
Male Sexual Differentiation Disorder and 5α-Reductase-2 Deficiency
Authors
INTRODUCTION
Disorders in male sexual differentiation result in an incompletely masculinized individual with a 46,XY karyotype and testes. Mutations in the isozyme 5α-reductase-2 result in a disorder of male sexual differentiation that has revealed the role of dihydrotestosterone in male sexual differentiation.
BIOCHEMISTRY AND MOLECULAR BIOLOGY OF 5α-REDUCTASE ISOENZYMES
Steroid 5α-reductase isozymes located in the microsomes of the cell are nicotinamide-adenine dinucleotide phosphate (NADPH)-dependent proteins that reduce the double bond at the 4-5 position of a variety of C19 and C21 steroids. These isozymes convert testosterone to the more potent androgen, dihydrotestosterone (DHT). Testosterone and DHT bind to the intracellular androgen receptor (a member of the nuclear steroid/thyroid hormone receptor superfamily) and interact with a cognate androgen DNA response element to regulate target gene expression.1, 2 Although testosterone and DHT interact with the same androgen receptor, they can produce distinct biological responses.3 The molecular mechanism for this remains unclear though DHT has been reported to bind to the androgen receptor more avidly than testosterone,4 and the DHT-receptor complex is more efficiently transformed to the DNA-binding state than is the testosterone-receptor complex.5
In the early 1960s, it was theorized that multiple 5α-reductase isozymes existed.6 In 1975 and 1976, Moore and Wilson7, 8 detected different pH optima for 5α-reductase activity in genital and nongenital skin. In genital skin, the major enzymatic activity had a narrow, acidic pH optimum of 5.5,8 and was low in the genital skin of affected subjects with 5α-reductase deficiency.7, 9 Enzymatic activity with a neutral to alkaline pH (pH 7 to 9) present in both normal genital and nongenital skin, was found to be normal in the genital skin of affected subjects with 5α-reductase deficiency. Kinetic analysis of 5α-reductase activity in the epithelium and stroma of the prostate also suggested that there were different 5α-reductase activities.10, 11 Pharmacologic studies of specific 5α-reductase inhibitors provided further evidence of more than one 5α-reductase isozyme.12 Although many attempts were made to purify 5α-reductases, they were not successful due to the extreme insolubility of the protein.
In the early 1990s, two genes encoding two isozymes13, 14, 15, 16 were cloned using expression cloning technology: steroid 5α-reductase type 1 (gene symbol: SRD5A1) and steroid 5α-reductase type 2 (gene symbol: SRD5A2). Mutations in the 5α-reductase-2 gene were found to be responsible for a disorder in male sexual differentiation.14, 17 The characteristics of the two 5α-reductase isozymes are summarized in Table 1.
Table 1. Comparison of human 5α-reductase isozymes
|
| Type 1 | Type 2 |
| Gene structure | 5 exons, 4 introns | 5 exons, 4 introns |
| Gene, chromosome location | SRD5A1, 5p15 | SRD5A2, 2p23 |
| Size | 259 amino acids, Mr=29,462 | 254 amino acids, Mr=28,398 |
| Tissue distribution | Liver, nongenital skin, prostate, brain, ovary, testis, breast | Prostate, epididymis, seminal vesicle, genital skin, liver, uterus, breast, hair follicle, placenta, testis |
| PH optima | Neutral to basic | Acidic or neutral |
| Prostate level | Low | High |
| Prostate cell distribution | Epithelial cells | Stromal and epithelial cells |
| Activity in 5a-reductase deficiency | Normal | Mutated |
| Finasteride inhibition | Ki > 300 nM | Ki = 3-5 nM |
Note: Data obtained from Wilson et al. (1993), Endocrine Reviews 14: 577–593; Russell et al. (1994). Annual Review of Biochemistry 63:25–61; Zhu et al. (1998), Bailliere’s Clinical Endocrinology and Metabolism 12:83–113.
The human 5α-reductase-2 gene has five exons and four introns, encodes a highly hydrophobic 254 amino acid protein with the molecular weight of approximately 28.4 kilodaltons (kd), and maps to the short arm of chromosome 2 band 23.14, 16, 18 The type 2 isozyme has a much higher affinity for testosterone (apparent Km = 4 to 50 nM) than the type 1 isozyme (Km = 1 to 5 μM); while, the apparent Km (3 to 10 μM) for NADPH cofactor is similar in both isozymes. The type 2 isozyme is sensitive to finasteride, a 5α-reductase inhibitor, and it has an acidic pH optimum in enzymatic assays.16 However, in its native state, it may have a neutral pH optimum.19 The type 2 isozyme is expressed in the external genital tissues early in gestation.20 In adulthood, its expression is relatively high in prostate, genital skin, epididymis, seminal vesicle, and liver, whereas it is quite low in testis, nongenital skin, uterus, and brain. It is also expressed in the ovary and hair follicles.21, 22
The 5α-reductase-1 gene is normal in affected subjects with 5α-reductase deficiency.14 It also has 5 exons and 4 introns and is located on the short arm of chromosome 5 band 15. This isozyme has 259 highly hydrophobic amino acids with a molecular weight approximately 29.5 kd.16 There is approximately 50% homology between human type-1 and type-2 isozymes in amino acid compositions. 5α-reductase-1 has a broad alkaline pH optimum, a lower substrate affinity, and a lower sensitivity to finasteride inhibition.16, 18 This isozyme is highly expressed in nongenital skin, liver, breast and certain brain regions, while its presence in the prostate, genital skin, epididymis, seminal vesicle, testis, adrenals, and kidney is low. Its expression is detected at birth in the liver and nongenital skin and is present throughout life, although its expression in embryonic tissues is low. The physiologic function of 5α-reductase-1 is still obscure. It may play a significant role in parturition and fetal growth as demonstrated in a knockout mouse model.23, 24
CLINICAL SYNDROME OF 5α-REDUCTASE-2 DEFICIENCY
An inherited defect in 5α-reductase-2 gene results in genital ambiguity in the male. This syndrome was first described clinically and biochemically in 1974 in studies of 24 affected subjects from 13 families in a large Dominican kindred,25 and in two siblings from Dallas.26 Two other large cohorts in New Guinea27 and Turkey28, 29, 30 have been described as well as many other cases around the world.18, 31, 32, 33
Most males who are homozygous for 5α-reductase-2 deficiency have striking ambiguity of the genitalia with a clitoral-like phallus, severely bifid scrotum, pseudovaginal perineoscrotal hypospadias, and a rudimentary prostate (Figure 1).18, 25, 33, 34, 35 Consequently, many affected males are assigned a female gender at birth and are reared as girls. On occasion, more masculinized subjects have been described; they may lack a separate vaginal opening,36 or have a blind vaginal pouch which opens into the urethra,37 penile hypospadias35 or even a penile urethra.38 Wolffian duct differentiation is normal with seminal vesicles, vasa deferentia, epididymides, and ejaculatory ducts, and there are no Müllerian structures. Cryptorchidism is frequently described; however, it is not invariably present. Testes may be located in the abdomen but are usually found in the inguinal canal or scrotum.
Affected 46,XY subjects with 5α-reductase-2 deficiency are clinical models for defining the major actions of testosterone and DHT during male sexual differentiation and development (see Figure 1 and Table 2). Testosterone secreted in utero by the testes acts directly on the Wolffian ducts to cause differentiation to the vas deferens, epididymis, and seminal vesicles. In the urogenital sinus and urogenital tubercle, testosterone functions as a prehormone, where its conversion to DHT results in differentiation of the external genitalia and prostate.25 This is consistent with the demonstration in human fetus that at the time of sexual differentiation, DHT formation occurs in the urogenital sinus, urogenital tubercle, and urogenital swellings, but does not occur in the Wolffian anlage until sexual differentiation is completed.39 Animal studies using a 5α-reductase-2 inhibitor provide further evidence supporting the differential roles of testosterone and DHT in male sexual differentiation.40, 41
With puberty, the affected males have increased muscle mass and deepening of the voice.25 The musculature is noted to be prominent in Dominican, New Guinean, and Turkish subjects.25, 27, 28, 29 Affected males in these kindreds are similar in height to their unaffected siblings, and there is no gynecomastia in adulthood.37, 42 There is substantial growth of the phallus with rugation and hyperpigmentation of the scrotum. Inguinal testes descending into the scrotum at puberty have been observed in some patients.36, 37, 42 Libido is intact, and patients can have erections.25, 43 Although patients are generally oligo- or azoospermic, normal sperm concentrations have been reported in patients with descended testes.44, 45, 46 Affected patients from the Dominican kindred46 and from Sweden47 have been reported to father children, suggesting that DHT does not play a major role in spermatogenesis and sperm function. These clinical findings suggest that pubertal events, including male sexual function and spermatogenesis, are primarily testosterone-mediated (see Table 2).
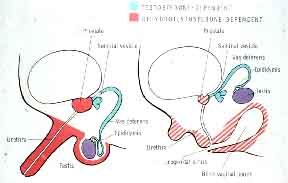 Figure 1. The specific roles of testosterone and DHT in male sexual differentiation in utero (adapted from Imperato-McGinley et al. (1974). Steroid 5a-reductase deficiency in man: An inherited form of male pseudohermaphroditism. Science, 186: 1213-1216).
Figure 1. The specific roles of testosterone and DHT in male sexual differentiation in utero (adapted from Imperato-McGinley et al. (1974). Steroid 5a-reductase deficiency in man: An inherited form of male pseudohermaphroditism. Science, 186: 1213-1216).
.
Table 2. Androgen action at puberty
| Testosterone | Dihydrotestosterone | ||
| I | Anabolic actions | I | Increased facial, body hair |
| Muscle mass increased | II | Scalp hair recession | |
| Enlargement penis | |||
| Enlargement scrotum | III | Prostate differentiation and growth | |
| Enlargement vocal cords | |||
| Skeletal maturation | IV | PSA expression | |
| growth spurt | |||
| epiphyseal closure | V | Pituitary-gonadal feedback | |
| II | Spermatogenesis | ||
| III | Male sex drive, performance | ||
| IV | Pituitary-gonadal feedback | ||
The prostate in the affected males is nonpalpable on rectal examination,25, 48 and is rudimentary on transrectal ultrasound and MRI visualization.43, 49 Prostatic volumes are one-tenth the size of normal age-matched controls. DHT replacement therapy results in an increase in prostate size.43, 50 These data provide clinical evidence that prostate differentiation and growth is mediated largely by DHT (see Figure 1 and Table 2). 5α-reductase inhibition as treatment for benign prostate hyperplasia evolved in part from the clinical observation that affected adult males with 5α-reductase-2 deficiency have rudimentary prostates caused by lifelong DHT deficiency. Prostate diseases such as prostate cancer and benign prostate hyperplasia have not been reported in affected subjects with 5α-reductase-2 deficiency.
Affected adult males have less facial and body hair than their unaffected male relatives, and male pattern baldness has never been observed in these subjects.25, 27, 28 The 5α-reductase-2 inhibitor, finasteride, is now used for treatment of male pattern baldness.51, 52
Androgen involvement in sebaceous gland secretion is suggested by the finding that no demonstrable sebum is produced in 46,XY subjects with complete androgen insensitivity.53 However, affected males with 5α-reductase-2 deficiency who rarely have acne produce normal amounts of sebum, suggesting that it is not regulated by the 5α-reductase-2 isozyme.53
Homozygous females with a 5α-reductase-2 gene mutation have normal to elevated plasma testosterone, with low DHT, and an elevated testosterone/DHT ratio.54 Urinary 5β/5α C19 and C21 steroid metabolites are elevated and similar to the ratios of homozygous males. As in affected males, despite elevated testosterone, decreased body hair is found and no facial hair is present. Sebum production is normal.54
BIOCHEMICAL FEATURES OF 5α-REDUCTASE-2 DEFICIENCY
The biochemical characteristics of 5α-reductase-2 deficiency have been well defined over the years.18, 25, 33, 37, 43 They include: (1) normal to elevated levels of plasma testosterone; (2) decreased levels of plasma DHT; (3) an increased testosterone to DHT ratio at baseline and/or following hCG stimulation; (4) decreased conversion of testosterone to DHT in vivo, with conversion ratios of testosterone to DHT of <1%; (5) reduced 5α-reductase activity in genital tissue and cultured fibroblasts; (6) normal metabolic clearance rates of testosterone and DHT; (7) decreased production of urinary 5α-reduced androgen metabolites with increased 5β/5α urinary metabolite ratios; (8) decreased plasma and urinary 3α-androstanediol glucuronide, a major metabolite of DHT;55 (9) a global defect in steroid 5α-reduction as demonstrated by decreased urinary 5α-reduced metabolites of both C21 steroids and C19 steroids other than testosterone (e.g. cortisol, corticosterone, 11β-hydroxy-androstenedione, and androstenedione).56 Although the defect of 5α-reduction of steroids is generalized, only the defective reduction of testosterone to DHT appears to be of clinical significance.
The affected subjects have increased plasma levels of LH, an increased LH pulse amplitude with normal LH frequency.57 The elevated mean LH level occurs despite an elevated mean plasma testosterone, suggesting a role for DHT in the negative feedback control of LH. Plasma FSH levels may be elevated. Although some of the elevation in FSH is undoubtedly attributable to cryptorchidism and seminiferous tubular damage, a role for DHT in the feedback control of FSH cannot be ruled out.58
MOLECULAR GENETICS OF 5α-REDUCTASE-2 DEFICIENCY
In the early 1990s, with the cloning of 5α-reductase isozyme genes, the genetic defect of 5α-reductase deficiency was defined. Our New Guinean kindred of affected 46,XY males who were clinically and biochemically characterized as having 5α-reductase deficiency were in the first genetic study.14, 27 A deletion of more than 20 kb in the 5α-reductase-2 gene was found in these patients by Southern blot analysis,14 whereas the 5α-reductase-1 gene was normal. To date, more than 61 mutations in the 5α-reductase-2 gene (Figure 2) have been identified,(references18, 33, 59 and Human Gene Mutation Database), including the three largest kindreds with 5α-reductase-2 deficiency in the world—the Dominican, New Guinean, and Turkish kindred. These mutations include 50 missense mutations, 6 small deletions, 3 splicing junction alterations, 1 single nucleotide insertion, and 1 large deletion involving the entire gene. As stated above, patients in the New Guinean kindred have a large deletion of 5α-reductase-2 gene.14 In the Dominican kindred, a missense mutation is found in exon 5 of the 5α-reductase-2 gene, substituting thymidine for cytosine and resulting in a substitution of the nonpolar amino acid tryptophan for the basic, polar amino acid arginine at position 246 of the isozyme.17, 60 This missense mutation causes a decrease in binding of the cofactor, NADPH, an altered pH optimum with a loss of enzymatic activity. In 5a-reductase-2 deficient subjects from the Turkish kindred, a single base deletion in exon 5 of the 5α-reductase-2 gene has been detected.30 This single-base deletion (adenine) results in a frame-shift at amino acid position 251 and an addition of 23 amino acids at the carboxyl-terminal of this 254 amino acid isozyme. This mutation in the isozyme results in a complete loss of enzymatic activity without an alteration in gene expression.
GENDER IDENTITY AND GENDER ROLE IN AFFECTED SUBJECTS WITH 5α-REDUCTASE-2 DEFICIENCY
Gender identity is the sense of being male or female, the awareness of knowing one's sex. Gender role is the expression of one's gender identity to the public. It is manifested by one's actions as either male or female.
Affected 46,XY subjects with 5α-reductase-2 deficiency raised as females often change gender role during or after puberty, providing a clinical model for defining the role of testosterone in the evolution of male gender identity. Since our original report in the Dominican community,25, 42, 63, 64 numerous groups have reported gender change in subjects with 5α-reductase-2 deficiency from many countries including individuals from the New Guinea, Turkey, Mexico, Cyprus, Algeria, Italy, Lebanon, Brazil, Pakistan, Saudi Arabia, UAE, Sweden, etc.18, 32, 33 It appears from these published observations that if puberty is permitted to occur spontaneously without surgical and hormonal reinforcement of the female sex of rearing, then a male gender identity, although discordant with the sex of rearing, will prevail. Under these circumstances, it appears that the extent of androgen (i.e. testosterone) exposure of the brain in utero, during the early postnatal period, and at puberty, has more of an effect in determining male gender identity than does sex of rearing and sociocultural influences.42, 64, 65
In the Dominican community, the female sex of rearing was never reinforced through castration and subsequent female hormone therapy. As such, at puberty a change in gender from female to male occurred in the vast majority of subjects from the older generation who were unambiguously reared as females.25, 42, 64 Interviews with affected 46,XY subjects showed that 17 of 18 subjects successfully changed gender identity from female to male. Between 7 and 12 years of age, patients who were raised as girls began to experience considerable anxiety over their lack of breast development and often began to be sexually attracted to girls. They became convinced of their male gender identity over the next several years and were masturbating and experiencing morning erections and nocturnal emissions. The change in gender role occurred on average at 16 years of age, with a range of 14 to 24 years. In three subjects, a gender role change did not occur until they were in their 20s. Admittedly, the fear of being stigmatized and anticipation of harassment by local villagers caused some subjects to hesitate at the prospect of changing gender roles, in some cases until they were confident of their ability to defend themselves.42, 64 We have continued to observe the behavioral characteristics of these subjects for more than 30 years and have no doubts as to their male behavioral pattern in adulthood.
Normally, the sex of rearing and testosterone imprinting of the brain act in unison to determine the complete expression of the male gender; however, subjects with 5α-reductase-2 deficiency demonstrate that in a laissez-faire environment, when rearing (female) is discordant with the testosterone-mediated biological sex, the biological sex prevails if normal testosterone activation of puberty is permitted to occur. From the data, it appears that the extent of testosterone exposure of the brain in utero, in the early postnatal period and at puberty, has greater impact in determining the male gender identity than the female sex of rearing. This experiment of nature emphasizes the importance of androgens, which act as inducers and activators in evolution of male gender identity in man.42, 64, 65
It has been proposed that the gender identity becomes fixed by 18 months to 4 years of age, at the time of language development.66, 67, 68 At this time, a child becomes aware of his or her gender; however, being aware of one’s gender and being unalterably fixed in that gender are two separate issues. The development of gender identity in man is continually evolving throughout childhood, becoming fixed with puberty.
The psychosexual studies in affected subjects with 5α-reductase-2 deficiency demonstrate that in humans, environmental or sociocultural factors are not solely responsible for the formation of a male gender identity; androgens make a strong and definite contribution. These data support the “hormone-influence theory” in the development of gender identity.42, 64, 65
DIAGNOSIS AND TREATMENT OF 5α-REDUCTASE-2 DEFICIENCY
5α-reductase-2 deficiency should be considered in 46,XY infants who are born with ambiguous external genitalia. Establishing the diagnosis of 5α-reductase-2 deficiency in infancy often requires that plasma testosterone/DHT ratios be determined following hCG administration.69 Measurement of urinary cortisol metabolite ratios, 5β-tetrahydrocortisol/5α-tetrahydrocortisol, is critical for the diagnosis of 5α-reductase-2 deficiency in infancy, because the amount of C19 androgen metabolites, etiocholanolone, and androsterone in the urine of neonates is insufficient for accurate measurement. In adult 46,XY subjects, an elevated testosterone/DHT ratio and markedly elevated urinary 5β/5α C19 androgen metabolites are essential to making the diagnosis. It should be noted that an elevation of the testosterone/DHT ratio can sometimes be found in subjects with androgen insensitivity. However, despite the elevated plasma testosterone/DHT ratio in some androgen insensitive subjects, the ratio of the urinary 5β/5α C19 androgen metabolites in androgen insensitivity are normal to only slightly elevated.70 Molecular genetic analysis to identify the 5α-reductase-2 gene mutation can be used to confirm the diagnosis.
Once the diagnosis of 5α-reductase-2 deficiency has been made in the newborn, a decision concerning the sex of rearing must be made. Our observations of the natural history of this disorder for more than 3 decades show that most affected subjects identify and behave as normal males in adulthood despite the trauma of having been raised in the wrong sex. We believe that it is desirable to raise children with 5α-reductase-2 deficiency as male, the gender compatible with their genetic and gonadal sex. They are psychosexually males and should be raised as male.42, 64, 65, 71 This necessitates early diagnosis of the condition followed by surgical correction of the external genitalia and correction of cryptorchidism if present.
Adequate genital correction in childhood is difficult because of the severity of the genital ambiguity, and the fact that the phallus is generally only slightly larger than a clitoris. To enlarge the phallus and facilitate hypospadias repair, newborns with this condition can be treated by administering DHT cream.71, 72, 73 Topical application of 2–2.5% DHT cream results in good phallic growth, facilitating surgical correction of penoscrotal hypospadias.71 The rationale behind the treatment is replacement of the deficient hormone DHT to induce phallic growth that theoretically would have occurred in utero and in the postnatal period. Administering DHT after the critical period of sexual differentiation in utero will stimulate phallic growth but will not correct the genital defect, as sexual differentiation occurs during a critical period in utero.
Most patients described in the literature have perineoscrotal hypospadias, and therefore surgical correction is more difficult. Despite this concern, surgical correction of the genitalia is feasible and made easier if enlargement of the phallus can be accomplished with DHT cream administration. Pseudovaginal perineoscrotal hypospadias has also been successfully repaired in adulthood in patients with 5α-reductase-2 deficiency. The quality of surgery, however, is dependent on the expertise of the surgeon and should therefore be attempted only by an experienced surgeon. Early correction of cryptorchism in infancy or early childhood could preserve fertility.47
If genital repair is successful, (1) the child will have a male puberty with normal male psychosexual development; (2) he will be equal in height to the normal males in his family; (3) there will be growth of the genitalia at puberty with an increase in muscle mass and deepening of the voice; (4) gynecomastia will not be a concern; and (5) fertility in this condition has been reported.46, 71
The most serious debate involves the management of subjects who are raised as females and diagnosed as having 5α-reductase-2 deficiency in the peri- and postpubertal period. After careful psychiatric evaluation, often subjects will be found to identify as males and should be encouraged to take their place as males in society. Occasionally, a patient is found to be unable to admit36 or acknowledge maleness; then a change in gender role should not be discussed at that time, and long-term evaluation is essential before gender decisions are made. Some subjects with this condition, who identify as male, will change gender role with time if they can deal with the social pressures of family, friends, etc. Thus, whether or not a gender role change will occur in an individual with 5α-reductase-2 deficiency at this time is obviously dependent on a host of social and cultural factors that might either consciously or subconsciously suppress or foster the change. All these factors must be considered by the patient's physician, as well as the psychiatrist working in concert with the patient and the family.71
SECONDARY CAUSES OF 5α-REDUCTASE DEFICIENCY
Secondary 5α-reductase deficiency has been described in subjects with androgen insensitivity, indicating that the 5α-reductase isozymes are regulated by androgens. Androgen-insensitive subjects often have decreased plasma DHT with elevated plasma testosterone/DHT ratios, similar to subjects with primary 5α-reductase-2 gene defects.70, 74 In contrast to the inherited 5α-reductase-2 deficiency in which a generalized severe defect of both hepatic and peripheral 5α-reductases is found, 5α-reductase activity in androgen insensitivity is preserved in the liver but deficient in the periphery. Thus, in androgen insensitivity syndrome, normal to minimally elevated urinary ratios of the 5β/5α metabolites of cortisol and corticosterone are found reflecting normal 5α-reduction metabolism in the liver, whereas increased ratios of the 5β/5α metabolites of C19 androgens reflect impaired peripheral 5α-reduction. This distinct steroid metabolite pattern can help distinguish between the two clinical entities.35, 70, 74 Clinical features of complete androgen insensitivity syndrome such as a female phenotype with gynecomastia and minimal to absent pubic and axillary hair highlight the diagnosis. Of course, molecular genetic identification of the gene defect will ultimately confirm the diagnosis.18, 33, 75
The defect in androgenic action from androgen receptor mutations may affect both type 1 and type 2 5α-reductase isozyme activity. Animal studies have shown that DHT upregulated 5α-reductase activity and both 5α-reductase type 1 and 2 mRNA levels in the rat prostate76, 77 and in primary cultures of rat or human scrotal skin fibroblasts.78
Hepatic 5α-reductase activity has also been found to be decreased in other clinical situations such as in porphyria,79 Cushing's syndrome,48 hypothyroidism, and anorexia nervosa.80 The reason of secondary 5α-reductase deficiency in these clinical conditions is unclear.
Studies of affected 46,XY subjects with 5α-reductase-2 deficiency over the last 3 decades has provided valuable information about male sexual development and has elucidated the roles of testosterone and DHT in human physiology and pathophysiology. Studies of subjects with this inherited condition have led to the development of specific 5α-reductase-2 inhibitors for the treatment of benign prostate hyperplasia and male pattern baldness.
REFERENCES
Evans RM: The steroid and thyroid hormone receptor superfamily. Science 240: 889, 1988 |
|
Zhu, Y. S. Molecular basis of steroid action in the prostate. Cellscience Reviews, 1: 27-55, 2005. |
|
Wilson JD: Sexual differentiation. Annu Rev Physiol 40: 270, 1978 |
|
Wilbert DM, Griffen JE, Wilson JD: Characterization of the cytosol androgen receptor of the human prostate. J Clin Endocrinol Metab 56: 113, 1983 |
|
Kovacs WJ, Griffin JE, Weaver DD et al: A mutation that causes lability of the androgen receptor under conditions that normally promote transformation to the DNA-binding state. J Clin Invest 73: 1095, 1984 |
|
McGuire JS, Tomkins GM: The heterogeneity of delta 4-3-ketosteroid reductase (5&b.alpha;). J Biol Chem 235: 1634, 1960 |
|
Moore RJ, Wilson JD: Diminished 5 alpha-reductase activity in extracts of fibroblasts cultured from patients with familial incomplete male pseudohermaphroditism, type 2. J Biol Chem 250: 7168, 1975 |
|
Moore RJ, Wilson JD: Steroid 5-alpha-reductase in cultured human fibroblasts: Biochemical and genetic evidence for two distinct enzyme activities. J Biol Chem 251: 5895, 1976 |
|
Wilson JD: Dihydrotestosterone formation in cultured human fibroblasts:Comparison of cells from normal subjects and patients with familial incomplete male pseudohermaphroditism, type 2. J Biol Chem 250: 3498, 1975 |
|
Bruchovsky N, Rennie PS, Batzold FH et al: Kinetic parameters of 5&b.alpha;-reductase activity in stroma and epithelium of normal, hyperplastic, and carcinomatous human prostates. J Clin Endocrinol Metab 67: 806, 1988 |
|
Hudson RW: Comparison of nuclear 5-alpha-reductase activities in the stromal and epithelial fractions of human prostatic tissue. J Steroid Biochem 26: 349, 1987 |
|
Jenkins EP, Andersson S, Imperato-McGinley J et al: Genetic and pharmacological evidence for more than one human steroid 5A-reductase. J Clin Invest 89: 293, 1992 |
|
Andersson S, Russell DW: Structural and biochemical properties of cloned and expressed human and rat steroid 5&b.alpha;-reductases. Proc Natl Acad Sci U S A 87: 3640, 1990 |
|
Andersson S, Berman DM, Jenkins EP et al: Deletion of steroid 5&b.alpha;-reductase-2 gene in male pseudohermaphroditism. Nature 354: 159, 1991 |
|
Labrie F, Sugimoto Y, Luu-The V et al: Structure of human type II 5-alpha-reductase gene. Endocrinology 131: 1571, 1992 |
|
Russell DW, Wilson JD: Steroid 5 alpha-reductase: Two genes/two enzymes. Annu Rev Biochem 63: 25, 1994 |
|
Thigpen, A. E., Davis, D. L., Gautier, T., Imperato-McGinley, J., and Russell, D. W. The molecular basis of steroid 5 alpha-reductase deficiency in a large Dominican kindred. N.Engl.J.Med., 327: 1216-1219, 1992. |
|
Zhu, Y. S., Katz, M. D., and Imperato-McGinley, J. Natural potent androgens: lessons from human genetic models. Baillieres.Clin.Endocrinol.Metab., 12: 83-113, 1998. |
|
Thigpen, A. E., Cala, K. M., and Russell, D. W. Characterization of chinese hamster ovary cell lines expressing human steroid 5 alpha-reductase isozymes. J.Biol.Chem., 268: 17404-17412, 1993. |
|
Thigpen, A. E., Silver, R. I., Guileyardo, J. M., Casey, M. L., McConnell, J. D., and Russell, D. W. Tissue distribution and ontogeny of steroid 5-reductase isozyme expression. J.Clin.Invest., 92: 903-910, 1993. |
|
Eicheler, W., Tuohimaa, P., Vilja, P., Adermann, K., Forssmann, W. G., and Aumuller, G. Immunocytochemical localization of human 5 alpha-reductase 2 with polyclonal antibodies in androgen target and non-target human tissues. J.Histochem.Cytochem., 42: 667-675, 1994. |
|
Eicheler, W., Dreher, M., Hoffmann, R., Happle, R., and Aumuller, G. Immunohistochemical evidence for differential distribution of 5 alpha-reductase isoenzymes in human skin. Br.J.Dermatol., 133: 371-376, 1995. |
|
Mahendroo, M. S., Cala, K. M., and Russell, D. W. 5-Reduced androgens play a key role in murine parturition. Mol.Endocrinol., 10: 380-392, 1996. |
|
Mahendroo, M. S., Cala, K. M., Landrum, C. P., and Russell, D. W. Fetal death in mice lacking 5-reductase type 1 caused by estrogen excess. Mol.Endocrinol., 11: 917-927, 1997. |
|
Imperato-McGinley, J., Guerrero, L., Gautier, T., and Peterson, R. E. Steroid 5-reductase deficiency in man: An inherited form of male pseudohermaphroditism. Science, 186: 1213-1216, 1974. |
|
Walsh, P. C., Madden, J. D., Harrod, M. J., Goldstein, J. L., MacDonald, P. C., and Wilson, J. D. Familial incomplete male pseudohermaphroditism, type 2. Decreased dihydrotestosterone formation in pseudovaginal perineoscrotal hypospadias. N.Engl.J.Med., 291: 944-949, 1974. |
|
Imperato-McGinley, J., Miller, M., Wilson, J. D., Peterson, R. E., Shackleton, C. H. L., and Gajdusek, D. C. A cluster of male pseudohermaphrodites with 5 alpha-reductase deficiency in Papua New Guinea. Clin.Endocrinol.(Oxf.), 34: 293-298, 1991. |
|
Akgun, S., Ertel, N., Imperato-McGinley, J., Sayli, B., and Shackleton, C. H. L. Familial male pseudohermaphroditism in a Turkish village due to 5-reductase deficiency. Am.J.Med., 81: 267-274, 1986. |
|
Imperato-McGinley, J., Akgun, S., Ertel, N. H., Sayli, B., and Shackleton, C. H. L. The coexistence of male pseudohermaphrodites with 17-ketosteroid reductase deficiency and 5-reductase deficiency within a Turkish kindred. Clin.Endocrinol.(Oxf.), 27: 135-143, 1987. |
|
Can, S., Zhu, Y. S., Cai, L. Q., Ling, Q., Katz, M. D., Akgun, S., Shackleton, C. H. L., and Imperato-McGinley, J. The identification of 5-reductase-2 and 17 -hydroxysteroid dehydrogenase-3 gene defects in male pseudohermaphrodites from a Turkish kindred. J.Clin.Endocrinol.Metab., 83: 560-569, 1998. |
|
Thigpen, A. E., Davis, D. L., Milatovich, A., Mendonca, B. B., Imperato-McGinley, J., Francke, U., Wilson, J. D., and Russell, D. W. The molecular genetics of steroid 5-reductase 2 deficiency. J.Clin.Invest., 90: 799-809, 1992. |
|
Wilson, J. D., Griffin, J. E., and Russell, D. W. Steroid 5-reductase 2 deficiency. Endocr.Rev., 14: 577-593, 1993. |
|
Zhu, Y. S. and Imperato-McGinley, J. Pseudohermaphroditism, male, due to 5-reductase-2 deficiency. In L. Martini (ed.), Encyclopedia Of Endocrine Diseases, pp. 131-135. San Diego, CA: Elsevier Inc., 2004. |
|
Imperato-McGinley, J. Disorders of sexual differentiation. In J. B. Wyngaarden, L. H. Smith, Jr., and J. C. Bennett (eds.), Cecil Textbook of Medicine, 19 ed, pp. 1320-1332. Philadelphia: W.B. Saunders, 1992. |
|
Fratianni, C. M. and Imperato-McGinley, J. The syndrome of 5-reductase deficiency. The Endocrinologist, 4(4): 302-314, 1994. |
|
Imperato-McGinley, J., Peterson, R. E., Leshin, M., Cooper, G., Draghi, S., Berenyi, M., and Wilson, J. D. Steroid 5-reductase deficiency in a 65-yar old male pseudohermaphrodite: The natural history, ultrastructure of the testes and evidence for inherited enzyme heterogeneity. J.Clin.Endocrinol.Metab., 50: 15-22, 1980. |
|
Imperato-McGinley, J., Peterson, R. E., Gautier, T., and Sturla, E. Male pseudohermaphroditism secondary to 5-reductase deficiency: A model for the role of androgens in both the development of the male phenotype and the evolution of a male gender identity. J.Steroid Biochem.Mol.Biol., 11: 637-645, 1979. |
|
Ng, W. K., Taylor, N. F., Hughes, I. A., and et al. 5-reductase deficiency without hypospadias. Arch.Dis.Child., 65: 1166-1167, 1990. |
|
Siiteri, P. and Wilson, J. D. Testosterone formation and metabolism during male sexual differentiation in the human embryo. J.Clin.Endocrinol.Metab., 38: 113-125, 1974. |
|
Imperato-McGinley, J., Sanchez, R. S., Spencer, J. R., Yee, B., and Vaughan, E. D. Comparison of the effects of the 5-reductase inhibitor finasteride and the antiandrogen flutamide on prostate and genital differentiation: Dose-response studies. Endocrinol., 131: 1149-1156, 1992. |
|
Spencer, J. R., Torrado, T., Sanchez, R. S., Vaughan, E. D., Jr., and Imperato-McGinley, J. Effects of flutamide and finasteride on rat testicular descent. Endocrinol., 129: 741-748, 1991. |
|
Imperato-McGinley, J., Peterson, R. E., Gautier, T., and Sturla, E. The impact of androgens on the evolution of male gender identity. In S. J. Kogan and E. S. E. Hafez (eds.), Pediatric andrology, pp. 99-108. The Hague: Martinus Nijhoff, 1981. |
|
Imperato-McGinley, J. and Zhu, Y. S. Androgens and male physiology - The syndrome of 5-reductase-2 deficiency. Mol.Cell.Endocrinol., 198: 51-59, 2002. |
|
Cantu, J. M., Hernandez-Montes, H., del Castillo, V., Cortés-Gallegos, V., Sandoval, R., Armendares, S., and Parra, A. Potential fertility in incomplete male pseudohermaphroditism type 2. Rev.Invest.Clin., 28: 177-182, 1976. |
|
Cai, L.-Q., Fratianni, C. M., Gautier, T., and Imperato-McGinley, J. Dihydrotestosterone regulation of semen in male pseudohermaphrodites with 5 alpha-reductase-2 deficiency. J.Clin.Endocrinol.Metab., 79(2): 409-414, 1994. |
|
Katz, M. D., Kligman, I., Cai, L. Q., Zhu, Y. S., Fratianni, C. M., Zervoudakis, I., Rosenwaks, Z., and Imperato-McGinley, J. Paternity by intrauterine insemination with sperm from a man with 5alpha-reductase-2 deficiency. N.Engl.J.Med., 336: 994-997, 1997. |
|
Nordenskjold, A. and Ivarsson, S. A. Molecular characterization of 5 alpha-reductase type 2 deficiency and fertility in a Swedish family. J Clin Endocrinol Metab, 83: 3236-3238, 1998. |
|
Peterson, R. E., Imperato-McGinley, J., Gautier, T., and Sturla, E. Male pseudohermaphroditism due to steroid 5-reductase deficiency. Am.J.Med., 62: 170-191, 1977. |
|
Imperato-McGinley, J., Gautier, T., Zirinsky, K., Hom, T., Palomo, O., Stein, E., Vaughan, E. D., Markisz, J., Ramirez de Arellano, E., and Kazam, E. Prostate visualization studies in males homozygous and heterozygous for 5-reductase deficiency. J.Clin.Endocrinol.Metab., 75: 1022-1026, 1992. |
|
Mendonca, B. B., Inacio, M., Costa, E. M., Arnhold, I. J., Silva, F. A., Nicolau, W., Bloise, W., Russel, D. W., and Wilson, J. D. Male pseudohermaphroditism due to steroid 5alpha-reductase 2 deficiency. diagnosis, psychological evaluation, and management. Medicine (Baltimore), 75: 64-76, 1996. |
|
Kaufman, K. D., Olsen, E. A., Whiting, D., Savin, R., DeVillez, R., Bergfeld, W., Price, V. H., Van Neste, D., Roberts, J. L., Hordinsky, M., Shapiro, J., Binkowitz, B., and Gormley, G. J. Finasteride in the treatment of men with androgenetic alopecia. Finasteride Male Pattern Hair Loss Study Group. J Am.Acad.Dermatol., 39: 578-589, 1998. |
|
Roberts, J. L., Fiedler, V., Imperato-McGinley, J., Whiting, D., Olsen, E., Shupack, J., Stough, D., DeVillez, R., Rietschel, R., Savin, R., Bergfeld, W., Swinehart, J., Funicella, T., Hordinsky, M., Lowe, N., Katz, I., Lucky, A., Drake, L., Price, V. H., Weiss, D., Whitmore, E., Millikan, L., Muller, S., Gencheff, C., and . Clinical dose ranging studies with finasteride, a type 2 5alpha- reductase inhibitor, in men with male pattern hair loss. J Am.Acad.Dermatol., 41: 555-563, 1999. |
|
Imperato-McGinley, J., Gautier, T., Yee, B., Cai, L.-Q., Epstein, J., and Pochi, P. The androgen control of sebum production: Studies of subjects with dihydrotestosterone deficiency and complete androgen insensitivity. J.Clin.Endocrinol.Metab., 76: 524-533, 1993. |
|
Katz, M. D., Cai, L.-Q., Zhu, Y. S., Herrera, C., DeFillo-Ricart, M., Shackleton, C. H. L., and Imperato-McGinley, J. The biochemical and phenotypic characterization of females homozygous for 5-reductase-2 deficiency. J.Clin.Endocrinol.Metab., 80: 3160-3167, 1995. |
|
Ertel, N. H., Akgun, S., Samojlik, E., Kirschner, M. A., and Imperato-McGinley, J. Decreased 3-androstanediol glucuronide levels in plasma and random urines in male pseudohermaphroditism caused by 5- reductase deficiency. Metabolism, 38: 817-821, 1989. |
|
Peterson, R. E., Imperato-McGinley, J., Gautier, T., and Shackleton, C. H. L. Urinary steroid metabolites in subjects with male pseudohermaphroditism due to 5-reductase deficiency. Clin.Endocrinol.(Oxf.), 23: 43-53, 1985. |
|
Canovatchel, W. J., Gautier, T., Volquez, D., and et al. LH pulsatility in subjects with 5-reductase deficiency and decreased DHT production. J.Clin.Endocrinol.Metab., 78(4): 916-921, 1994. |
|
Fratianni, C. M., Canovatchel, W. J., Gautier, T., Volquez, D., Huang, S., and Imperato-McGinley, J. FSH pulsatile secretion by RIA and IFMA in subjects with 5 alpha-reductase deficiency (Abstract #398). The Endocrine Society 75th Annual Meeting, Las Vagas. 1993. |
|
Baldinotti, F., Majore, S., Fogli, A., Marrocco, G., Ghirri, P., Vuerich, M., Tumini, S., Boscherini, B., Vetri, M., Scommegna, S., Rinaldi, R., Simi, P., and Grammatico, P. Molecular Characterization of 6 Unrelated Italian Patients With 5alpha-Reductase Type 2 Deficiency. J Androl, 29: 20-28, 2008. |
|
Cai, L.-Q., Zhu, Y. S., Katz, M. D., Herrera, C., Baez, J., DeFillo-Ricart, M., Shackleton, C. H. L., and Imperato-McGinley, J. 5-reductase-2 gene mutation in the Dominican Republic. J.Clin.Endocrinol.Metab., 81: 1730-1735, 1996. |
|
Wigley, W. C., Prihoda, J. S., Mowszowicz, I., Mendonca, B. B., New, M. I., Wilson, J. D., and Russell, D. W. Natural mutagenesis study of the human steroid 5 alpha-reductase 2 isozyme. Biochemistry, 33: 1265-1270, 1994. |
|
Imperato-McGinley, J., Peterson, R. E., Gautier, T., Shackleton, C. H. L., and Arthur, A. Decreased urinary C19 and C21 steroid 5-metabolites in parents of male pseudohermaphrodites with 5-reductase deficiency: Detection of carriers. J.Clin.Endocrinol.Metab., 60: 553-558, 1985. |
|
Imperato-McGinley, J. and Peterson, R. E. Gender identity and hermaphroditism. Science, 191: 872, 1976. |
|
Imperato-McGinley, J., Peterson, R. E., Gautier, T., and Sturla, E. Androgens and the evolution of male-gender identity among male pseudohermaphrodites with 5-reductase deficiency. N.Engl.J.Med., 300: 1233-1237, 1979. |
|
Imperato-McGinley, J. and Zhu, Y. S. Gender and behavior in subjects with genetic defects in male sexual differentiation. In D. W. Pfaff, A. P. Arnold, A. M. Etgen, S. E. Fahrbach, and R. T. Rubin (eds.), Hormones, Brain, Behavior, 1 ed, pp. 303-345. Orlando, Florida: Academic Press, 2002. |
|
Money, J., Hampson, J. G., and Hampson, J. L. Hermaphroditism: Recommendations concerning assignment of sex, change of sex and psychological management. Bull.Johns Hopkins Hosp., 97: 284-300, 1955. |
|
Money, J., Hampson, J. G., and Hampson, J. L. An examination of some basic sexual concepts: The evidence of human hermaphroditism. Bull.Johns Hopkins Hosp., 97: 301-319, 1955. |
|
Money, J. and Ogunro, C. Behavioral sexology: ten cases of genetic male intersexuality with impaired prenatal and pubertal androgenization. Arch.Sex.Behav., 3: 181-205, 1974. |
|
Imperato-McGinley, J., Gautier, T., Pichardo, M., and Shackleton, C. H. L. The diagnosis of 5-reductase deficiency in infancy. J.Clin.Endocrinol.Metab., 63: 1313-1318, 1986. |
|
Imperato-McGinley, J., Peterson, R. E., Gautier, T., Cooper, G., Danner, R., Arthur, A., Morris, P. L., Sweeney, W. J., and Shackleton, C. H. L. Hormonal evaluation of a large kindred with complete androgen insensitivity: Evidence for secondary 5-reductase deficiency. J.Clin.Endocrinol.Metab., 54: 931-941, 1982. |
|
Imperato-McGinley, J. 5 alpha-reductase-2 deficiency. Curr.Ther.Endocrinol.Metab., 6: 384-387, 1997. |
|
Carpenter, T. O., Imperato-McGinley, J., Boulware, S. D., Weiss, R. M., Shackleton, C. H. L., and Wilson, J. D. Variable expression of 5 alpha-reductase deficiency: Presentation with male phenotype in a child of Greek origin. .Clin.Endocrinol.Metab., 71(2): 318-322, 1990. |
|
Cantu, J. M. Product replacement therapy in steroid 5-reductase deficiency. Ann.Genet., 21: 120-121, 1978. |
|
Jukier, L., Kaufman, M., Pinsky, L., and Peterson, R. E. Partial androgen resistance associated with secondary 5 alpha- reductase deficiency: identification of a novel qualitative androgen receptor defect and clinical implications. J.Clin.Endocrinol.Metab., 59: 679-688, 1984. |
|
Zhu, Y. S., Cai, L. Q., Cordero, J. J., Canovatchel, W. J., Katz, M. D., and Imperato-McGinley, J. A novel mutation in the CAG triplet region of exon 1 of androgen receptor gene causes complete androgen insensitivity syndrome in a large kindred. J.Clin.Endocrinol.Metab., 84: 1590-1594, 1999. |
|
George, F. W., Russell, D. W., and Wilson, J. D. Feed-forward control of prostate growth: dihydrotestosterone induces expression of its own biosynthetic enzyme, steroid 5 alpha-reductase. Proc.Natl.Acad.Sci.U.S.A., 88: 8044-8047, 1991. |
|
Normington, K. and Russell, D. W. Tissue distribution and kinetic characteristics of rat steroid 5 alpha-reductase isozymes. evidence for distinct physiological functions. J.Biol.Chem., 267: 19548-19554, 1992. |
|
Horton, R., Pasupuletti, V., and Antonipillai, I. Androgen induction of steroid 5 alpha-reductase may be mediated via insulin-like growth factor-I. Endocrinol., 133: 447-451, 1993. |
|
Kappas, A., Bradlow, H. L., Gillette, P. N., and Gallagher, T. F. 3. Studies in porphyria. I. a defect in the reductive transformation of natural steroid hormones in the hereditary liver disease, acute intermittent porphyria. J.Exp.Med., 136: 1043-1053, 1972. |
|
Bradlow, H. L., Boyar, R. M., O'Connor, J., Zumoff, B., and Hellman, L. Hypothyroid-like alterations in testosterone metabolism in anorexia nervosa. J.Clin.Endocrinol.Metab., 43: 571-574, 1976. |