition, two unrelated females with anosmia and normal reproductive function were found to have FGFR1 mutations.8 Reversal of GnRH deficiency has even been reported in some male patients with FGFR1 mutations following therapy with testosterone. Overall, this reversal seems to occur in about 10% of subjects.5
Fibroblast growth factor 8 (FGF8) is a ligand for FGFR1. Mutations in FGF8 have been associated with Kallmann syndrome in humans and animal models. Mice with a homozygous mutation in FGF8 had no olfactory neurons in the hypothalamus, while heterozygous mice had markedly fewer hypothalamic olfactory neurons. Humans with FGF8 mutations have a range of phenotypes, including adult onset HH.9 This widely variable phenotype suggests that mutations at different sites on the genome cause different degrees in loss of function that in turn lead to a varying clinical presentation.
In addition to HH, patients with a mutation in FGFR1/FGF8 will have a cleft palate in 30% of cases.1 Ear, cartilage, and digital abnormalities have also been reported. The presence of these abnormalities in a patient with HH should raise suspicion for FGFR1/FGF8 mutation.
The PROKR2 is a G-protein coupled receptor that binds to the ligand prokinectin-2 (PROK2). Mice deficient in PROK2 have small and abnormally shaped olfactory bulbs and an accumulation of neurons in the rostral migratory stream (RMS) between the subventricular zone and the olfactory bulb.10 The clinical phenotype of PROK2/PROKR2 mutations in humans includes both Kallmann syndrome and normosmic hypogonadotropic hypogonadism. In addition to reproductive abnormalities, patients with these mutations can present with fibrous dysplasia, synkinesia, and epilepsy. Specifically, patients with more severe reproductive dysfunction tend to have biallelic mutations in PROK2/PROKR2 and fewer associated non-reproductive abnormalities. Patients with monoallelic mutations tend to have less severe reproductive dysfunction but more nonreproductive abnormalities.11
CHARGE SYNDROME
CHARGE syndrome is a phenotype associated with hypogonadotropic hypogonadism. This phenotype is characterized by colobomata, heart anomalies, choanal atresia, retardation, and genital and ear anomalies in addition to hypogonadotropic hypogonadism.1, 12 Multiple sets of diagnostic criteria for CHARGE syndrome have been proposed.12 The syndrome is caused by a mutation in the CHD7 gene.
The CHD 7 gene encodes chromodomain helicase DNA binding protein-7 and is involved in alteration of nucleosome structure. The CHD7 gene was first identified as the site responsible for CHARGE syndrome by Vissers in 2004.13 This initial study examined 17 patients and identified missense, nonsense, and splice mutations. Since that time, multiple mutations have been described in the CHD7 gene leading to both CHARGE syndrome and IHH without the additional abnormalities associated with CHARGE syndrome.12 Thus, CHD7 mutations can lead to a varied phenotype including Kallmann syndrome, nHH and CHARGE syndrome.
The involvement of multiple organ systems in CHARGE syndrome can be explained by the variety of tissues that express CHD7. CHD7 is expressed in the olfactory placode, spinal cord, nasopharynx and eye. In mouse models, CHD7 expression is temporally correlated with olfactory neuron development, although direct involvement has not been demonstrated definitively.14 Thus, the association between CHD7 mutations and CHARGE syndrome and Kallmann syndrome is both biologically plausible and clinically demonstrable.
NORMOSMIC HYPOGONADOTROPIC HYPOGONADISM
While Kallmann syndrome genes are primarily involved in the migration of neurons from the olfactory bulb to the hypothalamus, genes in which mutations lead to normosmic hypogonadotropic hypogonadism are involved in the function of the hypothalamic–pituitary–gonadal axis. The most obvious gene loci to examine regarding normosmic HH are those of the GnRH receptor (GnRHR) and GnRH itself. GnRHR mutations have been well described. They are responsible for roughly one-fifth cases of sporadic cases and about a half of autosomal recessive inherited cases of nIHH.15 Mutations in the gene for GNRH1 have proven elusive. It was not until 2009 that Bouligand et al. identified GNRH1 mutations in siblings with nIHH.16 A male and female pair of siblings with nIHH were found to be homozygous for mutations in the GNRH1 gene. Both parents and an unaffected sibling were heterozygous. The affected individuals were able to be treated by administration of pulsatile GnRH. A subsequent review by Chan et al. of over 300 subjects with nIHH established that GnRH1 mutations were a rare cause of nIHH,17 accounting for about 1% of cases.
Mutations in the leptin receptor (LEPR) are associated with HH in addition to obesity and hyperphagia. In 300 patients studied with severe early onset obesity and hyperphagia, 3% had a mutation in the LEPR. These subjects were characterized by altered immune systems and hypogonadotropic hypogonadism in addition to their obesity. Interestingly, they had relatively normal levels of leptin.18
Congenital adrenal hypoplasia (CAH) is also associated with normosmic HH. The DAX1 gene has been implicated in this relationship. DAX1 works during embryologic development to antagonize SRY and is therefore essential in sexual differentiation. Subjects with congenital adrenal hypoplasia due to DAX1 mutation will also suffer from normosmic HH.19 In this case it will almost certainly be the adrenal dysfunction that prompts investigation into the function of the hypothalamic–pituitary–gonadal axis as infants with CAH that is not recognized clinically are unlikely to survive until puberty.
Kisspeptin is a peptide encoded by the gene KISS1. It was originally described as a metastasis suppressor in melanoma and breast cancer. Kisspeptin binds to GPR54, which is encoded by the gene KISS1R.20 Kisspeptin is now known to be an important positive regulator of GnRH secretion.21 Mutations in the KISS1 receptor (KISS1R) have been reported as a rare cause of HH. Further study involving examining the KISS1R gene in affected humans and in knockout mice revealed that mutations in the gene resulted nIHH with an autosomal recessive mode of inheritance.22 While animal models have not conclusively demonstrated HH with loss of function in kisspeptin, a loss of function mutation leading to nIHH has been recently described in the kisspeptin gene KISS1 in humans.23
Neurokinin B (NKB) is another neuropeptide that is linked to nHH. It is highly expressed in the arcuate nucleus, a region of the brain that also expresses high levels of kisspeptin. Topaloglu studied four separate families with strong histories of nHH that did not have a known mutation identified. All affected individuals in this population were found to have homozygous mutations in either the TAC3 gene that encodes NKB or the TAC3R gene that encodes the NKB receptor.24
The anatomic and functional relationship between kisspeptin and NKB has led to a greater understanding of the physiologic regulation of GnRH release. The method of regulation of GnRH secretion based on nutritional and metabolic status has been elusive. The most likely candidate hormones, leptin and estrogen, do not have receptors on the GnRH neurons.25 Recent research has demonstrated that kisspeptin and NKB are directly involved in signaling between the arcutate nucleus and GnRH neurons. In this work by Young et al. a continuous kisspeptin infusion resulted in normal GnRH secretion in patients with loss of function mutations in TAC3 or TAC3R. This allowed the authors to conclude that both kisspeptin and NKB are essential parts of the endocrine cascade governing GnRH release. Furthermore they concluded that NKB was proximal to kisspeptin in this pathway and may, in fact, be a modulator of kisspeptin release.25
CLINICAL APPROACH
Hypogonadotropic hypogonadism is a rare condition. In females it is likely to present as primary amenorrhea that is subsequently found to be associated with low levels of FSH and LH. The first step in evaluation of these patients should be a thorough physical exam and family history. The physical exam should include an evaluation for the clinical features of CHARGE syndrome, renal and skeletal abnormalities, synkinesia, and cleft lip/palate. The family history should focus on the reproductive histories of male and female family members. Associated exam findings, clinical phenotype, and mode of inheritance can help to focus the laboratory evaluation. However, given the great degree of clinical overlap seen between mutations at different gene loci and the degree of heterogeneity seen with mutations in a single gene, it is unlikely that this initial evaluation will lead to a definitive diagnosis. The most appropriate test is most likely to be a microarray that will evaluate for hundreds of known mutations. If the microarray does not identify a mutation, the information from the clinical exam and history may allow the clinician to focus on a few specific genes to evaluate for less common or novel mutations.
REFERENCES
Topaloglu, A.K., and Kotan, L.D. (2010). Molecular causes of hypogonadotropic hypogonadism. Current opinion in obstetrics & gynecology 22, 264-270 |
|
Bick, D., Franco, B., Sherins, R.J., Heye, B., Pike, L., Crawford, J., Maddalena, A., Incerti, B., Pragliola, A., Meitinger, T., et al. (1992). Brief report: intragenic deletion of the KALIG-1 gene in Kallmann's syndrome. The New England journal of medicine 326, 1752-1755. |
|
itteloud, N., Quinton, R., Pearce, S., Raivio, T., Acierno, J., Dwyer, A., Plummer, L., Hughes, V., Seminara, S., Cheng, Y.Z., et al. (2007). Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. The Journal of clinical investigation 117, 457-463. |
|
Tsai, P.S., and Gill, J.C. (2006). Mechanisms of disease: Insights into X-linked and autosomal-dominant Kallmann syndrome. Nature clinical practice Endocrinology & metabolism 2, 160-171. |
|
Beate, K., Joseph, N., Nicolas de, R., and Wolfram, K. (2012). Genetics of isolated hypogonadotropic hypogonadism: role of GnRH receptor and other genes. International journal of endocrinology 2012, 147893. |
|
Hardelin, J.P., and Dode, C. (2008). The complex genetics of Kallmann syndrome: KAL1, FGFR1, FGF8, PROKR2, PROK2, et al. Sexual development : genetics, molecular biology, evolution, endocrinology, embryology, and pathology of sex determination and differentiation 2, 181-193. |
|
Pitteloud, N., Meysing, A., Quinton, R., Acierno, J.S., Jr., Dwyer, A.A., Plummer, L., Fliers, E., Boepple, P., Hayes, F., Seminara, S., et al. (2006). Mutations in fibroblast growth factor receptor 1 cause Kallmann syndrome with a wide spectrum of reproductive phenotypes. Molecular and cellular endocrinology 254-255, 60-69. |
|
Trarbach, E.B., Costa, E.M., Versiani, B., de Castro, M., Baptista, M.T., Garmes, H.M., de Mendonca, B.B., and Latronico, A.C. (2006). Novel fibroblast growth factor receptor 1 mutations in patients with congenital hypogonadotropic hypogonadism with and without anosmia. The Journal of clinical endocrinology and metabolism 91, 4006-4012. |
|
Falardeau, J., Chung, W.C., Beenken, A., Raivio, T., Plummer, L., Sidis, Y., Jacobson-Dickman, E.E., Eliseenkova, A.V., Ma, J., Dwyer, A., et al. (2008). Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. The Journal of clinical investigation 118, 2822-2831. |
|
Ng, K.L., Li, J.D., Cheng, M.Y., Leslie, F.M., Lee, A.G., and Zhou, Q.Y. (2005). Dependence of olfactory bulb neurogenesis on prokineticin 2 signaling. Science 308, 1923-1927. |
|
Sarfati, J., Guiochon-Mantel, A., Rondard, P., Arnulf, I., Garcia-Pinero, A., Wolczynski, S., Brailly-Tabard, S., Bidet, M., Ramos-Arroyo, M., Mathieu, M., et al. (2010). A comparative phenotypic study of kallmann syndrome patients carrying monoallelic and biallelic mutations in the prokineticin 2 or prokineticin receptor 2 genes. The Journal of clinical endocrinology and metabolism 95, 659-669. |
|
Kim, H.G., and Layman, L.C. (2011). The role of CHD7 and the newly identified WDR11 gene in patients with idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Molecular and cellular endocrinology 346, 74-83. |
|
Vissers, L.E., van Ravenswaaij, C.M., Admiraal, R., Hurst, J.A., de Vries, B.B., Janssen, I.M., van der Vliet, W.A., Huys, E.H., de Jong, P.J., Hamel, B.C., et al. (2004). Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nature genetics 36, 955-957. |
|
Wierman, M.E., Pawlowski, J.E., Allen, M.P., Xu, M., Linseman, D.A., and Nielsen-Preiss, S. (2004). Molecular mechanisms of gonadotropin-releasing hormone neuronal migration. Trends in endocrinology and metabolism: TEM 15, 96-102. |
|
Beranova, M., Oliveira, L.M., Bedecarrats, G.Y., Schipani, E., Vallejo, M., Ammini, A.C., Quintos, J.B., Hall, J.E., Martin, K.A., Hayes, F.J., et al. (2001). Prevalence, phenotypic spectrum, and modes of inheritance of gonadotropin-releasing hormone receptor mutations in idiopathic hypogonadotropic hypogonadism. The Journal of clinical endocrinology and metabolism 86, 1580-1588. |
|
Bouligand, J., Ghervan, C., Tello, J.A., Brailly-Tabard, S., Salenave, S., Chanson, P., Lombes, M., Millar, R.P., Guiochon-Mantel, A., and Young, J. (2009). Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation. The New England journal of medicine 360, 2742-2748. |
|
Chan, Y.M., de Guillebon, A., Lang-Muritano, M., Plummer, L., Cerrato, F., Tsiaras, S., Gaspert, A., Lavoie, H.B., Wu, C.H., Crowley, W.F., Jr., et al. (2009). GNRH1 mutations in patients with idiopathic hypogonadotropic hypogonadism. Proceedings of the National Academy of Sciences of the United States of America 106, 11703-11708. |
|
Farooqi, I.S., Wangensteen, T., Collins, S., Kimber, W., Matarese, G., Keogh, J.M., Lank, E., Bottomley, B., Lopez-Fernandez, J., Ferraz-Amaro, I., et al. (2007). Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. The New England journal of medicine 356, 237-247. |
|
Muscatelli, F., Strom, T.M., Walker, A.P., Zanaria, E., Recan, D., Meindl, A., Bardoni, B., Guioli, S., Zehetner, G., Rabl, W., et al. (1994). Mutations in the DAX-1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. Nature 372, 672-676. |
|
Ohtaki, T., Shintani, Y., Honda, S., Matsumoto, H., Hori, A., Kanehashi, K., Terao, Y., Kumano, S., Takatsu, Y., Masuda, Y., et al. (2001). Metastasis suppressor gene KiSS-1 encodes peptide ligand of a G-protein-coupled receptor. Nature 411, 613-617. |
|
Oakley, A.E., Clifton, D.K., and Steiner, R.A. (2009). Kisspeptin signaling in the brain. Endocrine reviews 30, 713-743. |
|
Seminara, S.B., Messager, S., Chatzidaki, E.E., Thresher, R.R., Acierno, J.S., Jr., Shagoury, J.K., Bo-Abbas, Y., Kuohung, W., Schwinof, K.M., Hendrick, A.G., et al. (2003). The GPR54 gene as a regulator of puberty. The New England journal of medicine 349, 1614-1627. |
|
. Topaloglu, A.K., Tello, J.A., Kotan, L.D., Ozbek, M.N., Yilmaz, M.B., Erdogan, S., Gurbuz, F., Temiz, F., Millar, R.P., and Yuksel, B. (2012). Inactivating KISS1 mutation and hypogonadotropic hypogonadism. The New England journal of medicine 366, 629-635. |
|
Topaloglu, A.K., Reimann, F., Guclu, M., Yalin, A.S., Kotan, L.D., Porter, K.M., Serin, A., Mungan, N.O., Cook, J.R., Ozbek, M.N., et al. (2009). TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction. Nature genetics 41, 354-358. |
|
Young, J., George, J.T., Tello, J.A., Francou, B., Bouligand, J., Guiochon-Mantel, A., Brailly-Tabard, S., Anderson, R.A., and Millar, R.P. (2012). Kisspeptin Restores Pulsatile LH Secretion in Patients with Neurokinin B Signaling Deficiencies: Physiological, Pathophysiological and Therapeutic Implications. Neuroendocrinology. |