This chapter should be cited as follows:
Nzioka A, Okiro P, Glob Libr Women's Med
ISSN: 1756-2228; DOI 10.3843/GLOWM.409323
The Continuous Textbook of Women’s Medicine Series – Obstetrics Module
Volume 10
Common obstetric conditions
Volume Editor: Professor Sikolia Wanyonyi, Aga Khan University Hospital, Nairobi, Kenya

Chapter
Pathology of the Placenta
First published: February 2021
Study Assessment Option
By answering four multiple-choice questions (randomly selected) after studying this chapter, readers can qualify for Continuing Professional Development points plus a Study Completion Certificate from GLOWM.
See end of chapter for details.

INTRODUCTION
The placenta is a maternofetal organ responsible for nutrition, waste elimination and gaseous exchange between the developing fetus and mother. The placenta also produces a number of hormones that are important during pregnancy. The placenta has two components: the fetal placenta, which develops from the same blastocyst that forms the fetus, and the maternal placenta, which develops from the maternal uterine tissue. The placenta begins to develop upon implantation of the blastocyst into the maternal endometrium. The outer layer of the blastocyst becomes the trophoblast, which forms the outer layer of the placenta. This outer layer is divided into two further layers: the underlying cytotrophoblast layer and the overlying syncytiotrophoblast layer. The syncytiotrophoblast is a multinucleated continuous cell layer that covers the surface of the placenta. The syncytiotrophoblast contributes to the barrier function of the placenta.
PLACENTAL PHYSIOLOGY
Maternal placental circulation
After ovulation, the uterine endometrium undergoes decidualization in preparation for implantation, due to progesterone secretion. This change can be seen as early as day 23, roughly 10 days following peak of luteinizing hormone.
Spiral arteries in decidua are remodeled so that they become less convoluted and their diameter is increased. Spiral artery remodelling begins in the first few weeks of pregnancy and modifies the arteries to high-flow low-resistance vessels capable of meeting the demands of the developing fetus. Remodelling of the spiral arteries probably begins in the late first trimester and is completed by 18–20 weeks of gestation. There is relatively high pressure as the maternal blood fills the intervillous space through these spiral arteries, and bathes the fetal villi in blood, allowing an exchange of gases to take place. As the pressure decreases between pulses, the deoxygenated blood flows back through the endometrial veins. Maternal blood flow is approximately 600–700 ml/min at term.
Fetoplacental circulation
Deoxygenated fetal blood passes through umbilical arteries to the placenta. At the junction of umbilical cord and placenta, the umbilical arteries branch radially to form chorionic arteries. Chorionic arteries, in turn, branch into cotyledon arteries. In the villi, these vessels eventually branch to form an extensive arterio-capillary-venous system, bringing the fetal blood extremely close to the maternal blood; but no intermingling of fetal and maternal blood occurs.
The human placenta performs the following physiological functions: respiratory, nutritive, excretory, production of enzymes, production of pregnancy associated plasma proteins, barrier and endocrine functions.
PLACENTAL ENDOCRINOLOGY
The human placenta and fetal membranes secrete a large number of hormones.1 These hormones are identical or similar in structure and function to hormones secreted by the hypothalamus, pituitary and ovaries. These hormones include the following.
Steroid hormones estrogens and progesterone
Estrogens are secreted initially by the corpus luteum and later by the fetoplacental unit (fetal adrenal cortex and the placenta). The syncytiotrophoblast produces estrogen in large quantities under hCG stimulation. Progesterone is initially produced by the corpus luteum and prepares the endometrium for implantation. Between week 5 and 8 postovulation, progesterone production is taken over by the placental syncytiotrophoblast.2 Progesterone hormone maintains a non-contractile uterus and champions development of an endometrium conducive for the pregnancy. By the end of the first trimester, placental production of estrogen and progesterone hormones replaces the corpus luteum.
Peptide hormones
Activin and inhibin are produced by the trophoblasts and regulate hCG production.
Cytokine growth factors (transforming growth factor (TGF)-alpha, TGF-beta, epidermal growth factor (EGF)) are produced by trophoblasts. They stimulate proliferation of trophoblast and production of fibronectin.
Human chorionic adrenocorticotropin (hACTH) is produced in small amounts. It is thought to have similar functions ACTH.
Human chorionic gonadotropin (hCG) is a glycoprotein similar in structure to pituitary luteinizing hormone. It is primarily synthesized by the villous syncytiotrophoblasts. Synthesis begins before implantation and is detectable 7–10 days after implantation. This forms the basis for early pregnancy tests. Peak levels are reached at 8–10 weeks' gestation. Its role is to maintain maternal corpus luteum that secretes progesterone and estrogens.
Human chorionic thyrotropin (hCT) is produced in small amounts by the syncytiotrophoblast. Its function is believed to be similar to thyroid stimulating hormone (TSH).
Human placental growth hormone has a similar structure to pituitary growth hormone except for 13 amino acids. It regulates maternal blood glucose levels so that the fetus has adequate nutrient supply.
Human placental lactogen (hPL) is a polypeptide similar to growth hormone synthesized by the villous syncytiotrophoblast. First detectable by 4 weeks with peak levels at end of third trimester. It acts as an insulin antagonist to influence growth, maternal mammary duct proliferation and lipid and carbohydrate metabolism.
Insulin-like growth factors stimulates proliferation and differentiation of cytotrophoblasts.
Placental alkaline phosphatase (PLAP) is an alkaline phosphatase normally produced by syncytiotrophoblast and primordial germ cells. May be involved in migration of primordial germ cells in developing fetus.
Relaxin is produced by the villous cytotrophoblast. It softens the cervix and pelvic ligaments in preparation for childbirth.
Pregnancy specific beta-1 glycoprotein (SP1) is present in syncytiotrophoblast and extravillous trophoblast.
PLACENTAL ANATOMY
Placental mean weight
As the fetus grows, the placental weight also increases as indicated in Table 1 below.3
1
Mean placental weight by gestational age.
Gestational age (weeks) | Mean placental weight (g) |
Prior to 28 | 253 |
28–32 | 314 |
33–36 | 391 |
37–40 | 456 |
>40 | 496 |
Parts of the placenta
The human placenta is composed of the following parts (Figure 1):4
- Disc composed of the fetal portion known as the chorionic plate and the maternal portion known as the basal plate decidua. The disc is divided into cotyledons from the primary stem villi and into lobules from secondary stem villi. The average size of the disc at term is 22 cm in diameter, 2.0–2.5 cm in thickness and 470 g in weight.
- Membranes composed of the amnion, exocelomic space, chorion and decidual capsularis. Membranes usually insert directly onto the placental edge:
- Amnion is the innermost lining of the amniotic cavity. It is composed of flat epithelial cells that rest on a thin basement membrane with underlying thin band of loose connective tissue. It may show squamous metaplasia, especially near cord or in pregnancies complicated by oligohydramnios.
- Exocelomic space is the potential space between the amnion and chorion that allows the membranes to slide against each other without friction.
- Chorion is a connective tissue membrane containing fetal vessels. It is internal to the amnion and external to the villi.
- Membranous chorion (chorion laeve) formed by the collapse of the intervillous space during development. It is composed of mononuclear, sometimes vacuolated, trophoblasts and scattered atrophic chorionic villi.
- Decidual chorion (chorion frondosum) located in the placenta proper.
- Umbilical cord which is the twisted cable that connects the fetus to the placenta and carries the two umbilical arteries and a single umbilical vein. The vessels branch out over the fetal surface to form the villous tree. The average size of the umbilical cord is 55–60 cm long and 2.0–2.5 cm in diameter.
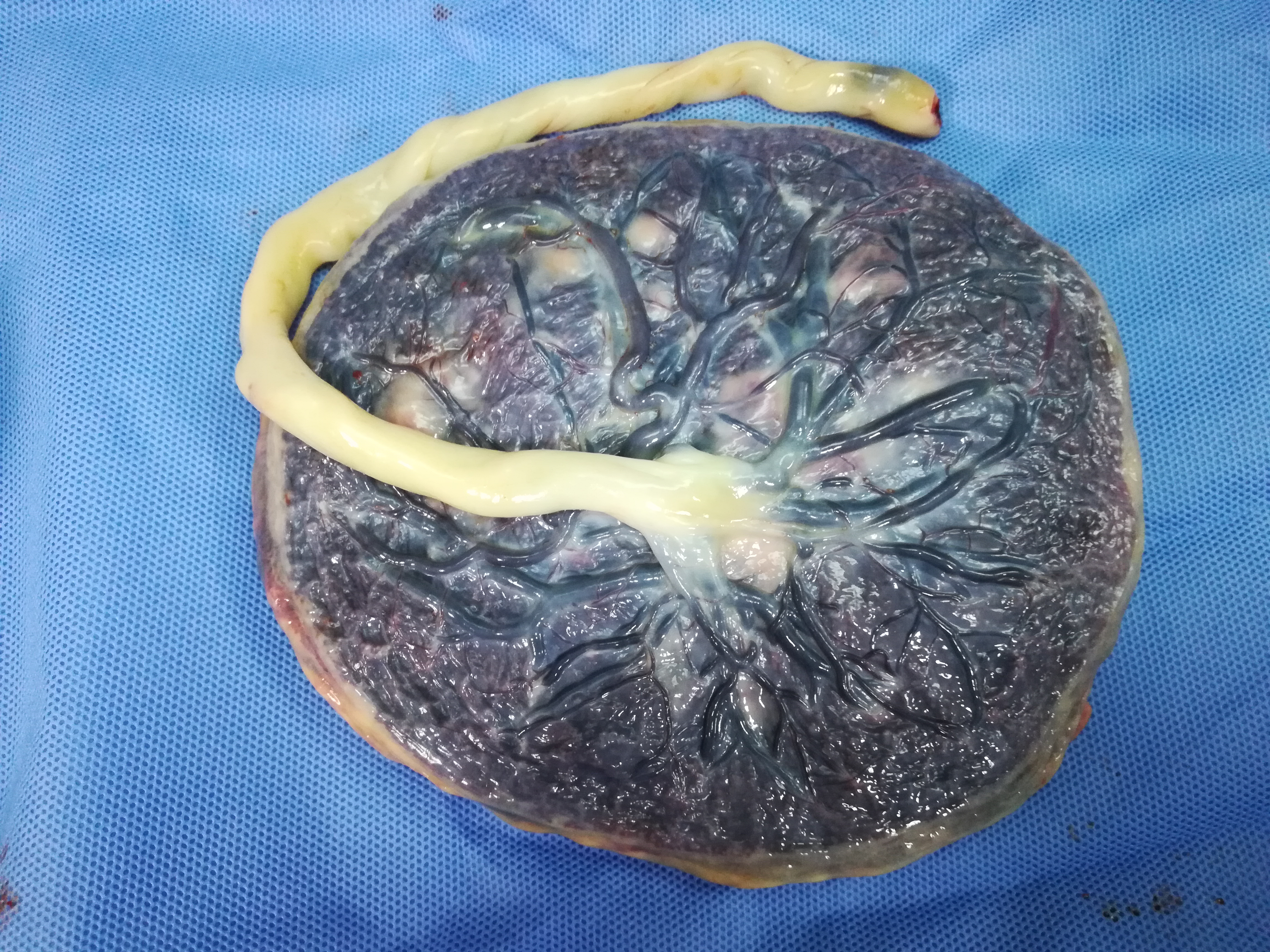
1
Gross placenta and umbilical cord.
PLACENTAL EXAMINATION
Placental pathology continues to be an underutilized and inadequately handled surgical subspecialty. No evaluation of a sick or demised neonate is complete without placental examination. Pathology of the placenta, umbilical cord, and/or placental membranes is causally ascribed for between 11% and 65% of stillbirth cases.5 Placental examination is a useful adjunct in defining etiology, prognosis, and risk of recurrence of pregnancy disorders, and is aided by the provision by the clinician of pertinent history to guide this process. Placental injury can result at different points in placental development and function. Early injury often results from abnormalities in maternal blood supply during developmental processes of placentation. Later insults result from compromise of fetal blood supply and external insults.
In 1997, the College of American Pathologists outlined clinical indications for placental pathological examination, highlighting maternal, fetal-neonatal and placental indicators of placental pathology,6 and highlighting the importance of relevant clinical history to accompany placentas submitted for pathological evaluation (Table 2). The minimum information required for placental evaluation are the gestational age, obstetric index, maternal history, baby weight, Apgar score, malformations and other pertinent findings.
2
Summary of the indications for comprehensive gross and microscopic placental examination by a pathologist.6
Maternal indications | Fetal/neonatal indications | Placental indications |
Preterm delivery at 36 weeks or less Fever or suspected infection Abnormal antenatal testing (abnormal fetal movements, stress and non-stress tests, sonographic tests, amniocentesis tests triple and quadruple blood tests) Severe oligohydramnios/polyhydramnios Hypertension/diabetes Vaginal bleeding Chronic diseases (cardiovascular, renal, autoimmune) Thick meconium with fetal distress) Unfavorable obstetric outcomes (defined as one or more previous stillbirths, spontaneous abortions, or premature births) Maternal history of substance abuse | Intrauterine fetal demise or early neonatal death NICU admission (e.g. prematurity, hyaline membrane disease, infections, congenital anomalies) SGA/LGA (<10th or >90th centile) Birth depression (pH <7.0/Apgar <7; assisted ventilation >10 min; Abnormal tone/neonatal seizures) Suspected infection Hydrops fetalis Multiple pregnancy Congenital abnormalities | Umbilical cord abnormalities Abnormal insertion site Short or long umbilical cord Other findings (hypercoiling, true knots, parenchymal masses, hemorrhages, abnormal shape) |
SGA, small for gestational age; LGA, large for gestational age
GROSS PLACENTAL LESIONS
Several gross placental lesions are described, with variable clinical significance. These are summarized in Table 3 below.7
3
Description of gross placental lesions.
Umbilical cord | Membranes | Parenchyma |
Color: (yellow: infection; greenish: meconium; brown: hemosiderin) Coiling: (hypocoiled: reduced activity/poor growth; hypercoiled: fetal distress) Insertion site: (velamentous/furcate – into membranes; marginal – risk of hemorrhage/compression) Length: (short: reduced fetal activity/malformations; long: multiple poor outcomes) Knots: false/true Vessels: (single umbilical artery: congenital malformations) Neoplasms: (cysts, teratomas, angiomyxomas) Hematomas | Color: (yellow: infection; greenish: meconium; brown: hemosiderin) Surface: (amnion nodosum) Insertion: (marginal: normal; circummarginate/circumvallate – see text below) | Weight: (fetal-placental weight ratios; small – FGR; heavy – hydrops placentalis, delayed villous maturation) Dimensions and appearance: (FGR, infarcts, hemorrhages; maternal floor infarction/perivillous fibrin deposition) Placental variants: (succenturiate (accessory lobe), membranacea (chorionic sac covered by placental tissue), duplex (bilobed), bipartite, tripartite (incompletely separate discs) |
FGR, fetal growth restriction
STORAGE AND PROCESSING OF PLACENTA FOR PATHOLOGICAL EVALUATION
Placentas should never be frozen. Freezing distorts the villi, obscures meconium, and compromises diagnosis. For fixation, the specimen should be placed at least 10 times its volume of 10% neutral buffered formalin. If samples are taken fresh for fixation before trimming, they should be no more than 2 cm thick. The rest of the processing is similar to other tissue specimen.
CLASSIFICATION OF PLACENTAL PATHOLOGIES
Major pathologies are evaluated by both gross examination and placental histopathology. Below, the major pathologically significant lesions are described. In the evaluation of lesions affecting the placenta, they are often outlined as those resulting from maternal/trophoblastic abnormalities, fetal vascular/stromal abnormalities and postplacental lesions. These may result from maldevelopment, malperfusion, loss of integrity, or secondary lesions.8,9
- Maternal maldevelopment/malperfusion lesions
Historically, emphasis was placed on gross placental abnormalities, including abnormal shape, size, and weight. This has remained an important indicator of placental function, especially in combination with other fetal parameters, such as fetoplacental weight ratios.10 More recently, the description of other lesions includes defective arterial remodeling and superficial implantation.11 These are the described pathogenetic mechanisms of disorders such as pre-eclampsia, and reflect maternal malperfusion of the placental bed. These lesions are marked grossly by placental hypoplasia (weight <10th centile; thin umbilical cord <8 mm), infarcts and hemorrhages (abruptio placenta, marginal/chronic abruptio) and microscopically by villous infarcts and accelerated villous maturation.9 - Fetal stromal-vascular lesions
Similar to maternal lesions, these can be divided into fetal maldevelopmental/malperfusion lesions, loss of integrity and secondary or extrinsic factors.11 Maldevelopmental lesions include delayed villous maturation, villous capillary lesions and dysmorphic villi. Malperfusion may be global or partial, highlighted by the lesions previously known as fetal thrombotic vasculopathy, and can affect a segment of or the entire placenta.12 Loss of integrity results in fetomaternal hemorrhages and villous edema, and secondary insults including meconium effects. - Inflammatory, infectious lesions
This category of disorders accounts for a large proportion of placental pathologies. It includes preterm deliveries and recurrent pregnancy losses.13,14 The spectrum of acute infections includes chorioamnionitis, acute villitis and acute intervillositis. Chronic lesions (chronic villitis, intervillositis, and deciduitis) and idiopathic lesions (villitis of unknown etiology (VUE), fetal eosinophilic/T cell lymphoplasmacytic vasculitis, and chronic histiocytic intervillositis (CHIV)) account for the remainder of lesions.15 - Others lesions
A final category includes lesions the pathogenesis of which remains incompletely understood. These include perivillous fibrin deposition, as well as other lesions such as placental malformations/deformations, placental disruptions, heterotopias, hydrops placentalis and genetic or chromosomal abnormalities.11
The major components of these lesions are described briefly below.
Maternal vascular malperfusion
Following the consensus of the Amsterdam placenta working group that aimed to standardize the reporting of placental pathologies, maternal vascular malperfusion is the preferred term for these lesions.9 It encompasses inadequate spiral artery remodeling and pathology, with resultant abnormal spiral artery flow. Its effects include fetal growth restriction and fetal demise.16 The clinical syndromes associated with these lesions are preeclampsia and preeclampsia-like syndromes (thrombophilia, renovascular disease) and diabetes mellitus.17,18 The gross findings resulting from these lesions are:
- Small placentas (weight less than 10th centile for gestation)
- Placental infarcts involving more than 5% of placental volume at term, away from the margin, or any infarcts in a preterm placenta
- Indenting retro placental hemorrhages (abruptio placenta).19
The resultant structural abnormalities include accelerated villous maturation and distal villous hypoplasia in partial or global obstruction, and infarcts in segmental/complete maternal vascular malperfusion, with areas overlying occluded spiral arteries showing ischemic necrosis.11 The molecules described to be involved in these lesions include, but are not limited to pro-angiogenic factors such as vascular endothelial growth factor (VEGF) and placental growth factor (PIGF), as well as anti-angiogenic factors, such as soluble fms-like tyrosine kinase 1 (sFlt-1/VEGFR-1), which are produced by the developing placenta.20,21 This is an area of ongoing research for biomarkers to predict risk of preeclampsia. Decidual arteriopathy, which is a lesion described in the basal plate of pre-eclamptic placentas, is an associated finding that includes acute atherosis of spiral arteries, fibrinoid necrosis with or without foamy macrophages, muscularized spiral arteries and chronic perivasculitis.19 Rupture of these poorly remodelled/artheromatous vessels results in loss of maternal vascular integrity, and can present acutely as abruptio placenta.22,23 Other etiologies include vasoactive drugs, trauma and uterine rupture.24 This lesion is distinguished from marginal abruption, a more chronic process resulting from rupture of decidual veins in a background of premature rupture of membranes, cervical insufficiency, low lying placentation, anatomical abnormalities and chorioamnionitis,19,20 resulting in recurrent marginal hemorrhages and circumvallation/circummargination of placental membranes and chorioamnionic hemosiderosis.
Fetal vascular malperfusion
Previously termed fetal thrombotic vasculopathy (FTV), this is an uncommon but significant lesion that is likely secondary to obstruction due to cord lesions, hypercoagulable states and fetal cardiac dysfunction.12 It has been associated with significant neurological impairment and adverse fetal outcome.25 Three forms of obstruction have been described:8
- Thrombosis of chorionic plate and stem villous vessels leading to degeneration and eventual loss of capillaries in downstream villi12
- Mechanical obstruction (recurrent or intermittent cord compression)
- Vessel wall damage.26
Multiple histologic findings have been described, including thrombosis of the fetal chorionic plate or umbilical cord vessels which may be visible grossly, mural organizing thrombosis with lamination, with or without muscular degeneration and ectasia of the vessel walls, and calcification of older thrombi.26 Distal avascular villi can be seen.
The Amsterdam consensus group suggested a grading system with low and high grade lesions, representing segmental and global forms of fetal vascular malperfusion, respectively, and possibly better predicting subsequent clinical outcome and complications.9,26
Inflammatory/infectious lesions
Acute chorioamnionitis accounts for a large proportion of this group of lesions, and is a frequent forerunner of preterm birth and a major contributor to neonatal complications.13 Infections commonly occur in early gestation (23–24 weeks) as compared to term,14,27 and are characterized by neutrophilic infiltrates of the fetal membranes and chorionic plate. A three-tier staging and two-tier grading system has been proposed by the Amsterdam group, as indicators of duration and intensity of inflammation. An equally important component is the fetal inflammatory response, which is an indicator or adverse neonatal outcomes.8 Other patterns of acute inflammation are acute villitis, commonly seen in E. coli and group B streptococcal infections, and acute intervillositis with intervillous microabcesses, as exemplified by Listeria monocytogenes infections. A host of other organisms from the gastrointestinal, genitourinary, oral and skin have been implicated in causation of acute chorioamnionitis.13 The described routes of infection are most often ascending, rarely hematogenous and transamniotic. Chronic chorioamnionitis is an infrequent finding and is often seen with other inflammatory lesions, including villitis of unknown etiology (VUE) and longstanding ascending inflammation (see below).28 Chronic infection, characterized by lymphohistiocytic infiltrates, presents as chronic villitis (associated with TORCH (toxoplasmosis, rubella cytomegalovirus, herpes simplex, and HIV) infections) and/or intervillositis (described in malaria).29 Other forms of inflammation of uncertain etiology include VUE, a cause of recurrent pregnancy loss thought to represent a graft versus host disease (GVHD) type of reaction,30 chronic histiocytic intervillositis (CHIV), also associated with recurrent pregnancy loss and fetal growth restriction31 and eosinophilic/T cell vasculitis, an inflammatory lesion of fetal origin and uncertain clinical significance.32
Finally, other important lesions include:
- Delayed villous maturation (previously villous maturation defect or villous dysmaturity), seen often in placentas of diabetic mothers and characterized by large placentas, with abundant villus stroma and poor formation of vasculosyncytial membranes between fetal capillaries and maternal blood.33
- Fetal hydrops/hydrops placentalis, also resulting in large placentas, which may be immune (ABO/Rhesus incompatibility) or non-immune (cardiovascular, fetal anemias).34
- Perivillous fibrin deposition/maternal floor infarction: rare, related idiopathic lesions characterised by deposition of fibrinoid material in the placental resulting in fetal growth restriction, and risks of recurrence with fetal death.35,36
PLACENTAL MALARIA
Malaria in pregnancy is substantial cause of morbidity and mortality for the pregnant woman and her fetus and newborn. The general immune suppression during pregnancy makes women more susceptible to malarial infection especially during first trimester. Initially low cell-mediated immune response in placenta makes it a preferred site for the malaria parasites to hide from host immune responses. However, as malarial infection progresses, there is an increase in cell-mediated immune response resulting in massive recruitment of macrophages to intervillous spaces of placenta causing oxidative stress and apoptotic cell death in placenta. This can lead to poor pregnancy outcome, such as abortions, still birth, IUGR, and LBW.37
Placental microscopic (histologic) findings in malaria include:
- Active disease shows free and intraerythrocytic parasites in intervillous space with minimal amounts of coarse hemozoin brown pigment.
- Chronic infection shows intervillositis with trophoblast basement membrane thickening and increased syncytial knots has been noted. In chronic infections, parasites coexist with pigment covered with fibrin.
- In inactive infections, only pigment is identified. Half of the patients with parasites in placenta have no parasites in peripheral blood.38
PRACTICE RECOMMENDATIONS
- Placental examination is a significant part of evaluation a sick or demised neonate.
- Adequate and relevant history is mandatory for placental evaluation and generation of a clinically useful report.
- Certain gross lesions are indicators for possible significant microscopic lesions.
- Placental pathologies may originate preplacentally (maternal vascular malperfusion/maldevelopment), intraplacentally (fetal vascular malperfusion) or postplacentally (infection, inflammation), and may be predictors of long-term life outcomes of the affected infants.
- Certain significant but rare conditions result in recurrent pregnancy loss, and should be born in mind in patients with poor obstetric histories.
CONFLICTS OF INTEREST
The author(s) of this chapter declare that they have no interests that conflict with the contents of the chapter.
Feedback
Publishers’ note: We are constantly trying to update and enhance chapters in this Series. So if you have any constructive comments about this chapter please provide them to us by selecting the "Your Feedback" link in the left-hand column.
REFERENCES
Parry S, Strauss JF. III: Placental hormones. In: DeGroot LJ, Jameson JL (eds.) Endocrinology, 4th edn. Philadelphia, WB Saunders, 2001:Chap 178;2379. | |
Roberts V. Placental development and physiology. In: UpToDate, Post TW (Ed), UpToDate, Waltham, MA. (Accessed on May 19, 2020.) | |
Ziadie M. Placental development. PathologyOutlines.com website. http://www.pathologyoutlines.com/topic/placentaplacentaldevel.html. | |
Ziadie M. Anatomy of the placenta. PathologyOutlines.com website. http://www.pathologyoutlines.com/topic/placentaplacentaldevel.html. | |
Ptacek I, Sebire NJ, Man JA, et al. Systematic review of placental pathology reported in association with stillbirth. Placenta 2014;35(8):552–62. | |
Langston C, Kaplan C, Macpherson T, et al. Practice guideline for examination of the placenta: developed by the Placental Pathology Practice Guideline Development Task Force of the College of American Pathologists. Archives of Pathology & Laboratory Medicine 1997;121(5):449–76. | |
Roberts DJ. Gross examination of the placenta, 2018. | |
Redline RW. Placental pathology: a systematic approach with clinical correlations. Placenta 2008;29 Suppl A:S86–91. | |
Khong TY, Mooney EE, Ariel I, et al. Sampling and Definitions of Placental Lesions: Amsterdam Placental Workshop Group Consensus Statement. Archives of Pathology & Laboratory Medicine 2016;140(7):698–713. | |
Molteni RA, Stys SJ, Battaglia FC. Relationship of fetal and placental weight in human beings: fetal/placental weight ratios at various gestational ages and birth weight distributions. The Journal of Reproductive Medicine 1978;21(5):327–34. | |
Redline RW. Classification of placental lesions. American Journal of Obstetrics and Gynecology 2015;213(4 Suppl):S21–8. | |
Redline RW, Ariel I, Baergen RN, et al. Fetal vascular obstructive lesions: nosology and reproducibility of placental reaction patterns. Pediatric and developmental pathology: the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society, 2004;7(5):443–52. | |
Redline RW. Inflammatory response in acute chorioamnionitis. Seminars in Fetal & Neonatal Medicine 2012;17(1):20–5. | |
Mueller-Heubach E, Rubinstein DN, Schwarz SS. Histologic chorioamnionitis and preterm delivery in different patient populations. Obstetrics and Gynecology 1990;75(4):622–6. | |
Redline RW. Placental inflammation. Seminars in neonatology: SN, 2004;9(4):265–74. | |
Redline RW, Boyd T, Campbell V, et al. Maternal vascular underperfusion: nosology and reproducibility of placental reaction patterns. Pediatric and developmental pathology: the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society, 2004;7(3):237–49. | |
Mestan KK, Check J, Minturn L, et al. Placental pathologic changes of maternal vascular underperfusion in bronchopulmonary dysplasia and pulmonary hypertension. Placenta 2014;35(8):570–4. | |
Karumanchi SA, Maynard SE, Stillman IE, et al. Preeclampsia: a renal perspective. Kidney International 2005;67(6):2101–13. | |
Hawfield A, Freedman BI. Pre-eclampsia: the pivotal role of the placenta in its pathophysiology and markers for early detection. Ther Adv Cardiovasc Dis 2009;3(1):65–73. | |
Roberts JM, Taylor RN, Goldfien A. Clinical and biochemical evidence of endothelial cell dysfunction in the pregnancy syndrome preeclampsia. American Journal of Hypertension 1991;4(8):700–8. | |
Maynard SE, Min JY, Merchan J, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 2003;111(5):649–58. | |
Harris BA, Jr. Peripheral placental separation: a review. Obstetrical & Gynecological Survey 1988;43(10):577–81. | |
Aftolin F, Khudr G, Benirschke K, et al. The syndrome of chronic abruptio placentae, hydrorrhea, and circumvallate placenta. American Journal of Obstetrics andGynecology 1973;116(3):347–50. | |
Schmidt P, Raines DA. Placental Abruption (Abruptio Placentae). StatPearls. Treasure Island (FL): StatPearls Publishing StatPearls Publishing LLC, 2018. | |
Redline RW. Severe fetal placental vascular lesions in term infants with neurologic impairment. AmericanJournal of Obstetrics and Gynecology 2005;192(2):452–7. | |
Heider A. Fetal Vascular Malperfusion. Archives of Pathology & Laboratory Medicine 2017;141(11):1484–9. | |
Salafia CM, Weigl C, Silberman L. The prevalence and distribution of acute placental inflammation in uncomplicated term pregnancies. Obstetrics and Gynecology 1989;73(3 Pt 1):383–9. | |
Jacques SM, Qureshi F. Chronic chorioamnionitis: a clinicopathologic and immunohistochemical study. Human Pathology 1998;29(12):1457–61. | |
Jacques SM, Qureshi F. Chronic intervillositis of the placenta. Archives of Pathology & Laboratory Medicine 1993;117(10):1032–5. | |
Chan JS. Villitis of Unknown Etiology and Massive Chronic Intervillositis. Surgical Pathology Clinics 2013;6(1):115–26. | |
Boyd TK, Redline RW. Chronic histiocytic intervillositis: a placental lesion associated with recurrent reproductive loss. Human Pathology 2000;31(11):1389–96. | |
Redline RW. Clinically and biologically relevant patterns of placental inflammation. Pediatric and developmental pathology: the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society, 2002;5(4):326–8. | |
Evers IM, Nikkels PG, Sikkema JM, et al. Placental pathology in women with type 1 diabetes and in a control group with normal and large-for-gestational-age infants. Placenta 2003;24(8–9):819–25. | |
Kraus FT, Redline RW, Gersell DJ, et al. Placental Pathology. King DW (ed.) Washington DC: American registry of Pathology, in collaboration with the Armed Forces Institute of Pathology, 2004;2. | |
Faye-Petersen OM, Ernst LM. Maternal Floor Infarction and Massive Perivillous Fibrin Deposition. Surgical Pathology Clinics 2013;6(1):101–14. | |
Chen A, Roberts DJ. Placental pathologic lesions with a significant recurrence risk – what not to miss! APMIS: acta pathologica, microbiologica, et immunologica Scandinavica, 2017. | |
Sharma L, Shukla G. Placental Malaria: A New Insight into the Pathophysiology. Front Med (Lausanne) 2017;4:117. | |
Mamudo R, Ismail, Jaume Ordi, et al. Placental pathology in malaria: A histological, immunohistochemical, and quantitative study. Hum Pathol 2000;31(1):85–93. |
Online Study Assessment Option
All readers who are qualified doctors or allied medical professionals can automatically receive 2 Continuing Professional Development points plus a Study Completion Certificate from GLOWM for successfully answering four multiple-choice questions (randomly selected) based on the study of this chapter. Medical students can receive the Study Completion Certificate only.
(To find out more about the Continuing Professional Development awards program CLICK HERE)