Sex Cord-Stromal, Steroid Cell, and Germ Cell Tumors of the Ovary
Authors
INTRODUCTION
Although most benign and malignant ovarian tumors are derived from the surface epithelium of the ovary, a variety of often morphologically complex tumors originate from the most highly specialized elements of the organ, namely, the sex cords, the stroma or mesenchyme, and the germ cells.1, 2 These tumors potentially pose many problems in differential diagnosis for the pathologist because of their varied microscopic features. They are often of particular interest clinically because of endocrine function and in malignant cases a sometimes prolonged course. This chapter deals primarily with the pathologic and clinical aspects of these tumors. It follows the histologic classification and nomenclature of the World Health Organization (WHO) and International Society of Gynecological Pathologists, which have attempted to formulate internationally acceptable terminology for ovarian tumors.3
SEX CORD-STROMAL TUMORS
This category includes tumors that are purely composed of epithelial (sex cord) elements, mesenchymal (stromal) elements, or both. The descriptive term sex cord-stromal tumors avoids any controversy about the origin of the neoplastic constituents and, at the same time, acknowledges the frequent presence of both sex cord (e.g., granulosa and Sertoli cells) and stromal derivatives (e.g., fibroblasts, theca cells, and Leydig cells) within these tumors.
Most sex cord-stromal tumors are readily identifiable as belonging in either the granulosa-stromal cell or the Sertoli-stromal cell category, but a minority have patterns or cell types that are intermediate morphologically between the two. These tumors are designated sex cord-stromal tumors, unclassified. One tumor in this intermediate category with distinctive pathologic features and clinical associations has been given a specific designation: sex cord tumor with anular tubules. A rare tumor contains significant amounts of easily recognizable testicular and ovarian elements, warranting the diagnosis of a mixed form of sex cord-stromal tumor, or gynandroblastoma. That tumor is exceedingly rare if one uses the stringent criteria for its diagnosis adopted by WHO. Including the nonfunctioning fibroma, a pure stromal neoplasm that accounts for more than half of the sex cord-stromal tumors, the latter are responsible for only approximately 6% of all ovarian neoplasms.
Granulosa-stromal cell tumors
These tumors are composed of granulosa cells and stromal derivatives, that is, fibroblasts and theca cells or both, with each type of cell occurring singly or in various combinations. Granulosa cell tumors are defined as neoplasms containing more than a minor component of granulosa cells with or without accompanying theca cells or fibroblasts. The designation granulosa-theca cell tumor has often been used for those specimens containing both cell types but granulosa cell tumor is probably more appropriate because it is difficult to be certain whether the theca element is truly neoplastic or reflects a response of the ovarian stroma to the proliferation of the neoplastic granulosa cells in many cases. The second group of tumors within the granulosa-stromal cell category has been designated generically as “tumors in the thecoma-fibroma group” because of the morphologic overlap between the thecoma and the fibroma and the occasional difficulty in distinguishing them when the morphologic features are intermediate.
GRANULOSA CELL TUMOR
The granulosa cell tumor is the most common form of ovarian neoplasm associated with overt endocrine manifestations, which are almost always estrogenic. The tumor occurs in all age groups but is most common between 50 and 55 years of age. Fewer than 5% of the patients are younger than the age of normal puberty, and approximately three quarters of these children exhibit isosexual pseudoprecocity.4 In adult patients, menstrual disturbances such as menometrorrhagia, oligomenorrhea, and amenorrhea, which may last for months to years, are commonly encountered. The typical clinical manifestation in the postmenopausal woman is uterine bleeding. The pathologic background for these menstrual disorders comprises a spectrum of endometrial changes, which range from cystic hyperplasia to atypical hyperplasia to invasive adenocarcinoma. Carcinoma develops in approximately 5% of all adult patients with granulosa cell tumors (or thecomas), is twice as frequent in postmenopausal as in premenopausal women with these neoplasms, and is of low grade and amenable to curative therapy in the majority of cases.5 Granulosa cell tumors may be associated with additional estrogenic manifestations, such as swelling and tenderness of the breasts and a feeling of rejuvenation in older women. A rare granulosa cell tumor is virilizing.6 In approximately 10% of cases, the presenting manifestations are because of rupture of the tumor with hemoperitoneum.
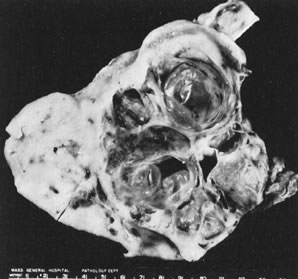
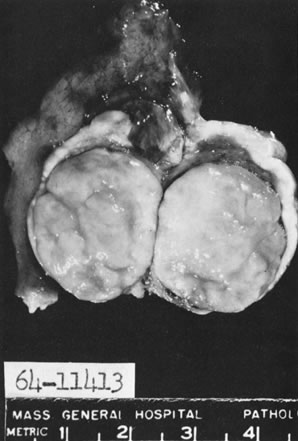
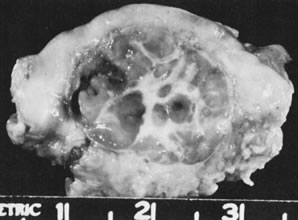
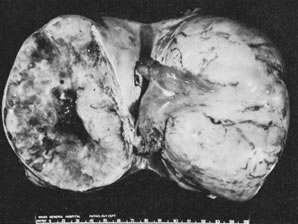
In 10–15% of the cases, the granulosa cell tumor does not cause palpable ovarian enlargement, and unless ultrasound examination is performed before surgery, is not detected until the time of a hysterectomy for another indication, such as atypical endometrial hyperplasia or carcinoma. More often, the tumor is palpable on pelvic or abdominal examination. At operation, it may appear solid or have both solid and cystic components (Fig. 1). On sectioning, the solid tissue may be gray, white, or yellow, reflecting the amount of intracellular fat it contains, and may be soft or firm depending on the relative quantities of granulosa cells and fibrothecomatous components. Two types of gross presentation are highly suggestive of granulosa cell tumor. One is a solid, pale yellow mass associated with extensive hemorrhage, and the other is a multicystic tumor with the locules filled with fluid or clotted blood. A rare appearance is that of a large unilocular or oligolocular thin-walled cystic tumor, resembling a serous cystadenoma; curiously, this gross appearance unusually often is associated with virilization instead of estrogenic changes.6 In summary, the granulosa cell tumor can simulate a variety of other types of ovarian tumor on gross examination, but the diagnosis may be suggested by the gross features, particularly in some clinical settings.
|
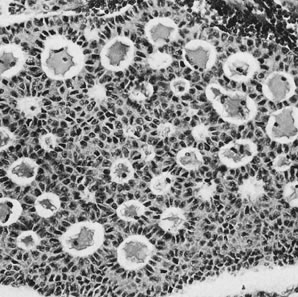
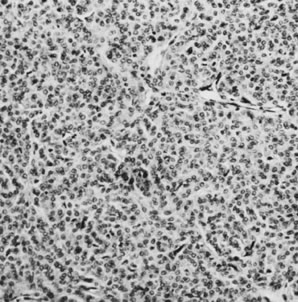
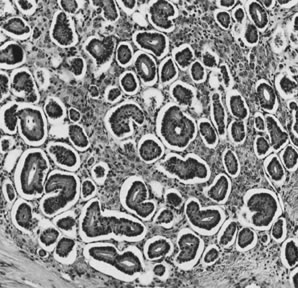
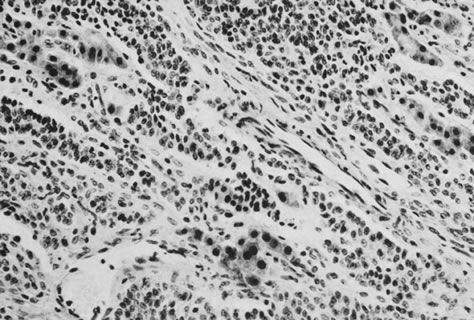
On microscopic examination, the granulosa cell tumor can be divided into two major subtypes, adult form and juvenile form, which differ strikingly in their patterns and cytologic features. The adult granulosa cell tumor is occasionally composed almost entirely of granulosa cells, with few or no accompanying thecal or fibroblastic elements, but more often the latter are present in some amount and may be substantial. The granulosa cell component of the tumor has a variety of patterns. The well-differentiated form most commonly has a microfollicular (Fig. 2), trabecular (Fig. 3), or insular pattern, or a combination of the three. Rarely, the pattern is macrofollicular, with the tumor composed of large follicles resembling normally developing follicles. Less well-differentiated tumors typically have a diffuse or sarcomatoid pattern, characterized by a sea of cells with little or no intervening stroma (Fig. 4); this type of tumor is particularly apt to rupture. Sometimes a watered-silk or zigzag (gyriform) pattern is seen. The stromal element of the tumor may consist of fibroblasts that have laid down considerable collagen, as well as cells that resemble theca externa, theca interna, or theca lutein cells.
|
|

|
Lipid stains and a variety of histochemical reactions, which, when positive, are considered characteristic of steroid hormone-producing cells, typically color the theca cell component of the tumor but not the granulosa cells, suggesting that the theca cells are responsible for the hormone secretion. It is possible, however, that the theca cells secrete androgens, which are aromatized to estrogens by the granulosa cells in tumors containing both elements, as in the normal follicle. Some estrogenic granulosa cell tumors, in contrast, appear to have only a minor theca cell component, and the mechanism of estrogen production by such tumors is not clear.
The juvenile granulosa cell tumor is so named because more than 95% of the cases occur within the first 3 decades, accounting for the majority of granulosa cell tumors occurring during that age period.4 Microscopic examination generally shows a nodular pattern of granulosa cell proliferation with the nodules typically containing follicles intermediate in size between the microfollicles and macrofollicles of the adult form of the tumor. The follicles are typically more variable in size and shape than those of the adult granulosa cell tumor. The neoplastic granulosa cells typically have abundant cytoplasm (i.e., are luteinized) unlike the more commonly nonluteinized cells of the adult tumor. They usually exhibit brisk mitotic activity. In addition, the nuclei of the juvenile tumor are not pale and grooved as in the adult granulosa cell tumor and may be strikingly pleomorphic. As a result, some juvenile granulosa cell tumors have a highly malignant appearance microscopically and are sometimes confused with yolk sac tumors or embryonal carcinomas by inexperienced pathologists.
The treatment of granulosa cell tumors depends on their stage and the age of the patient.7 The majority of these tumors are stage I. Because bilaterality is encountered in fewer than 5% of the cases, unilateral tumors can be treated effectively by simple salpingo-oophorectomy when the patient is young and the preservation of fertility is an important consideration. For the older woman, the appropriate treatment is almost always hysterectomy with bilateral salpingo-oophorectomy, even in stage IA cases. Recurrences are mostly in the pelvis and lower abdomen, but distant metastases have been reported at numerous sites. Recurrences can often be treated effectively by another surgical procedure followed by chemotherapy or radiation therapy, to which some adult granulosa cell tumors have been very sensitive. Various chemotherapeutic regimens used in the treatment of malignant germ cell tumors, such as vincristine, actinomycin D, and cyclophosphamide; cisplatin, vinblastine, and bleomycin; and bleomycin, etoposide, and cisplatin, have proved effective in the treatment of extraovarian disease in cases of adult granulosa cell tumor. There has been insufficient experience with juvenile granulosa cell tumors to draw conclusions about the value of chemotherapy or radiation therapy.
The survival rate of patients with adult granulosa cell tumors is approximately 90% at 10 years but drops to approximately 50% at 30 years because of the frequency of late recurrences, which have been reported more than 30 years after surgery.8, 9, 10 Despite their more malignant histologic appearance, juvenile granulosa cell tumors are associated with a better prognosis, with a survival rate of approximately 90%, and a rarity of late recurrences.4 Inhibin11 is available to aid in detecting the presence of recurrent granulosa cell tumor, and immunostaining for inhibin may be of great value in the pathologic differential diagnosis of granulosa cell tumors and, indeed, other sex cord-stromal tumors.12
TUMORS IN THE THECOMA-FIBROMA GROUP
This category includes fibromas and thecomas, as well as tumors with intermediate features. Included in the last category is a distinctive subtype: the sclerosing stromal tumor.
Fibroma
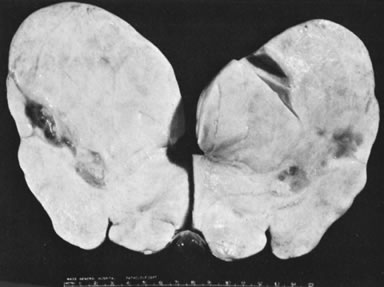
Ovarian fibromas occur at an average age of 48 years; fewer than 10% are encountered in females younger than the age of 30 years. These tumors are solid and have a fat, chalky-white, trabeculated appearance on their sectioned surfaces (Fig. 5). They are bilateral in approximately 10% of the cases.13 Microscopic examination shows intersecting fascicles of spindle cells producing collagen (Fig. 6); a storiform or pinwheel pattern is occasionally present. Hyaline plaques and foci of calcification are sometimes encountered, with calcification occurring, particularly in the bilateral tumors that are found in young women with the basal cell nevus (Gorlin's) syndrome.14 Intercellular edema is often present; a marked degree of edema has been correlated with the presence of ascites, which accompanies 40% of fibromas over 10 cm in diameter and with Meigs' syndrome (ascites and hydrothorax relieved by the removal of a benign ovarian tumor with fibromatous features), which occurs in fewer than 1% of the cases.15 Cellular fibromas with minor mitotic activity may recur, especially if they are found to be ruptured or adherent at the time of operation; even in the absence of rupture or adhesion, cellular fibromas occasionally recur many years after their removal.16 The rare fibrosarcoma is characterized by significant nuclear atypicality and mitotic activity that is generally greater on the average than three per 10 high-power fields; these tumors are highly malignant.
|
|
Thecoma
Thecomas are divided into two subtypes: the typical and the luteinized forms.17, 18 Both tumors form solid masses composed of firm tissue that may be yellow throughout or white with focal areas of yellow coloration (Fig. 7). Microscopic examination of the typical thecoma shows the exclusive presence or marked predominance of pale, vacuolated, lipid-rich cells that resemble theca interna cells (Fig. 8). A fibromatous component may be present, and occasionally a minor number of granulosa cells are observed. The luteinized thecoma has a background of typical fibroma or thecoma but contains, in addition, large lutein cells (i.e., cells resembling those of the corpus luteum) (Fig. 9). The lutein cells may contain variable amounts of lipid in their cytoplasm.
|
|
|
The typical thecoma occurs rarely before puberty and is seen in somewhat older patients on the average than the granulosa cell tumor. The clinical manifestations, which are almost always estrogenic, are similar to those of the granulosa cell tumor, except that a malignant behavior is extremely rare. Among the cases that have been described as malignant in the literature are endocrinologically inert fibrosarcomas and diffuse granulosa cell tumors misinterpreted as thecomas.19 Luteinized thecomas occur at an average younger age than typical thecomas; although they are most frequent in postmenopausal women, 30% of them have been encountered in patients younger than 30 years of age. The luteinized thecoma is estrogenic in approximately half the cases and androgenic in 11%. Because the thecoma is almost invariably benign, it can be treated by a conservative operation when it occurs in a young woman. A unique ovarian tumor that appears to fall in the luteinized thecoma group is associated with sclerosing peritonitis.20 The tumor is often bilateral and typically exhibits brisk mitotic activity. Despite the latter, it has not exhibited metastatic potential, but rare tumors have been fatal as a result of complications of the sclerosing peritonitis they cause by currently unknown mechanisms.
Unclassified
Because the theca cell and fibroblast, both in the normal ovary and in ovarian tumors, are both derivatives of the ovarian stromal cell, it is not surprising that occasional tumors exist in an intermediate zone between the thecoma and the fibroma. Such tumors may contain an intermediate quantity of cytoplasmic lipid and be unaccompanied by clear-cut evidence of steroid hormone production. The designation of tumors in this category as thecomas by some observers and fibromas by others probably accounts for the considerable variation in the reported frequency of the thecoma relative to the granulosa cell tumor.
The sclerosing stromal tumor belongs in the intermediate category between the thecoma and fibroma but possesses distinctive clinical and pathologic features.21 Unlike the fibroma and the typical thecoma, it has been encountered in the first 3 decades in more than 80% of personally observed cases. Ascites has complicated the sclerosing stromal tumor rarely, and exceptionally, estrogenic or androgenic manifestations have been described. The tumors have been unilateral. On gross examination, the neoplasm is typically predominantly solid with areas of edema and often cyst formation; an occasional tumor is predominantly cystic. The neoplastic tissue is white with foci of yellow coloration. The microscopic picture is characterized by the presence of pseudolobules of cellular tissue separated by cell-poor fibrous or edematous areas. The pseudolobules often contain a rich network of thin-walled vessels. The tumor cells are of two types: spindle cells producing collagen and rounded lutein-type cells with shrunken nuclei and vacuolated cytoplasm that contains considerable lipid. No case of malignant sclerosing stromal tumor has been reported. A rare cellular stromal tumor that contains signet-ring-like cells that do not contain mucin or lipid has been designated signet-ring stromal tumor.22
Sertoli-stromal cell tumors (androblastomas)
These tumors,23 which were originally designated arrhenoblastoma, have been divided into three categories according to their degrees of differentiation; a fourth category, characterized by the presence of foreign (heterologous) elements; and a fifth category with an architecture resembling that of the rete testis.
Sertoli-stromal cell tumors occur predominantly in young women in the reproductive age group, with an average age incidence of 25 years, but may be encountered occasionally in children and older women. Typically, these tumors are associated with a more-or-less abrupt onset of defeminization, with amenorrhea followed by progressive virilization, as a result of the production of a variety of androgens. A return of the menses to normal in approximately 1 month and usually some regression of hirsutism follows the complete removal of the tumor, but a decrease in size of an enlarged clitoris and restoration of normal female pitch occur less often. Although one third to two fifths of Sertoli-stromal cell tumors are virilizing, some are nonfunctioning and still others are associated with estrogenic manifestations. The latter may be the result of estrogen production by Sertoli cells or Leydig cells or possibly, in some cases, by secretion of androstenedione by Leydig cells and its conversion to estrone in the peripheral tissues.
The gross features of Sertoli-stromal cell tumors vary greatly from one specimen to another, resembling those of granulosa cell tumors, but the former tumors are cystic less often than the latter.
WELL-DIFFERENTIATED FORMS
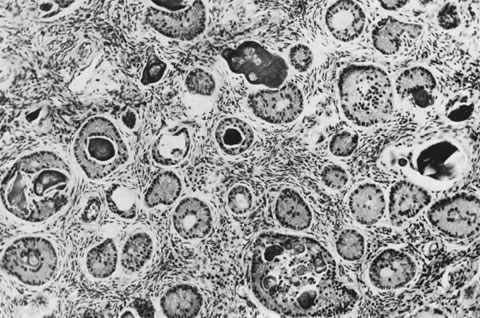
These tumors may be composed entirely or almost entirely of either Sertoli cells or Leydig cells or may contain both elements in considerable numbers; pure Sertoli cell tumors are rare.24 Well-differentiated Sertoli-stromal cell tumors of the mixed variety are characterized by hollow or solid tubules separated to varying extents by mature Leydig cells (Fig. 10), which occasionally contain rod-shaped eosinophilic bodies known as crystals of Reinke.25 These structures are more or less specific for Leydig cells. Varying amounts of lipid may be found within the cytoplasm of the Sertoli cells of the tubules as well as in the Leydig cells. These tumors may be associated with androgenic or estrogenic manifestations. Tumors composed mainly of Sertoli cells are often estrogenic. The pure Leydig cell tumor belongs nosologically in the Sertoli-stromal cell category but is discussed with steroid cell tumors because it is closest morphologically to neoplasms in that category.
|
TUMORS OF INTERMEDIATE DIFFERENTIATION
These tumors, which are the most common form of Sertoli-stromal cell tumor, are characterized by immature Sertoli cells, typically containing small oval-to-round nuclei and growing in a variety of patterns, and by a stromal component that varies in its cellularity, most typically being relatively acellular and often edematous, and contains at least some well-differentiated Leydig cells (Fig. 11). A lobular growth is often conspicuous on low power. The Sertoli cells may be disposed in round, oval, or elongated aggregates; in masses or within tubules, but most characteristically, they lie in short, single rows that simulate the sex cords of the developing testis. Varying amounts of lipid may be present in the neoplastic cells. The tumors are usually associated with androgenic manifestations but may be estrogenic or nonfunctioning.
|
POORLY DIFFERENTIATED TUMORS
These tumors were originally called sarcomatoid because one of the most frequent forms contains areas resembling a fibrosarcoma. However, the Sertoli cell, as well as the stromal component of the tumor, may be poorly differentiated; and simulate an undifferentiated carcinoma, not otherwise specified;a poorly differentiated Sertoli cell tumor may be estrogenic. Minor better-differentiated areas may be crucial in categorizing these tumors.
TUMORS WITH HETEROLOGOUS ELEMENTS
Heterologous elements are found mainly in tumors that have a background of intermediate or poor differentiation.2627 The most commonly encountered component is mucinous epithelium resembling that of the gastrointestinal tract, containing goblet cells and even argentaffin cells. The mucinous component may be benign, borderline, or carcinomatous and rarely is the predominant element of the tumor on both gross and microscopic examinations. Occasionally, microcarcinoid tumors develop from the argentaffin cells. Islands of immature cartilage and rhabdomyoblasts are the mesenchymal heterologous element that may be encountered. They may be prominent components of the sarcomatoid form of the neoplasm.
RETIFORM SERTOLI-LEYDIG CELL TUMOR
This tumor28 differs from the other forms clinically and pathologically, occurring at an average patient age of 15 years, 10 years younger than the average age of patients with Sertoli-stromal cell tumors as a group. Microscopic examination shows a network of tubules and papillae simulating the structure of the rete testis. Because of the presence of tubules, cysts, and papillae, the tumor is often misinterpreted by pathologists as a borderline or invasive serous tumor. The usual young age of the patient is a clinical clue that the tumor is not a serous tumor.
The prognosis of Sertoli-stromal cell tumors is related to both their stage and their histologic subtype.23 The well-differentiated tumors are benign. The rare tumors of other types that present at a stage higher than stage I are associated with a very poor prognosis. Patients with stage I tumors of intermediate differentiation have a survival rate of almost 90%; those with poorly differentiated tumors, 40%; and those with tumors containing mesenchymal heterologous elements such as skeletal muscle, cartilage, or both, approximately 30%. The presence of mucinous epithelium and carcinoid tumorlets does not appear to affect the prognosis of the underlying homologous component of the tumor. Too few cases of retiform Sertoli-stromal cell tumor with adequate follow-up for evaluation of prognosis have been reported, but these tumors appear to be more malignant clinically than otherwise similar Sertoli-stromal cell tumors without a retiform component.
Sertoli-stromal cell tumors are almost always unilateral, and therefore, unilateral salpingo-oophorectomy is the appropriate procedure for a stage IA tumor in a young patient in whom the preservation of fertility is an important consideration. The poor prognosis associated with poorly differentiated stage IA tumors, whether homologous or heterologous, raises the possibility of adjuvant chemotherapy to improve survival. In addition, because the survival of patients with ruptured intermediate tumors is approximately 20% lower than that of patients with intact tumors, adjuvant chemotherapy is a consideration for those tumors as well. There is very little experience with the chemotherapy of high-stage or recurrent Sertoli-stromal cell tumors, but partial and complete responses have been recorded occasionally with the use of regimens similar to those used for recurrent or metastatic granulosa cell tumors.
Gynandroblastomas
The term gynandroblastoma was coined to describe those tumors that contain cellular elements of both ovarian and testicular types.1 Unfortunately, the cases reported in the literature collectively comprise a hodgepodge of tumors that have little in common morphologically. In addition, it must be emphasized that extensive sampling of a granulosa cell tumor often discloses small areas that are more compatible morphologically with a Sertoli-stromal cell tumor and vice versa. For these reasons, the WHO has recommended that the term gynandroblastoma be restricted to those specimens in which there are substantial amounts of mature and indisputable ovarian and testicular cellular components. There are no consistent endocrine manifestations of these very rare tumors.
Sex Cord-Stromal Tumors, Unclassified
These tumors lie in the indeterminate zone between granulosa-stromal cell tumors and Sertoli-stromal cell tumors, with cells and patterns of growth that are compatible with, but not specific for, a diagnosis of either type of tumor. These neoplasms account for approximately 10% of all sex cord-stromal tumors. Sex cord-stromal tumors from pregnant patients are particularly apt to be hard to classify and often must be placed in the unclassified group.29
The sex cord tumor with annular tubules30 is a relatively rare neoplasm that exists in two forms: as a large solitary mass, which often metastasizes to lymph nodes, and, more remarkably, as a common feature of the Peutz-Jeghers syndrome (gastrointestinal polyposis with oral and cutaneous melanin pigmentation). In patients with this syndrome, the tumors are typically small, multiple, and focally calcified. The largest tumor associated with the Peutz-Jeghers syndrome was 3 cm in diameter. Microscopic examination shows solid tubules with a central mass of cytoplasm and peripheral nuclei; the tubules are arranged in the form of a simple ring with an eosinophilic hyaline body at the center or, more often, a complex ring with the tubule rotating around multiple hyaline spheres (Fig. 12). The small tumors in patients with the Peutz-Jeghers syndrome may be associated with estrogenic manifestations. The large tumors, which are not associated with the syndrome, are usually nonfunctioning but may have estrogenic or, rarely, androgenic manifestations and often secrete progesterone in large amounts, resulting in a decidual reaction in the endometrium. In addition to an elevation of progesterone in the serum, there may be an increase in müllerian inhibiting substance. In one reported case, it served as a helpful tumor marker, becoming elevated before clinical evidence of recurrent tumors over a period of 22 years.31
|
STEROID CELL TUMORS
These tumors, which were called lipid cell tumors and lipoid cell tumors, are composed exclusively of large cells with abundant cytoplasm and central nuclei, resembling lutein, Leydig, and adrenocortical cells (Fig. 13). When pathologists encounter a rare tumor of this type, they should use all the clinical, biochemical, topographic, and cytologic evidence available to determine the cell of origin. In most cases, this determination is unsuccessful and the tumor is designated “steroid cell tumor, not otherwise specified.”32 In approximately one quarter of the cases, a small number of the neoplastic cells contain crystals of Reinke, identifying the tumor as a Leydig cell tumor (Figs. 14 and 15). This tumor, which is almost always benign and typically secretes testosterone and virilizes the patient, has two possible origins. It may arise from hilus cells (hilar Leydig cells), which can be identified in the ovarian hilus in more than 80% of the female population.33 The second origin is from ovarian stromal cells, which rarely exhibit focal differentiation into Leydig cells. When the tumor lies entirely in the hilus, it is usually referred to as a “hilus cell tumor.” When it is confined within the ovarian stroma it is called “Leydig cell tumor, nonhilar type.” Some Leydig cell tumors arise at the junction of the medullary stroma and hilus or are so large that their site of origin is not possible to determine.
|

|
|
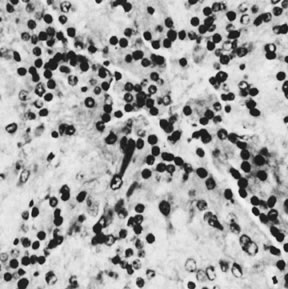
A second type of steroid cell tumor of identifiable origin is the stromal luteoma, which is composed of a uniform population of steroid-type cells and is situated within the ovarian stroma, often on a background of stromal hyperthecosis (stromal hyperplasia with the formation of lutein cell nests).34 Stromal luteomas are small benign tumors that are usually associated with estrogenic manifestations but may be nonfunctioning or occasionally androgenic.
Steroid cell tumors not otherwise specified are often large neoplasms of which the site of origin is indeterminable because of their size and the absence of identifiable crystals of Reinke in the cytoplasm of their cells. It is assumed that they arise either from the ovarian stroma or from hilus cells. These tumors are usually androgenic, but occasional examples are estrogenic or are associated with Cushing's syndrome. Microscopic examination shows steroid-type cells that contain variable amounts of intracellular lipid, which is often abundant, imparting a yellow color to the tumor tissue on gross examination. The terms lipid cell tumor and lipoid cell tumor are not appropriate for all the tumors in this category because approximately 25% contain little or no intracellular lipid and are red to brown on gross examination.
At least 40% of steroid cell tumors not otherwise specified are clinically malignant.31 Such tumors typically have one or more of the following features: a diameter of 7 cm or greater; loci of hemorrhage or necrosis or both; significant nuclear atypicality; and a mitotic rate of two or more per 10 high-power fields. 32 A malignant behavior has not been reported in children, in whom these tumors are very rare; in older women, the frequency of malignancy is greater after than before menopause. Most of the tumors that have been associated with Cushing's syndrome have been malignant. In these cases, although an origin in adrenal rest tissue has been considered a possibility and has given rise to the term adrenal rest tumor of the ovary, such a derivation is unlikely since adrenal rests occur almost exclusively in the broad ligament or ovarian hilus, and the tumors associated with Cushing's syndrome have been intraovarian. It appears more likely that the clinical manifestations are caused by ectopic production of adrenal hormones by steroid cells of gonadal origin.
Steroid cell tumors not otherwise specified are almost always unilateral and in young females can be treated by salpingo-oophorectomy if the tumor is unilateral. No effective treatment has been reported for steroid cell tumors that have extended beyond the ovary or recurred after oophorectomy.
Pregnancy luteomas are tumor-like nodules composed of lipid-free steroid cells occurring during pregnancy.1 They are detected almost always during the third trimester in multipara and undergo involution in the puerperium, indicating that they are dependent on human chorionic gonadotropin for their structural integrity. These lesions are multiple in two thirds of the cases and bilateral in half of the cases, appearing as large, red-to-brown, discrete, solid tumor-like nodules. They are usually incidental findings at the time of Cesarean section or tubal ligation, but in one quarter of the cases, they produce enough androgenic hormone to virilize the mother and also any female offspring. It is important to differentiate the pregnancy luteoma from a steroid cell tumor because the former requires no therapy and may be diagnosed by biopsy or simple excision. During the puerperium, the lesional cells degenerate with nuclear pyknosis and cytoplastic lipid accumulation, eventually forming a scar, which ultimately disappears.
GERM CELL TUMORS
Dysgerminoma
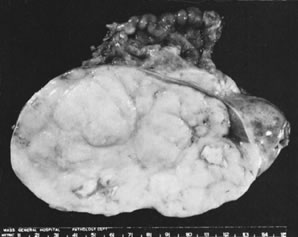
The dysgerminoma, like the testicular seminoma, is composed of large cells resembling the primordial germ cells of the embryo in their rich cytoplasmic content of glycogen and the staining of their cell membranes for alkaline phosphatase.35 This neoplasm is the single most common form of primitive malignant germ cell tumor of the ovary but accounts for less than 1% of ovarian cancers in females of all ages. It is a relatively common form of ovarian cancer, however, in young women in the reproductive age group; it is encountered occasionally in older women and in young children but is very rare after the age of 50 years and does not arise after the menopause. On gross examination, the dysgerminoma forms a solid mass that occasionally shows areas of caseation-like necrosis or cystic degeneration. The external and sectioned surfaces are characteristically lobulated, and the neoplastic tissue is pale brown to pink to cream-colored and soft to firm depending on its relative content of tumor cells and stroma (Fig. 16). Microscopic examination shows a diffuse array of large, rounded cells with clear cytoplasm and central, rounded nuclei that are flattened along their margins and contain one or a few prominent nucleoli (Fig. 17). The fibrous stroma varies from scanty to abundant and almost always is infiltrated by lymphocytes, which may be sprinkled singly throughout the tumor or may be present in large numbers, occasionally forming lymphoid follicles. In a minority of cases, noncaseating sarcoid-like granulomas are also present in the stroma. Among malignant germ cell tumors, the dysgerminoma is most often bilateral. Gross bilaterally is observed in approximately 10% of the cases and microscopic evidence of bilaterality in an additional estimated 10% of the cases.
|
|

Approximately 10% of dysgerminomas contain isolated syncytiotrophoblast cells, which can be stained immunohistochemically for human chorionic gonadotropin (hCG).36 These tumors are associated with elevated levels of hCG in the serum and sometimes are accompanied by hormonal changes that are usually estrogenic and rarely androgenic. The clinical manifestations of this form of dysgerminoma may be isosexual pseudoprecocity, irregular bleeding in the reproductive age period, and rarely virilization. Serum tumor markers of the dysgerminoma include lactic dehydrogenase and neuron-specific enolase, but are not entirely reliable in monitoring the course of the patient.37, 38
The dysgerminoma has an excellent prognosis because it is confined to the ovaries in three quarters of the cases and to a single ovary in two thirds of the cases. In addition, because of the dysgerminoma's marked sensitivity to radiation therapy and chemotherapy, the patient is cured in a high percent of the cases, even when spread beyond the ovary or recurrence has taken place.3, 39, 40[4142 The stage IA dysgerminoma is currently treated by salpingo-oophorectomy and lymph node sampling to exclude metastasis. Bilateral oophorectomy is not performed unless the contralateral ovary is replaced by tumor. If tumor is present in lymph nodes or has spread elsewhere, combination chemotherapy is preferred over radiation therapy because of a lower frequency of complications of the former. The survival rate for a dysgerminoma should approach 100%, and the majority of patients with higher stage tumors should also be cured by chemotherapy.
Yolk sac tumor
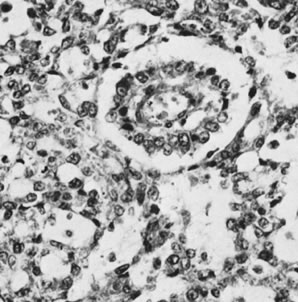
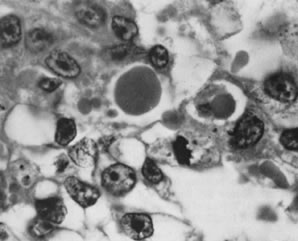
This tumor, which was originally called endodermal sinus tumor, is characterized by a variety of patterns and cell types that have been interpreted as simulating those of yolk sac epithelium and its derivatives.43, 44 The neoplasm accounts for approximately 20% of primitive germ cell tumors of the ovary. The endodermal sinus tumor is the first described and the most common subtype of yolk sac tumor. It is characterized by a network of spaces lined by immature epithelial cells (reticular pattern) and occupied focally by single papillary projections, which have been designated Schiller-Duval bodies (Fig. 18). These structures, which contain single central blood vessels and are lined by neoplastic epithelium, resemble to some extent downgrowths of yolk sac epithelium into the extraembryonic mesenchyme of the labyrinthine placenta of the rodent, known as endodermalsinuses. Schiller-Duval bodies are present in most endodermal sinus tumors but may be rare or absent; in such cases, the tumor may have a pure reticular pattern, which merges in many cases with microcystic foci. Another microscopic feature of the endodermal sinus tumor is the presence of intracellular hyaline bodies (Fig. 19). The yolk sac nature of the tumor is confirmed by the immunohistochemical demonstration of alpha-fetoprotein in some of the tumor cells and by the presence of elevated levels of this oncofetal protein in the serum. A more mature form of yolk sac tumor is characterized by a polyvesicular vitelline pattern, in which vesicles lined by columnar-to-flattened epithelium are present in a cellular stroma. These vesicles often show an eccentric constriction, which has been interpreted as a recapitulation of the formation of the secondary or definitive yolk sac from the primary yolk sac (Fig. 20). In the normal embryo, the primary yolk sac becomes vestigial, and the secondary yolk sac develops into the gastrointestinal tract and its appendages, such as the liver. The polyvesicular vitelline pattern is usually a minor component of a yolk sac tumor, but in rare cases, it predominates. Other patterns are papillary and solid. In addition, in keeping with the role of the yolk sac in the formation of the gastrointestinal tract and its appendages, rare yolk sac tumors have a distinctive glandular pattern with cribriform areas and are composed of cells that resemble those of the primitive intestinal tract. These tumors are called glandular yolk sac tumors;45 some of them simulate closely endometrioid adenocarcinomas with or without squamous differentiation (endometrioid-like yolk sac tumors) and can be distinguished from true endometrioid tumors by the demonstration of alpha-fetoprotein in the cytoplasm of their neoplastic cells.46 A final form of yolk sac tumor is the hepatoid form, which is indistinguishable from a hepatocellular carcinoma of the liver and is also associated with alpha-fetoprotein production.47 Gross examination of a yolk sac tumor reveals a solid mass that typically shows areas of necrosis, gelatinous degeneration, hemorrhage, and often cyst formation (Fig. 21). Rupture has been reported in approximately one third of cases.
|
|
|
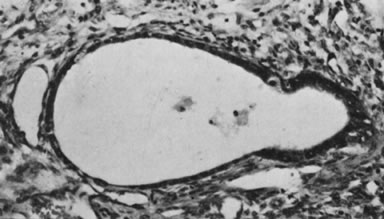
|
The yolk sac tumor is almost always encountered in children or young women and occurs very rarely after the age of 50 years. The alpha-fetoprotein level is elevated in the serum in almost all cases. At surgery, approximately 70% of the patients are stage I. Because bilaterality is very rare, the unilateral tumor can be treated by simple salpingo-oophorectomy, but since such treatment by itself has usually been followed by recurrence and death of the patient, adjuvant chemotherapy, such as cisplatin, vinblastine, and bleomycin, is now administered after surgery.3, 40, 41, 42 With the use of modern chemotherapy, the majority of patients with stage I tumors are cured of their disease, and most patients with more advanced stages of disease also survive after similar chemotherapy, in some cases after a cytoreductive surgical procedure.
Embryonal carcinoma
The embryonal carcinoma is a very rare form of malignant germ cell tumor in the ovary in contrast to its relative frequency in the testis.48 The tumor is composed of cells that are thought to be totipotent, growing in one or more of several patterns, including solid, glandular, and papillary. The neoplastic cells resemble those of the germ disc of the embryo. The reported examples have contained syncytiotrophoblast cells, typically in proximity to blood-filled spaces (Fig. 22). Immunoperoxidase studies have shown the presence of hCG in these cells. Alpha-fetoprotein is also demonstrable in almost all the cases, raising the question of whether the tumor is a mixed embryonal carcinoma and yolk sac tumor instead of a pure embryonal carcinoma.
|
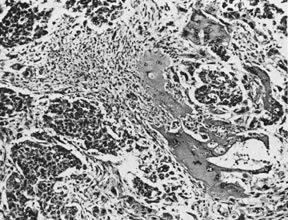
Almost half the patients in the reported cases have been prepubertal, and many of these children have had sexual precocity due to hCG secretion. Postpubertal patients have vaginal bleeding in approximately one third of the cases and, rarely, androgenic manifestations. The treatment and prognosis are similar to those of the yolk sac tumor.34, 35, 36, 37, 38, 39, 40, 41, 42
Choriocarcinoma
The very rare pure choriocarcinoma of the ovary may arise on the background of a primary ovarian gestation, may be metastatic from a uterine primary tumor, or may be of germ cell derivation.49, 50 It is difficult to be certain of the last type of origin unless the patient is prepubertal. Choriocarcinoma of germ cell origin is a component of a mixed germ cell tumor more often than a pure neoplasm.
As elsewhere, the choriocarcinoma is grossly hemorrhagic and friable. Microscopic examination shows cytotrophoblast or intermediate trophoblast or both growing in combination with syncytiotrophoblast, typically with nests of the first two cell types enclosed by rims of syncytiotrophoblast cells (Fig. 23). Occasionally, the tumor cells are more poorly differentiated, with blurring of the distinction between the various cell types.
|

The choriocarcinoma has endocrine manifestations similar to those of the embryonal carcinoma. The treatment of the stage IA tumor is surgical with adjuvant chemotherapy. The use of antitrophoblastic chemotherapeutic agents has resulted in long-term remissions in some patients with metastatic disease.
Polyembryoma
The extremely rare polyembryoma is composed exclusively or predominantly of embryos or embryoid bodies, which contain not only totipotential germ disk but also amnion, yolk sac, extraembryonic mesenchyme, and trophoblast.5152 Scattered foci of teratomatous, typically endodermal differentiation, also may be observed. Endocrine manifestations may result from the presence of syncytiotrophoblast cells in the embryoid bodies.
Teratomas
Teratomas are divided into two major groups, immature and mature, according to the degree of differentiation of their components, and a third category, which comprises tumors with large components that reflect exclusive differentiation into a specialized tissue of monodermal origin.
IMMATURE TERATOMA
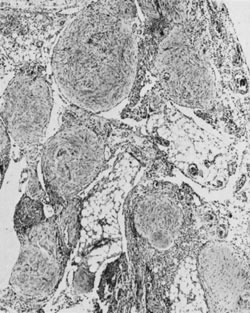
If any component of a teratoma is embryonic in its morphologic features, a diagnosis of immature teratoma is warranted. In the majority of cases, the predominant immature element is neuroectodermal, consisting of cellular glial tissue and neuroepithelial rosettes (Fig. 24).53, 54 The immature tissue should appear embryonal rather than simply fetal to be considered immature.
|
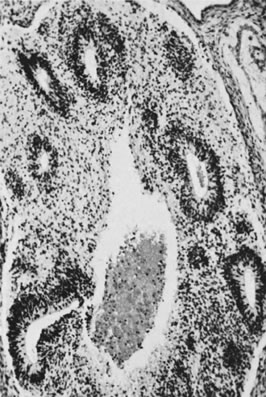
Immature teratomas have been graded by several investigators on the basis of the quantity of embryonic tissue that they contain. According to the criteria used in the largest reported series of cases,53 the presence of immature neural tissue occupying less than one 40× microscopic field results in the assignment of grade 1; grade 2 corresponds to the presence of one to four 40× fields with immature neural tissue; and in grade 3 tumors, four or more such fields are involved. More recently, a binary divide into low-grade and high-grade groups (combining grade 2 and grade 3 as high grade) has been recommended.55 On gross examination, the immature teratoma is almost always predominantly solid, with a variegated appearance on its sectioned surfaces (Fig. 25). Hard areas may be cartilaginous or bony, whereas soft areas may correspond to the immature, cellular portions of the tumor of neuroectodermal derivation. Areas of necrosis and hemorrhage are often encountered. Small cysts may contain serous or mucinous fluid or hair and sebaceous material.
|
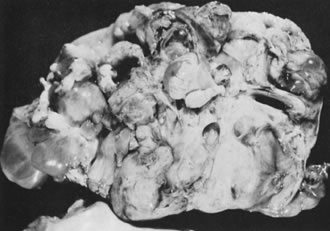
The immature teratoma typically presents as a pelvic or abdominal mass in a child or young woman, occurring very rarely after the age of 50 years. At surgery, 70% of the tumors are stage IA. A tumor is also present in the contralateral ovary in 10% of the cases and is almost always a dermoid cyst. The prognosis of the immature teratoma depends on its stage and grade. The significance of extraovarian spread, however, is not as great as in cases of surface epithelial cancers. The reason for the difference is that occasional immature teratomas spread to the peritoneum in the form of mature implants, typically composed of glial tissue (Fig. 26).56 The presence of these cytologically benign implants does not appear to affect the prognosis, except that rarely, the mature implants continue to grow and form large tumor masses. Finally, there is a clear correlation of prognosis with the grade of the tumor.
|
The treatment of choice for the stage IA immature teratoma in young females is unilateral salpingo-oophorectomy. If the tumor is grade 2 or 3, postoperative combination chemotherapy of the type used for yolk sac tumors is warranted, but if it is only grade 1, the prognosis is so good with surgery alone that chemotherapy is generally withheld. Similar combination chemotherapy, in some cases after a cytoreductive surgical operation, has also been used in cases of recurrent or metastatic disease with beneficial results.
Mature solid teratoma
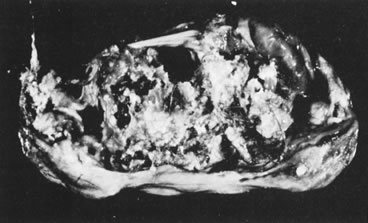
The mature solid teratoma is a rare tumor characterized by the exclusive presence of mature tissues in a predominantly solid mass that has been sampled carefully to exclude an immature component (optimally, one microscopic section for every centimeter of diameter of the tumor) (Fig. 27). Gross appearance of the mature teratoma resembles that of the immature solid teratoma, except that necrosis and hemorrhage are encountered less often. The mature solid teratoma may also be associated with mature implants composed predominantly of glial tissue on the peritoneum. The survival rate should be 100%, although rare examples in the literature have been associated with a fatal outcome. The question arises in such cases, however, of whether the tumor had been sampled adequately to exclude areas of immature teratoma.
|

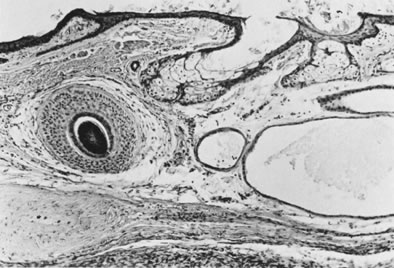
DERMOID CYST
The mature cystic teratoma is almost always a dermoid cyst lined by skin with its appendages (Figs. 28 and 29), although this type of lining may alternate with respiratory epithelium or mature glial tissue.58 Rarely, a mature cystic teratoma is lined entirely by one or the other of those tissue types. Most dermoid cysts contain mesodermal and endodermal, as well as ectodermal derivatives, and almost every type of tissue has been identified in individual cases. Occasional, otherwise typical, dermoid cysts contain microscopic foci of immature tissue, which merits only a comment and not a diagnosis of immature teratoma.58
|
|
The dermoid cyst is bilateral in 12% of the cases. Usually, it can be shelled out of the ovary if preservation of fertility is an important consideration. Unless the contralateral ovary is enlarged and suspected of containing a tumor, incision and biopsy are not warranted because of the rarity of finding a dermoid cyst under those circumstances. The many unusual complications of dermoid cysts include torsion, rupture into the peritoneal cavity or hollow viscera, infection, Coombs-positive autoimmune hemolytic anemia that disappears after removal of the tumor, and the development of malignant change.57, 59, 60, 61, 62
Almost 2% of dermoid cysts undergo a secondary malignant change, which is typically encountered in women in the cancer age group, but is occasionally seen in younger patients as well.59, 60, 61, 62 More than 15% of dermoid cysts in women 70 years of age or older contain a secondary malignant tumor. Squamous cell carcinoma in situ has been described, but the most common type of malignancy is invasive squamous cell carcinoma, which may present as a nodule in the wall of the cyst or a fungating mass within its lumen. Other malignant tumors that have arisen in association with dermoid cysts include basal cell carcinoma, malignant melanoma, malignant sweat gland tumors, malignant struma, adenocarcinomas, carcinoid tumors, and a variety of sarcomas. All the malignant tumors associated with dermoid cysts have been unilateral. The survival rate of patients with stage IA squamous cell carcinoma is good, but that for patients with higher stage tumors and with sarcomas and various forms of adenocarcinoma is poor.
MONODERMAL TERATOMAS
This category includes a variety of rare neoplasms, such as primitive neuroectodermal tumors, retinal anlage tumors, and sebaceous gland tumors. The most common of these neoplasms, however, are those that contain exclusively or predominantly thyroid epithelial cells, neuroendocrine cells, or both.
Struma Ovarii
Although thyroid elements have been reported on microscopic examination in almost 20% of dermoid cysts, the term struma is used only when thyroid tissue is the predominant component of the neoplasm or is extensive enough to be detectable on gross examination.1, 2 The struma is most often pure or almost so but may appear as a mass in the wall of a dermoid cyst or, rarely, a mucinous cystadenoma. It has been encountered in all age groups.
On microscopic examination, the struma appears as a nonencapsulated mass of thyroid epithelial cells that may simulate normal thyroid glandular tissue or more often an embryonal, microfollicular, or macrofollicular thyroid adenoma (Fig. 30). Immunohistochemical staining for thyroglobulin confirms the diagnosis. Rarely, a follicular or papillary carcinoma develops in the thyroid tissue.
|
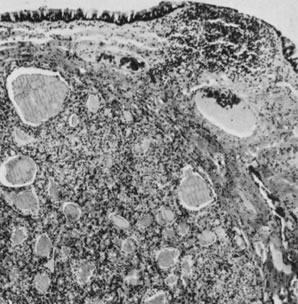
A struma may form a solid, brown, gelatinous mass having an appearance similar to that of cervical thyroid tissue or may form a unilocular or multilocular, thin-walled cystic mass with its compartments filled with glairy brown to greenish brown fluid. When a struma is associated with a dermoid cyst, its presence is often suspected on gross examination, but when it is the exclusive component of the specimen, usually it is not recognized as such or is misdiagnosed as a mucinous cystadenoma. The struma is almost always unilateral, although a dermoid cyst is occasionally present in the contralateral ovary.
In the majority of cases, struma is benign but rarely, it extends beyond the ovary.6364 It may implant in the form of mature thyroid tissue on the peritoneum. This condition, known as “benign strumosis,” does not appear to be clinically malignant and is compatible with longevity. Conversely, tumors that have an appearance similar to that of embryonal adenoma or papillary carcinoma of the thyroid gland can metastasize to various organs, such as lung, liver, and bone and to pelvic and para-aortic lymph nodes. Differentiation of benign from malignant struma ovarii is difficult in the absence of spread beyond the ovary. Tumors that prove to be malignant have a diameter greater than 5 cm, typically have an embryonal or solid pattern, and frequently are adherent to surrounding structures. Recurrence often occurs many years after surgery.
Very rarely, struma ovarii is accompanied by clinical evidence of hyperthyroidism, but almost all the putative cases of hyperfunctioning struma have been reported in the older literature, when sophisticated tests of thyroid function had not been available. In addition, the role of the cervical thyroid gland was not evaluated adequately in many of those cases.
Carcinoid tumor
Although an ovarian carcinoid tumor is often metastatic from a primary tumor of the small intestine or rarely another organ, primary carcinoid tumors of germ cell origin also occur. These tumors can be divided into four categories: the insular (midgut) type; the trabecular (foregut hindgut) type; the strumal carcinoid tumor; and the goblet cell or mucinous carcinoid tumor. Most carcinoid tumors in the germ cell category are associated with the presence of other teratomatous elements, at least on microscopic examination, but some of them are encountered in pure or almost pure form.
Insular carcinoid tumor
This tumor is characterized by rounded islands of epithelial cells and small glands (Fig. 31), with some of the cells containing neuroendocrine granules.65, 66 The granules are stainable by silver techniques and by the immunohistochemical demonstration of chromogranin. The pattern of the insular carcinoid tumor is similar to that of carcinoid tumors of the appendix and small intestine. The ovarian tumor occurs in adults of all ages but has not been reported in children. Up to one third of the cases are associated with the carcinoid syndrome, which occurs in the absence of metastasis because the hormone-containing venous effluent of the tumor bypasses the portal circulation. Removal of the tumor typically relieves the syndrome, with long-term survival unless the patient has metastatic disease or has already experienced irreversible cardiac valvular damage as a result of the hormonal changes.
|
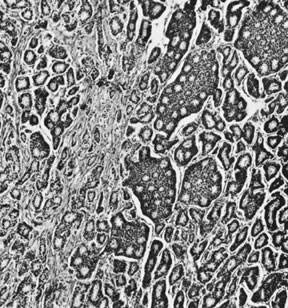
All the reported primary insular carcinoid tumors of the ovary have been unilateral, without evidence of spread at the time of operation. The tumor is typically solid and white or yellow and may resemble a fibroma because of an abundance of fibromatous stroma. Because the tumor rarely metastasizes, the 5-year survival rate is more than 95%.
Trabecular carcinoid tumor
This tumor, which resembles hindgut as well as foregut carcinoid tumors, is characterized predominantly by long, winding ribbons of epithelial cells that contain fewer neuroendocrine granules than those of the insular form.6768 The tumor has not been reported to be associated with the carcinoid syndrome, but hyperinsulinemic hypoglycemia and chronic constipation on a hormonal basis have been reported.69, 70 The trabecular carcinoid is also unilateral, resembling the insular carcinoid tumor in its macroscopic features. The prognosis is similar to that of the latter tumor.
Strumal carcinoid tumor
This tumor, which was often referred to in the earlier literature as “malignant struma,” is characterized by a combination of struma and carcinoid tumor, which, at least in some portion of the specimen, appear to be intimately admixed (Fig. 32).71 Strumal carcinoid tumors have been reported in adults from the 3rd to the 8th decades. Only a single case has been associated with the carcinoid syndrome; in several cases, however, there has been suggestive evidence of hypersecretion of thyroid hormone. At operation, all the tumors have been unilateral, although occasionally another type of neoplasm, usually a dermoid cyst, has occupied the contralateral ovary. The gross appearance of the tumor depends on the relative amounts of struma and carcinoid tumor it contains, as well as the presence or absence of recognizable teratomatous elements of other types, which often occur in the form of a dermoid cyst. Microscopic examination shows both struma and carcinoid tumor, which may be intimately mixed throughout much of the tumor or may appear as discrete components with an intimate mixture only at their interface. The carcinoid component is most often trabecular, occasionally mixed trabecular and insular, and, rarely, purely insular. Neuroendocrine granules have been shown within the carcinoid component in many cases, and thyroglobulin can be shown immunohistochemically. The rare finding of amyloid in the stroma of a strumal carcinoid tumor;72 the immunohistochemical identification of calcitonin in several cases; the presence of neuroendocrine granules in medullary carcinoma of the thyroid gland; and the rare association of the latter tumor with the carcinoid syndrome all suggest a kinship of these two neoplasms. In view of its almost invariable unilaterality, the strumal carcinoid tumor can be treated by simple salpingo-oophorectomy in young women. A clinically malignant behavior is very rare.
|

MUCINOUS CARCINOID TUMOR
This tumor, also referred to as “goblet cell carcinoid” and “adenocarcinoid,”73 is encountered most often in the appendix, from which it may metastasize to the ovary. Very rarely, it is a form of primary germ cell tumor arising in the ovary. It is characterized by features of a carcinoid tumor with an admixture of nonargentaffin cells, the most conspicuous of which are goblet cells.73 Occasional tumors have atypical features or have a frankly carcinomatous component.73
Malignant mixed germ cell tumors
These tumors, which account for approximately 10% of primitive germ cell tumors of the ovary, may contain any combination of the malignant forms of germ cell tumor described above.74, 75 The most frequently encountered tumor types are dysgerminoma and yolk sac tumor, which are typically sharply demarcated from one another. The probability of bilaterality and the gross appearance of individual mixed tumors reflect the nature of their various components. For example, tumors that contain an element of dysgerminoma are occasionally associated with the presence of dysgerminoma in the contralateral ovary, but those tumors that are devoid of a dysgerminoma component are very rarely bilateral. According to one study, the prognosis of a malignant mixed primitive germ cell tumor depends not only on its stage but also on its size (those tumors less than 10 cm in diameter were associated with an excellent prognosis) and the relative quantity of its most malignant components (i.e., yolk sac tumor, immature teratoma, grade 3, and choriocarcinoma).74 If any one of the last three neoplastic types occupied less than one third of the specimen, the prognosis was far better than if it occupied a larger fraction of the tumor. The treatment and prognosis of these tumors depend, to a large extent, on the accurate identification and possibly, to some extent, on the quantification of their neoplastic components by the pathologist, although the importance of quantification has decreased in recent years because of the effectiveness of combination chemotherapy.3, 40, 41, 42
GONADOBLASTOMA AND RELATED TUMORS
The gonadoblastoma is a hamartomatous neoplasm composed of germ cells, sex cord derivatives, and, in two thirds of the cases, stromal derivatives resembling lutein cells and Leydig cells without crystals of Reinke.76 The germ cells are similar to those of the ovarian dysgerminoma and testicular seminoma, and the sex cord elements have the appearance of immature Sertoli or granulosa cells. In one case, bundles of Charcot-Böttcher filaments, more or less specific for Sertoli cells, were identified in the sex cord element of a gonadoblastoma. The germ cells and sex cord derivatives grow characteristically in sharply demarcated aggregates, in which the most common pattern is one of germ cells distributed among more numerous smaller sex cord elements. The latter are typically oriented around hyaline bodies, creating a microfollicular appearance. The hyaline bodies are composed of dense basement membrane material, which is occasionally observed to be continuous with the basement membranes surrounding the discrete aggregates. Foci of calcification, which typically appear first in the hyaline (bodies), are present in most of the cases and often become confluent (Fig. 33). The calcification may be so extensive that it is recognizable on an x-ray film of the pelvis or abdomen. In approximately half the cases, the germ cells abandon the discrete aggregates to invade the stroma, forming a germinoma (dysgerminoma or seminoma). Occasionally, a more highly malignant form of germ cell tumor, such as a yolk sac tumor, choriocarcinoma, or embryonal carcinoma, develops in association with a gonadoblastoma. The gonadoblastoma may be microscopic or may form a large mass, which can be soft, fleshy, and cream colored in the presence of overgrowth by germinoma; yellow and extensively calcified; or of mixed color and consistency.
|
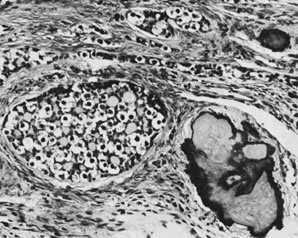
Almost all gonadoblastomas are found in patients with abnormal gonads. A few examples have been reported, however, in the gonads of otherwise apparently normal males and females. Although many of the smaller tumors can be shown to originate in streak gonads or dysgenetic testes, the nature of the underlying gonad is usually impossible to establish because it has been replaced completely by the neoplasm. The sexual disorder of the patient with a gonadoblastoma appears to be pure or mixed gonadal dysgenesis in most cases, but an occasional patient has had Turner's syndrome or true hermaphroditism. Most of the patients have been chromatin-negative. The most common karyotypes have been 46,XY and 45,XO/46,XY; other types of mosaicism have also been reported, and a rare patient has been 46,XX. The tumor may be discovered in childhood or during adult life. Approximately four fifths of the patients are phenotypic females, who are usually virilized to some degree. Although in many cases it may be impossible to determine whether associated endocrine abnormalities are attributable directly to the gonadoblastoma or to the underlying gonadal disorder, there has been both clinical and biochemical evidence, including in vitro studies of the neoplastic tissue, that the stromal derivatives of the tumor are capable of androgen production, resulting in masculinization. In vitro evidence of estrogen secretion has also been reported, and some patients have had hot flashes after the removal of the tumor.
Although the gonadoblastoma without an associated malignant germ cell tumor has not been shown to metastasize and can be regarded as an in situ form of malignancy, the common development of invasive germinoma (Fig. 34) and the occasional association of more virulent forms of germ cell tumor indicate that a gonad containing a gonadoblastoma is a potential hazard to the patient and should be removed. Because the tumor is bilateral in more than one third of the cases and almost always develops in a patient with abnormal gonads, the removal of the contralateral adnexa is also indicated in almost every case, even though neoplastic involvement is not evident at the time of operation. Because many gonadoblastomas have not been palpable on pelvic examination and have been incidental findings during an operation to determine the nature of a sexual disorder, and because patients with gonadal dysgenesis and a Y chromosome have been reported to harbor gonadoblastomas or other malignant germ cell tumors in approximately one quarter of the cases, routine gonadectomy is indicated in cases of gonadal dysgenesis with a Y chromosome. Although no large series of cases of malignant germ cell tumors arising in gonadoblastomas has been reported with adequate follow-up data, germinomas originating in gonadoblastomas are capable of metastasis, and the occasionally associated more malignant forms of germ cell tumors have been shown to exhibit a malignant behavior similar to that of the same types of tumor occurring in otherwise normal patients.
|
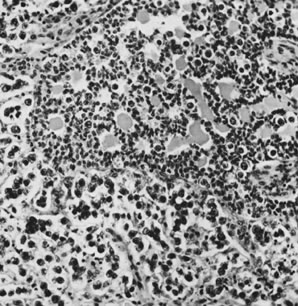
A small number of mixed germ cell and sex cord tumors resembling gonadoblastomas have been reported in individuals with normal sexual development and have been associated occasionally with endocrine manifestations.7879 Such tumors vary in their relative content of germ cells and sex cord elements, as well as the degree of maturity of the latter. There has been little or no hyaline body formation or calcification in the cases that have been observed, which generally permits microscopic differentiation from the typical gonadoblastoma.
REFERENCES
Scully RE, Young RH, Clement PB: Tumors of the ovary, maldeveloped gonads, fallopian tube and broad ligament. Atlas of Tumor Pathology, 3rd series, No. 23. Armed Forces Institute of Pathology, Washington, DC, 1999 |
|
Russell P, Farnsworth A: Surgical Pathology of the Ovaries, 2nd ed. Edinburgh, Churchill Livingstone, 1997 |
|
Scully RE: Histological typing of ovarian tumours. International Histological Classification of Tumours, Geneva, World Health Organization, Berlin, Springer, 1999 |
|
Young RH, Dickersin GR, Scully RE: Juvenile granulosa cell tumor of the ovary. A clinicopathologic analysis of 125 cases. Am J Surg Pathol 8: 575, 1984 |
|
Gusberg SB, Kardon P: Proliferative endometrial response to theca granulosa cell tumors. Am J Obstet Gynecol 111: 633, 1971 |
|
Nakashima N, Young RH, Scully RE: Androgenic granu1osa cell tumors of the ovary. A clinicopathologic analysis of 17 cases and review of the literature. Arch Pathol Lab Med 108: 786, 1984 |
|
Price FV, Schwartz PE: Management of ovarian stromal tumors. In Rubin SC, Sutton GP (eds): Ovarian Cancer, 405–423, New York, McGraw-Hill, 1993 |
|
Stenwig JT, Hazekamp JT, Beecham JB: Granulosa cell tumors of the ovary. A clinicopathological study of 118 cases with long term follow-up. Gynecol Oncol 7: 136, 1979 |
|
Bjorkholm E, Silfversward C: Prognostic factors in granulosa cell tumors. Gynecol Oncol 11: 261, 1981 |
|
Fox H, Agrawal K, Langley FA: A clinicopathologic study of 92 cases of granulosa cell tumor of the ovary with special reference to the factors influencing prognosis. Cancer 35: 231, 1975 |
|
Lappohn R, Burger H, Bouma J et al: Inhibin as a marker for granulosa cell tumors. N Engl J Med 329: 1539, 1993 |
|
McCluggage WG: Value of inhibin staining in gynecologic pathology. Int J Gynecol Pathol 20: 79, 2001 |
|
Dockerty MB, Masson JC: Ovarian fibromas: A clinical and pathologic study of two hundred and eighty-three cases. Am J Obstet Gynecol 47: 741, 1944 |
|
Gorlin RJ: Nevoid basal cell carcinoma syndrome. Medicine 66: 98, 1987 |
|
Samanth KK, Black WC: Benign ovarian stromal tumors associated with free peritoneal fluid. Am J Obstet Gynecol 107: 538, 1970 |
|
Prat J, Scully RE: Cellular fibromas and fibrosarcomas of the ovary: A comparative clinicopathologic analysis of seventeen cases. Cancer 47: 2663, 1981 |
|
Bjorkholm E, Silfversward C: Theca-cell tumors. Clinical features and prognosis. Acta Radiol Oncol Radiat Phys Biol 19: 241, 1980 |
|
Zhang J, Young RH, Arseneau J et al: Ovarian stromal tumors containing lutein or Leydig cells (luteinized thecomas and stromal Leydig cell tumors)—a clinicopathological analysis of fifty cases. Int J Gynecol Pathol 1: 270, 1982 |
|
Waxman M, Vuletin JC, Urcuyo R et al: Ovarian low grade stromal sarcoma with thecomatous features. A critical reappraisal of the so-called “malignant thecoma.” Cancer 44: 2206, 1979 |
|
Clement PB, Young RH, Hanna W et al: Sclerosing peritonitis associated with luteinized thecomas of the ovary. A clinicopathological analysis of six cases. Am J Surg Pathol 18: 1, 1994 |
|
Chalvardjian A, Scully RE: Sclerosing stromal tumors of the ovary. Cancer 31: 664, 1973 |
|
Dickersin GR, Young RH, Scully RE: Signet-ring stromal and related neoplasms of the ovary. Ultrastruct Pathol 19: 401, 1995 |
|
Young RH, Scully RE: Ovarian Sertoli-Leydig cell tumors: A clinicopathological analysis of 207 cases. Am J Surg Pathol 9: 543, 1985 |
|
Young RH, Scully RE: Ovarian Sertoli cell tumors. A report of 10 cases. Int J Gynecol Pathol 2: 349, 1984 |
|
Young RH, Scully RE: Well differentiated ovarian Sertoli-Leydig cell tumors. A clinicopathological analysis of 23 cases. Int J Gynecol Pathol 3: 277, 1984 |
|
Young RH, Prat J, Scully RE: Ovarian Sertoti-Leydig cell tumors with heterologous elements (i). Gastrointestinal epithelium and carcinoid: A clinicopathologic analysis of thirty-six cases. Cancer 50: 2448, 1982 |
|
Prat J, Young RH, Scully RE: Ovarian Sertoli-Leydig cell tumors with heterologous elements (ii). Cartilage and skeletal muscle: A clinicopathological analysis of twelve cases. Cancer 50: 2465, 1982 |
|
Young RH, Scully RE: Ovarian Sertoli-Leydig cell tumors with a retiform pattern: A problem in histopathologic diagnosis. Am J Surg Pathol 7: 755, 1983 |
|
Young RH, Dudley AG, Scully RE: Granulosa cell, Sertoli-Leydig cell and unclassified sex cord-stromal tumors associated with pregnancy: A clinicopathologic analysis of thirty-six cases. Gynecol Oncol 18: 181, 1984 |
|
Young RH, Welch WR, Dickersin GR et al: Ovarian sex cord tumor with annular tubules: Review of 74 cases including 27 with Peutz-Jeghers syndrome and four with adenoma malignum of the cervix. Cancer 50: 1384, 1982 |
|
Gustafson ML, Lee MM, Scully RE et al: Müllerian inhibiting substance: A novel tumor marker for ovarian sex cord tumor with annular tubules. N Engl J Med 326: 466, 1992 |
|
Hayes MC, Scully RE: Ovarian steroid cell tumors, not otherwise specified (lipid cell tumors): A clinicopathological analysis of 63 cases. Am J Surg Pathol 11: 835, 1987 |
|
Paraskevas M, Scully RE: Hilus cell tumor of the ovary. A clinicopathological analysis of 12 Reinke crystal-positive and nine crystal-negative cases. Int J Gynecol Pathol 8: 299, 1989 |
|
Hayes MC, Scully RE: Stromal luteoma of the ovary: A clinicopathological analysis of 25 cases. Int J Gynecol Pathol 6: 313, 1987 |
|
Bjorkholm E, Lundell M, Gyftodimos A et al: Dysgerminoma: The Radiumhemmet series 1927-1984. Cancer 65: 38, 1990 |
|
Zaloudek CJ, Tavassoli FA, Norris HJ: Dysgerminoma with syncytiotrophoblastic giant cells. A histologically and clinically distinctive subtype of dysgerminoma. Am J Surg Pathol 5: 361, 1981 |
|
Kavata M, Sekiya S, Hatakeyama R et al: Neuron-specific enolase as a serum marker for immature teratoma and dysgerminoma. Gynecol Oncol 32: 191, 1989 |
|
Pressley RH, Muntz HG, Falkenberry S et al: Serum lactic dehydrogenase as a tumor marker in dysgerminoma. Gynecol Oncol 44: 281, 1992 |
|
Buskirk SJ, Schray MF, Podratz KC et al: Ovarian dysgerminoma: A retrospective analysis of results of treatment, sites of treatment failure, and radiosensitivity. Mayo Clin Proc 62: 1149, 1987 |
|
Schwartz PE, Chambers SK, Chambers JT et al: Ovarian germ cell malignantics: The Yale University experience. Gynecol Oncol 45: 26, 1992 |
|
Gershenson DM: Update on malignant ovarian germ cell tumors. Cancer 71: 1581, 1993 |
|
Williams SD: Ovarian germ cell tumors. In Rubin SC, Sutton GP (eds): Ovarian Cancer. New York, McGraw-Hill, 1993 |
|
Teilum G: Endodermal sinus tumors of the ovary and testis: Comparative morphogenesis of the so-called mesonephroma ovarii (Schiller) and extra-embryonic (yolk sacallantoic) structures of the rat's placenta. Cancer 12: 1092, 1959 |
|
Kurman RJ, Norris HJ: Endodermal sinus tumor of the ovary. A clinical and pathologic analysis of 71 cases. Cancer 38: 2404, 1976 |
|
Cohen MB, Friend DS, Molnar JJ et al: Gonadal endodermal sinus (yolk sac) tumor with pure intestinal differentiation. A new histologic type. Pathol Res Pract 182: 609, 1987 |
|
Clement PB, Young RH, Scully RE: Endometrioid-like variant of ovarian yolk sac tumor. A clinicopathological analysis of eight cases. Am J Surg Pathol 11: 767, 1987 |
|
Prat J, Bhan AK, Dickersin GR et al: Hepatoid yolk sac tumor of the ovary (endodermal sinus tumor with hepatoid differentiation). A light microscopic, ultrastructural and immunohistochemical study of seven cases. Cancer 50: 2355, 1982 |
|
Kurman RI, Norris HJ: Embryonal carcinoma of the ovary. A clinicopathologic entity distinct from endodermal sinus tumor resembling embryonal carcinoma of the adult testis. Cancer 38: 2420, 1976 |
|
Jacobs AJ, Newland JR, Green RK: Pure choriocarcinoma of the ovary. Obstet Gynecol Surv 37: 603, 1982 |
|
Vance RP, Geisinger KR: Pure nongestational choriocarcinoma of the ovary, Report of a case. Cancer 56: 2321, 1985 |
|
Beck JS, Fulmer HF, Lee ST: Solid malignant ovarian teratuma with “embryoid bodies” and trophoblastic differentiation. J Pathol 99: 67, 1969 |
|
Prat J, Matias-Guiu X, Scully RE: Hepatic yolk sac differentiation in an ovarian polyembryoma. Surg Pathol 2: 147, 1987 |
|
Norris HJ, Zirkin HJ, Benson WL: Immature (malignant) teratoma of the ovary. A clinical and pathologic study of 58 cases. Cancer 27: 2359, 1976 |
|
Gershenson DM, Del Junco G, Silva EG et al: Immature teratoma of the ovary. Obstet Gynecol 68: 624, 1986 |
|
O'Connor DM, Norris HJ: The influence of grade on the outcome of Stage I ovarian immature (malignant) teratomas and the reproducibility of grading. Int J Gynecol Pathol 13: 283, 1994 |
|
Robboy SJ, Scully RE: Ovarian teratoma with glial implants on the peritoneum. Hum Pathol 1: 643, 1970 |
|
Pantoja E, Noy MA, Axtmayer RW et al: Ovarian dermoids and their complications. Obstet Gynecol Surv 30: 1, 1975 |
|
Yanai-Inbar I, Scully RE: Relation of ovarian dermoid cysts and immature teratomas: An analysis of 350 cases of immature teratoma and 10 cases of dermoid cyst with microscopic foci of immature tissue. Int J Gynecol Pathol 6: 203, 1987 |
|
Peterson WF: Malignant degeneration of benign cystic teratomas of the ovary. A collective review of the literature. Obstet Gynecol Surv 12: 793, 1957 |
|
Krumerman MS, Chung A: Squamous carcinoma arising in benign cystic teratoma of the ovary. A report of four cases and review of the literature. Cancer 39: 1237, 1977 |
|
Hirakawa T, Tsuneyoshi M, Enjoji M: Squamous cell carcinoma arising in mature cystic teratoma of the ovary. Clinicopathologic and topographic analysis. Am J Surg Pathol 13: 397, 1989 |
|
Pins MR, Young RH, Daly WJ, Scully RE. Primary squamous cell carcinoma of the ovary: Report of 37 cases. Am J Surg Pathol 20: 823, 1996 |
|
Zakmen A, Aftimos G, Kreidy R et al: Malignant struma ovarii: Report of two cases and selected review of the literature. J Surg Oncol 43: 61, 1990 |
|
Devaney K, Snyder R, Norris HJ et al: Proliferative struma ovarii and histologically malignant struma ovarii clinicopathological study of 54 cases. Int J Gynecol Pathol 12: 333, 1993 |
|
Robboy SJ, Norris HJ, Scully RE: Insular carcinoid primary in the ovary. A clinicopathologic analysis of 48 cases. Cancer 36: 404, 1975 |
|
Hilton P, Tweddel A, Wright A: Primary insular argentaffin carcinoma of ovary. Case report and literature review. Br J Obstet Gynaecol 95: 1324, 1988 |
|
Robboy SJ, Scully RE, Norris HJ: Primary trabecular carcinoid of the ovary. Obstet Gynecol 49: 202, 1977 |
|
Talerman A: Carcinoid tumors of the ovary. J Cancer Res Clin Oncol 107: 125, 1984 |
|
Morgello S, Schwartz E, Horwith M et al: Ectopic insulin production by a primary ovarian carcinoid. Cancer 62: 800, 1988 |
|
Motoyama T, Katayama Y, Watanabe H et al: Functioning ovarian carcinoids induced severe constipation. Cancer 70: 513, 1992 |
|
Robboy SJ, Scully RE: Strumal carcinoid of the ovary: An analysis of 50 cases of a distinctive tumor composed of thyroid tissue and carcinoid. Cancer 46: 2019, 1980 |
|
Morgan K, Wells M, Scott JS: Ovarian strumal carcinoid tumor with amyloid stroma report of a case with 20-year follow-up. Gynecol Oncol 22: 121, 1985 |
|
Baker PM, Oliva E, Young RH et al: Mucinous carcinoids of the ovary. A clinicopathologic study of 17 cases including some with a carcinomatous component. Am J Surg Pathol 25: 557, 2001 |
|
Kurman RJ, Norris HJ: Malignant mixed germ cell tumors of the ovary: A clinical and pathologic analysis of 30 cases. Obstet Gynecol 48: 579, 1976 |
|
Gershenson DM, Del Junco G, Copeland LJ et al: Mixed germ cell tumors of the ovary. Obstet Gynecol 64: 200, 1984 |
|
Scully RE: Gonadoblastoma: A review of 74 cases. Cancer 25: 1340, 1970 |
|
Manuel M, Katayama KP, Jones HW Jr: The age of occurrence of gonadal tumors in intersex patients with a Y chromosome. Am J Obstet Gynecol 124: 293, 1976 |
|
Jacobsen GK, Braendstrup O, Talerman A: Bilateral mixed germ cell sex-cord stroma tumor in a young adult woman. Case report. APMIS Suppl 23: 132, 1991 |
|
Talerman A, van der Harten JJ: A mixed germ cell-sex cord stroma tumor of the ovary associated with isosexual precocious puberty in a normal gift. Cancer 40: 889, 1977 |