This chapter should be cited as follows:
Contreras M, Kumpel B, et al., Glob Libr Women's Med
ISSN: 1756-2228; DOI 10.3843/GLOWM.418843
The Continuous Textbook of Women’s Medicine Series – Obstetrics Module
Volume 16
The prevention and management of Rh disease
Volume Editors:
Professor Gerard HA Visser, Department of Obstetrics and Gynaecology, University Hospital of Utrecht, Heidelberglaan 100, Utrecht 3584EA, The Netherlands
Professor Gian Carlo Di Renzo, PREIS International School, Florence, Italy

Chapter
Anti-RhD Immunoglobulin for the Prevention of Hemolytic Disease of the Fetus and Newborn: Polyclonal versus Monoclonal Antibodies
First published: January 2023
Study Assessment Option
By answering four multiple-choice questions (randomly selected) after studying this chapter, readers can qualify for Continuing Professional Development points plus a Study Completion Certificate from GLOWM.
See end of chapter for details.

FOREWORD
Throughout this chapter we use the terms D and anti-D, rather than RhD and anti-RhD, for the sake of simplicity.
BRIEF HISTORY OF RHD HEMOLYTIC DISEASE OF THE FETUS AND NEWBORN (HDFN)
Prior to the late 1960s, numerous studies in white Caucasians in high-income countries have shown that about 1% of D-negative mothers delivering an ABO-compatible D-positive infant developed anti-D by the end of the first pregnancy; six months after delivery, this figure rose to about 7–9%. Immunization correlated with the volume of transplacental hemorrhage (TPH), or fetomaternal hemorrhage (FMH), with paternal D antigens on the fetal cells. By the end of a second pregnancy with a D-positive infant, the frequency of anti-D immunization was of the order of 16–17%.1,2 Moreover, in the absence of immunosuppressive therapy with anti-D immunoglobulin (Ig), about 6% of all D-negative women, pregnant for the second time with a D-positive infant will have an infant affected with hemolytic disease. These figures are lower than expected because ABO incompatibility between mother and fetus is highly protective (55–90%) against D-immunization.3,4,5 and because approximately 30% of D-negative women will be non-responders to immunization by D-positive RBCs in a TPH.
POLYCLONAL ANTI-D IG
Suppression of the anti-D immune response that would otherwise follow pregnancy
From the suggestion that anti-D immunoglobulin might prevent immunization in D-negative mothers delivering a D-positive infant, it took about 6 years of numerous clinical trials in male volunteers and then in thousands of post-partum D-negative women, mostly primiparae, delivering ABO compatible infants, for anti-D Ig to be introduced into routine clinical use and become the great success it has proven to be in modern medicine.
The early experiments took place independently in Liverpool, UK, and New York, USA. Considering the protection against D-immunization afforded by ABO feto-maternal incompatibility, Finn, in 1960,6 suggested, that it might be possible to destroy any fetal RBCs in the maternal circulation by means of a suitable antibody. After experiments with IgM anti-D, the Liverpool group went on to demonstrate suppression of D-immunization in D-negative volunteers by means of plasma containing “incomplete” (IgG) anti-D (Figure 1); suppression was attributed to a better clearance of the injected chromium-labelled RBCs to the spleen.7 Stern et al. in 19618 had suggested that primary immunization to D could be suppressed if D-positive RBCs were coated in vitro with anti-D and then injected several times into D-negative subjects. None of the 16 men thus injected, produced anti-D, but when 10 of them were later given uncoated D-positive RBCs, five produced anti-D. These experiments were not pursued. Freda and colleagues, also in the USA, were the first to demonstrate that an anti-D Ig concentrate, given intramuscularly, could suppress D-alloimmunization in D-negative male prisoners and then in D-negative mothers delivering D-positive infants.9,10
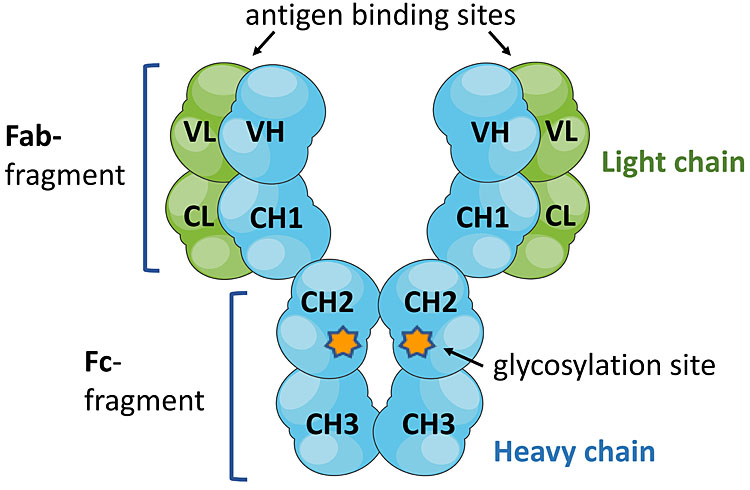
1
IgG structure. IgG molecules are comprised of two Fab fragments with two variable regions that bind antigen and the Fc fragment, which mediates effector activities by interacting with receptors (FcγR) on mononuclear phagocytic cells.
A large number of extensive clinical trials followed in many countries, administering anti-D Ig postnatally to over 50,000 D-negative women, mostly primiparae, delivering ABO compatible infants, in many cases measuring the extent of the TPH.1,11,12 Anti-D immunoglobulin was approved for clinical use in 1968 in the US and UK and then in several other countries.
Definite proof that D-immunization can be prevented by the administration of anti-D Ig after delivery of a D-positive infant must rest on the reduction of immunization of the mothers after delivery of a subsequent D-positive infant. Many women in the trials were followed up for years. The failure rate of anti-D prophylaxis at the end of second D-positive pregnancies was found to be about 1.5%, with 0.7% in each pregnancy due to primary immunization during pregnancy and an additional 0.2% due to large TPHs not covered by the anti-D dose given (Woodrow, et al. 1971,2 Combined Study, 1966,13 197114). Hence, the administration of anti-D Ig after sensitizing episodes during pregnancy (e.g. amniocentesis, CVS, molar or ectopic pregnancy) or abortion and after delivery of D-positive infants, is 90% effective in preventing maternal RhD immunization. Most of the remaining 10% could be covered by the routine administration of antenatal anti-D Ig prophylaxis (RAADP). The procedure had been shown to be safe by Bowman and colleagues,15 16 in Canada and by Tovey et al.17 in primiparae in the UK. Both groups showed that <0.1%15 or 0.16%17 of RhD negative mothers delivering a second RhD-positive infant had anti-D at full term, thus reducing D-alloimmunization significantly from the 1.5% (0.9%15–1.8%17) reported in untreated mothers. Bowman (1978)15 found that antenatal anti-D Ig does not harm the fetus and that it should be given as early as 28 weeks of gestation.
Dose of anti-D Ig for prophylaxis
Since it was unclear what dose of anti-D was necessary to suppress immunization by a given volume of fetal RBCs, meticulous dosage studies were done in volunteers18,19 and it was concluded that the minimum effective dose of anti-D Ig to suppress immunization by D-positive RBCs lies between 10 and 25 micrograms anti-D per ml of RBCs. To err on the side of safety, the WHO20 has recommended 25 μg (125 international units = i.u.) as the suppressive dose of anti-D Ig per ml of RBCs. The UK seems to be one of the few countries that measures anti-D Ig in international units.
Postnatal prophylaxis and for sensitizing events during pregnancy
The preparations of anti-D Ig available in different countries have varied. In the USA and many other countries, it has been customary to use 300 micrograms (1500 i.u.), covering up to 12 ml of foetal D-positive RBCs (up to 30 ml of blood) whereas in the UK, until recently 100 micrograms (500 i.u.) has been the dose used routinely for postpartum prophylaxis. Anti-D Ig should be administered within 72 hours of delivery, although it will still be partially effective if given within a week of exposure to the D antigen.21 Regardless of the dose used, it is recommended to measure the size of TPH at delivery to determine if additional anti-D Ig should be given (Fetomaternal Hemorrhage and Laboratory Methods for its Determination). Obviously, the lower dose of 100 micrograms will only cover TPHs up to 4 ml of RBCs (8–10 ml of blood), but will save considerable amounts of much needed anti-D Ig. For potentially sensitizing events up to 20 weeks' gestation, smaller doses of anti-D Ig are required; in the UK, a minimum dose of 50 μg (250 i.u.) is recommended. For such events after 20 weeks’ gestation, the standard postnatal dose is recommended within 72 hours and assessment of TPH size is required (see Fetomaternal Hemorrhage and Laboratory Methods for its Determination).
Routine antenatal anti-D prophylaxis (RAADP)
Data from several published studies show that there is general agreement that RAADP is safe for the fetus and significantly superior, when compared with no RAADP, in reducing the incidence of maternal D immunization. Various modes of administration of RAADP are followed in different countries, from a single dose of 300 micrograms at 28 weeks’ gestation, to 100 or 125 micrograms at 28 and 34 weeks or 300 μg at 28 and 34 week.16,22,23,24 The jury is still out for the recommended regime of RAADP, as there are not enough studies to compare different modalities and outcomes (Chilcott et al. 2004;25 Xie et al. 202026). It is likely that a single dose of 300 μg at 28 weeks might not be sufficient if we assume that there should be at least 25 μg of anti-D Ig in the maternal blood at delivery. Hence, two doses of 100 μg (500 i.u.) of anti-D at 28 and 34 weeks, respectively, will result in a higher concentration of anti-D in maternal plasma immediately before delivery.27 Ideally, the RHD status of the fetus should be determined before 28 weeks' gestation, thus reserving antenatal prophylaxis for the approximately 60–65% of women carrying D-positive fetuses (see Non-Invasive Prenatal Testing of Rh Fetal Status).
Success of anti-D Ig prophylaxis
Perinatal deaths due to HDFN have fallen dramatically since 1969/70, with the introduction of anti-D Ig prophylaxis. With just postnatal prophylaxis and anti-D administration for sensitizing episodes during pregnancy (e.g. amniocentesis, CVS) or abortion, mortality fell 100-fold in the UK, but it was still about 50 deaths a year. The decision to introduce antenatal prophylaxis in the western world has reduced mortality even further; in the UK, the reduction in immunization to under 1% of D-negative women carrying a D-positive fetus has paralleled a reduction in mortality associated with HDFN from the original 46/100,000 births to 1.6/100,000.28
Possible mechanism of action – fast clearance of D-positive RBCs by passive anti-D Ig and effectiveness of suppression of the immune response
There is clear evidence that the speed, though not the rate, of clearance of D-positive RBCs by anti-D correlates with its immunosuppressive effects and that the interaction between anti-D coated RBCs and mononuclear phagocytic cells in the spleen might explain this relationship.29 The precise mechanism of immunosuppression by anti-D is still unclear, although it is suggested that binding of the Fc part of IgG to low-affinity FcγR receptors on the splenic macrophages is essential for the accelerated clearance of RBCs from the circulation and their destruction by phagocytosis or extracellular cytotoxicity by the macrophages.27
The immune response to the D antigen can be suppressed by very small amounts of antibody, thus not needing blocking or masking of all antigen sites. If 20–25 μg of anti-D Ig can suppress immunization by 1 ml of D-positive RBCs in a D-negative adult, considering that the antibody will be distributed between the plasma and the extravascular space, at equilibrium only about 5% of anti-D and about 1% of D antigen sites will be bound.30
Source and safety of polyclonal anti-D Ig
Except for those countries that are totally or partially self-sufficient in anti-D Ig, such as The Netherlands, France, Finland, Australia and others, most of the source plasma pooled for the manufacture of anti-D Ig comes from extensive plasmapheresis of USA subjects hyperimmunised with especially selected and matched D-positive RBCs. The IgG anti-D is purified from plasma, which has been screened for all the mandatory microbial markers required for routine blood donors and the manufacturing process includes an inactivation procedure that kills enveloped and non-enveloped viruses. The preparations available for clinical use are very safe and no proven cases of transmission of infectious agents have been documented for anti-D Ig preparations for intramuscular use. Currently approved anti-D Igs prepared by ion exchange and virally inactivated, for intravenous use, have a good safety record and have less anti-A, -B and IgA than anti-D intended for intramuscular use.
Anti-D Ig preparations are polyclonal in nature, containing all subclasses of IgG (IgG1, 2, 3, 4) and minor contamination of IgA. D antibodies account for less than 1% of the total IgG in the preparation, and are predominantly IgG1 with variable amounts of IgG3.31
Anti-D Ig has been administered to hundreds of thousands of women. The reported rate of adverse events, mostly not serious, is <1 in 80,000 doses of anti-D Ig.28 The most serious adverse events have been cases of anaphylaxis in IgA-deficient women with anti-IgA, as most intramuscular anti-D Ig preparations contain traces of IgA; such women should be treated with the newer intravenous anti-D Ig preparations.
Due to its human origin, anti-D Ig is expensive and it is doubtful whether the USA plasma industry would be able to supply all the needs of those countries where anti-D prophylaxis is not in routine use.
Licensing requirements for polyclonal anti-D immunoglobulin preparations for clinical use
Before it came into routine clinical use, evidence of efficacy and safety of polyclonal anti-D Ig had been proven beyond doubt in numerous clinical trials in tens of thousands of D-negative women, delivering ABO compatible D-positive infants.15,30 32,33,34 However, the regulatory authorities demand strict licensing requirements before a new polyclonal anti-D Ig preparation is introduced into the markets of North America, Europe, Australasia and many other countries. The requirements that need to be met by polyclonal human anti-D Ig are for antenatal and postnatal prophylaxis of D immunization as well as for the treatment of D-negative persons transfused inadvertently with D-positive RBCs.
As an example, although the European Medicines Agency (2007)35 acknowledges the historical fact that D immunization following pregnancy can be prevented in the majority of cases by giving an adequate dose of polyclonal anti-D Ig to D-negative mothers after delivery of a D-positive infant or after potentially sensitizing events during pregnancy, it still demands stringent requirements prior to the marketing authorization of any new polyclonal anti-D Ig preparation. Verbatum, such requirements include the following:
- Biological data:
- In vitro potency assays addressing red-cell destruction to assess product comparability.
- Antibody content against different RhD phenotypes on red cells.
- In vivo and/or in vitro quantification of anti-RhD.
- IgG subclasses, in particular IgG1 and IgG3.
- Pharmacodynamics/pharmacokinetics:
- These data are essential to support the pharmacological activity and efficacy of the product and must be provided in each application dossier.
- The pharmacodynamic effect is established by the biological data for the product and the clinical efficacy studies.
- Single-dose pharmacokinetics studies should be carried out in 15 RhD negative subjects after intravenous and/or intramuscular administration. Serum clearance, volume of distribution, area under the curve and mean serum half-life should be measured.
- Efficacy
- Clinical data should be provided to demonstrate the efficacious prevention of RhD-isoimmunization in RhD-negative women who are pregnant with a RhD-positive fetus. The study should investigate at least 200 non-immunized patients and should include both ante-partum and post-partum administration, depending on the circumstance and indication for use, and should utilize an accepted dosage regimen via the desired route of administration. Blood samples should be collected just before treatment and at 72 hours and 3–6 months after treatment with the anti-RhD Ig. The incidence of anti-D at 3 and 6 months should be reported.
- Safety
- Adverse events from all subjects followed in clinical studies should be recorded and reported according to guidelines.
- Viral safety: compliance with CHMP recommendations with regard to viral safety is required. A pre-treatment serum sample from each patient included in the clinical trials should be stored at −70C for future testing.
MONOCLONAL ANTI-D
As stated above, polyclonal anti-D Ig is too expensive and possibly in insufficient supply to satisfy the world’s needs. Therefore, it would be ideal if an effective monoclonal anti-D was available for clinical use because it could be made in potentially unlimited quantities.
Monoclonal antibodies (mAbs) to many diseases, especially cancers, infections and immunological disorders are now clinically available, although they are expensive, requiring high therapeutic doses. They are made by harvesting the supernatant fluid from cultures of cells that have been manipulated to express the genes for a single IgG, which codes for the secreted protein. Most of these immortalized cell lines are derived from animal cells. Why is not monoclonal anti-D (mAb-D) also being used clinically?
The answer is becoming apparent, by understanding the immune reactions involved in anti-D prophylaxis.
Immunological basis of the mechanism of anti-D prophylaxis
Prophylactic anti-D may prevent the immune response to fetal D-positive RBCs by causing their destruction by macrophages in the spleen. While the hypervariable regions of the Fab portion of IgG anti-D bind to the D antigens on RBCs, the Cγ2 domain of the constant portion (Fc) of anti-D engages with IgG Fc receptors, mainly FcγRIIIa, on the macrophages in the splenic red pulp (Figure 2A).36 Here, due to hemo-concentration there is less free plasma IgG and macrophages are in direct contact with the IgG coated RBCs.37 This triggers phagocytosis of the RBCs and their destruction in cytoplasmic lysosomes38 without inflammatory responses.39
It is also likely that long-term tolerance to the D antigen may occur in some women who have had prophylaxis. After successful prophylaxis, the likelihood of D-immunization in subsequent unprotected pregnancies was greatly reduced.40,41 In this case, D-specific B lymphocytes may be inactivated by co-crosslinking (a) the B-cell receptor (antigen-specific IgM) binding the D antigen on that RBC with (b) the inhibitory FcγRIIb on the B lymphocytes binding Fc of anti-D bound to the RBC42 (Figure 2B). Because B cells are not adjacent to macrophages in the splenic red pulp,38 RBCs may infiltrate the B-cell-rich area of the splenic white pulp or lymph nodes for these interactions to occur.
A |
|
B |
|
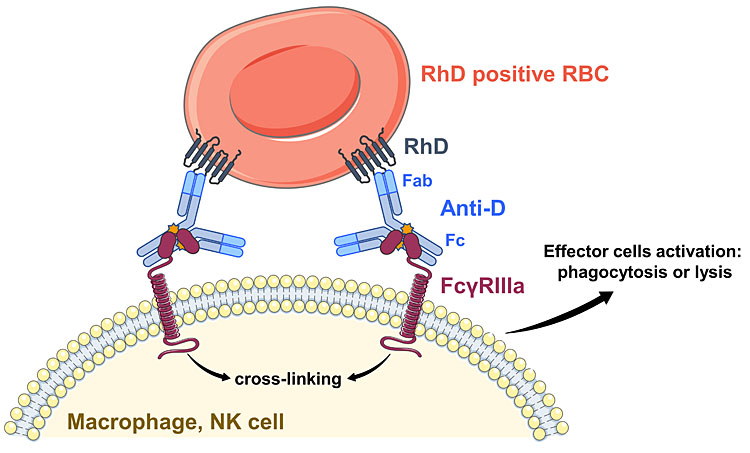
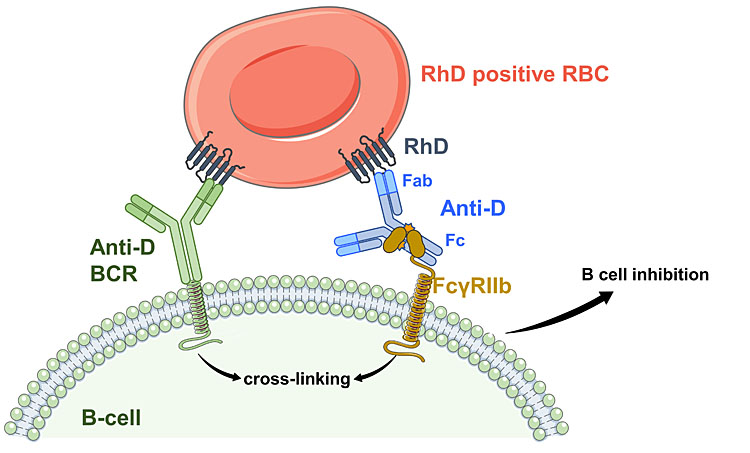
2
Recognition of anti-D-coated red blood cells (RBCs) by macrophages or B cells leads to different responses. (A) The central role of ADCC in vitro and in vivo lies with the FcγRIIIA, which binds to IgG anti-D at the Fc region when the IgG is bound to the RhD antigen. Multiple FcγRIIIA-antibody complexes lead to cross-linking and activation of the cellular response. (B) When a B-cell receptor (BCR; monomeric IgM) with specificity for the D antigen (anti-D BCR) binds to a RBC and FcγRIIB interacts with the Fc region of an antibody bound to any antigen on the same RBC, the B cell is inhibited and the anti-D response is suppressed. Again, multiple interactions are required for the antigen-specific response.
Measuring efficacy of mAb-Ds
Testing mAb-Ds in vitro is similar to the requirements for polyclonal anti-D Igs described above, but may include affinity measurements. The potency assay normally used is antibody-dependent cell-mediated cytotoxicity (ADCC)27 using effector cells expressing FcγRIIIa. Instead of splenic macrophages (not practical), natural killer (NK) cells from peripheral blood, which also bear FcγRIIIa are used. NK cells will lyse D-positive RBCs in the presence of anti-D; lysis is enhanced with proteolytic pre-treatment of the RBCs. The extent of lysis is determined by measuring the free hemoglobin over a range of anti-D concentrations.43 Normally the mAb-D is compared with a polyclonal anti-D Ig. Other in vitro assays have utilized peripheral blood monocytes, which express FcγRI and FcγRIIa but not FcγRIIIa. The activity in monocyte ADCC, phagocytosis44,45 and chemiluminescence46 assays of 45 IgG1 mAb-Ds correlated highly with the amount of IgG1 on RBCs, though IgG3 mAb-Ds had greater activity. Blocking studies showed FcγRI mediated all these effector activities; FcγRIIa was not utilized.46 In these multicenter studies, only a few IgG1 mAb-Ds exhibited activity in the NK ADCC assays, which are much more discriminating for biological efficacy.45,46
If in vitro tests of a mAb-D are deemed successful, in vivo studies with human volunteers can be carried out. Pharmacokinetic studies including the anti-D half-life are performed in D-negative volunteers. The rate of clearance of RBCs in D-positive volunteers can be measured by injection of radiolabeled (51Chromium) autologous RBCs pre-coated with anti-D followed by serial blood sampling for up to 7 days. This method avoids the risk of alloimmunization but does not approximate the situation in vivo.47
To determine the effectiveness of a mAb-D at preventing D-immunization, it must be injected into D-negative subjects, along with D-positive RBCs. Estimates of the RBC clearance, as above, will be informative because the speed of clearance is related to efficacy. This is followed by serial blood sampling for months and serological screening for IgG anti-D formation by the recipients. Primary anti-D responses are normally low titer, very slow to develop and by 6 months may not be detectable serologically (this is often the situation in primiparous, responder D-negative women post-partum, who rapidly form anti-D at the next D-positive pregnancy).2 This is known as “sensibilization”27 or priming. It can be misleading because if an anti-D test is negative, it may be assumed that the individual is not immunized, but if they are primed and later receive D-positive blood, transfusion reactions and/or D-immunization may occur. Because of this, subjects in a clinical trial must be challenged with a second injection of D-positive RBCs at about 6 months and a third at 9 months to induce immunization. A rising anti-D titer after the original (first) RBC injection or a rapid anti-D response to the second injection indicates failed prophylaxis. If after the second or third injection with D-positive RBCs, anti-D takes 6 weeks or more to develop, this is a primary response to the second or third injection and shows prophylaxis was successful. Thus only after a year can the subjects be categorized as (a) those with failed prophylaxis, (b) those with successful prophylaxis and (c) non-responders who did not produce anti-D and are not informative for the study.48 Chromium survival curves of D-positive RBCs in D-negative volunteers will also be informative about responsiveness or lack of responsiveness to D; collapse curves are indicative of responsiveness.18,27
Cell platforms for mAb-D production: the biological activity of antibodies depends on the producer сell line
All the mAb-Ds were derived from normal human IgG anti-Ds of D-immunized donors or patients, having their original protein sequences.
The species origin of immortalized cell line used for making mAb-Ds has changed with time. Four types of cell lines have been used by different groups and their mAb-Ds tested in autologous clearance, allogeneic clearance and/or prophylactic studies, usually in comparison with a polyclonal anti-D Ig.47 It is apparent that the type of cell line has a strong characteristic effect on the biological activity of the mAb-Ds.
From 1980 onwards, B lymphocytes from D-negative immunized donors were immortalized with Epstein-Barr virus (EBV) to form human B lymphoblastoid cell lines (B-LCL). These were cloned to select those secreting IgG anti-D.49 An autologous RBC clearance study was performed in London in 1990 with mAb BRAD-3, mediating rapid clearance.50 A pharmacokinetic study showed BRAD-3 (IgG3) and BRAD-5 (IgG1) had half-lives characteristic of their IgG subclass, 10 and 22 days respectively, and were well tolerated.51 This was followed by an allogeneic clearance and prophylaxis study of BRAD-3 and BRAD-5 in Bristol, as described above. BRAD-5, with high ADCC activity, cleared RBCs faster than BRAD-3, both showing a dose-response effect like the reference polyclonal anti-D Ig. Both mAb-Ds were effective at preventing D-immunization in volunteers, 22% were protected responders and 78% were non-responders.48
A similar result was then obtained in a large trial of 400 μg blended BRAD-3/BRAD-5 injected intramuscularly 24 hours after injection of 5 mL D-positive RBCs. The second, third and fourth RBC booster injections were again essential to detect the anti-D responders. Of the 92 D-negative subjects, 24 were protected responders with successful prophylaxis, 66 were non-responders and two were failures of prophylaxis, one apparent by 3 months and the other 4 weeks after the second RBC challenge (Figure 3). The success rate was 92%. Most (22) of the responders received homozygous D-positive RBC.52
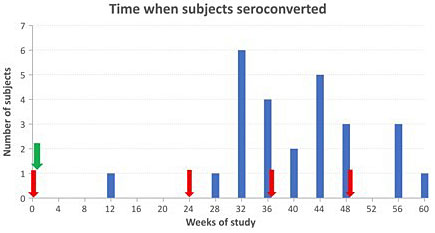
3
Time when anti-D was first detected in D-negative subjects. Red arrows indicate injections of D-positive RBC. Green arrow indicates injection of BRAD-3/BRAD-5 mAb-Ds on the following day. The height of the bars shows the number of responders (subjects with anti-D antibodies). Each subject with anti-D is represented only once.
By the turn of the 21st century, technology had favored the fusion of immunized human B cells with mouse myeloma cells to produce heterohybridoma cell lines (HH), which grew faster and secreted higher levels of mAb-D than B-LCL. One of these, FOG-1, with low ADCC activity, was also tested in vivo in 1990 but had slower RBC clearance than BRAD-3.50 HH mAb-Ds made in Edinburgh, Lille and Moscow also demonstrated variable, slow clearance compared to polyclonal anti-D Ig in both autologous and allogeneic studies. In the largest study, comprising 37 volunteers, nearly 60% of the D-negative subjects rapidly developed anti-D within 3 months, often with IgM as well (unusual for an anti-D response), indicating that the HH mAb-Ds had probably enhanced rather than prevented D-immunization.53
HH cell lines are, however, used for the manufacture of monoclonal antibodies for diagnostic reagents. These products have greatly improved blood grouping and testing.
About 20 years ago, Chinese Hamster Ovary (CHO) cells started to be used for recombinant/monoclonal antibodies to treat some diseases, and remain the cell line of choice for them. In Bern, MonoRho-CHO anti-D was given to 31 D-negative subjects in various doses after D-positive RBCs; clearance of the RBCs was extremely variable and much slower than with polyclonal anti-D (Rhophylac). Rhophylac was then given to seven subjects who had received MonoRho as “rescue medication” to protect them from becoming D-immunized (thus negating the aim of the study). On serologic testing after 6 months, anti-D was not detected in any subject.54 No challenge RBC injections were then given to determine the numbers of failures, responders and non-responders. Plasma levels of anti-D 48 hours after i.v. injection were 40 times lower for MonoRho than Rhophylac. No further results of MonoRho activity were published. BRAD-3 and BRAD-5 were also produced by CHO cells because the regulatory agencies then required them to be derived from a non-human cell line. These CHO recombinant mAb-Ds were less active in ADCC assays and slower at RBC clearance than those from the human B-LCL;55 the project was halted. Finally, Rozrolimupab (Sym001), another CHO anti-D comprised of a mixture of 25 recombinant mAb-Ds was developed in Copenhagen for prevention of HDFN and treatment of patients with immune thrombocytopenia (ITP) over 10 years ago; no data was released for HDFN but platelet count increases were variable and modest in 39% of primary ITP patients. Fever and/or chills occurred in 20% of patients and many had elevated inflammatory cytokines, interleukin-6 and tumor necrosis factor (TNF)α, on the day of treatment. Rescue medication of IVIG and steroids was given to 34% of the patients.56 Thus the CHO mAb-Ds are unreliable, can be inflammatory and lack effectiveness.
Then a rat myeloma cell line (YB2/0) known to secrete recombinant antibodies with high FcγRIIIa-mediated ADCC was used by two groups for the production of mAb-Ds. First the Cambridge mAb-D, FOG-1, was expressed in this cell line and showed markedly different characteristics to the earlier HH form. Clearance of autologous D-positive RBCs by FOG-1-YB2/0 was extremely rapid. The anti-D coated RBCs were completely cleared by 3–4 hours, with hemolysis peaking at 1–2 hours, some RBC clearance to the liver and mild febrile reactions in two of the five subjects.57 Then a mAb-D, R297-YB2/0, made in Lille, France, was also tested in an autologous clearance study; it promoted rapid and complete RBC clearance and was at least as potent as polyclonal anti-D Ig. Interestingly, at equal amounts of anti-D on the RBCs, twice as much TNFα was in plasma 4 hours after injection with R297-YB2/0 compared with polyclonal anti-D Ig.58 Then, Roledumab (R297-YB2/0 with one amino acid change) was tested and found to have a half-life of 18–22 days; it was safe and well tolerated in D-negative volunteers.59 In 2014, a Phase 2 clinical trial was registered in France with 300 μg Roledumab being administered i.m. or i.v. as antenatal and postnatal prophylaxis to 62 D-negative pregnant women carrying a D-positive fetus. The study was completed in 2017. Information on pharmacokinetics and safety was subsequently released but with incomplete data on five women and no reports of serious adverse events. The data on alloimmunization was withheld.60
A mAb-D, G12, originally developed as a B-LCL was expressed as recombinant in several cell lines and all were compared in ADCC assays. The HH had similar low activity to the B-LCL form but the YB2/0 recombinant had high ADCC activity. A novel G12 recombinant in a human non-lymphoid cell line, PER.C6, had very low ADCC activity.61
Many studies of mAb-Ds in humans have either not been published or reported only briefly in review papers or abstracts. This suggests that these anti-Ds did not function properly or did not prevent D-immunization. A clinical trial “failure” sheds light on what may be going wrong and does not reflect the competency of the scientists involved. Broadcasting information about negative results is essential for the advancement of science and medicine.
There is a mAb-D registered in India and widely used in India and several other countries – Rhoclone, mAb-D HG-92 of HH origin, manufactured by Bharat Serums and Vaccines Limited (Navi Mumbai). It does not mediate lysis of red cells in ADCC (authors’ data). Two publications reported clinical data.62,63 Recently, a recombinant form of Rhoclone was made in CHO cells and a similar clinical study published.64 Of a total of 359 women in the three studies who were given 300 micrograms of Rhoclone i.m. within 72 hours of delivery, none had anti-D by indirect antiglobulin (Coombs) test at 180 days. Control groups of women received polyclonal anti-D under the same conditions; of 202 women, two had anti-D at 180 days (at least one of them had anti-D on days 3 (1 : 2), 10 and 75 (1 : 64), so the woman had already been primed). Only 33% of the women were primigravidae. There was no data on feto-maternal ABO compatibility. Unfortunately, 13.6% and 20.8% of the monoclonal and polyclonal groups, respectively, were lost to follow up and excluded from analysis. No in vitro bioassay studies, nor in vivo pharmacokinetic or clearance studies of either Rhoclone preparations have been published. None of these studies included the follow-up of women during their subsequent pregnancy. Long-term use of Rhoclone in clinical practice could provide objective information about its real efficacy. Unfortunately, despite Rhoclone’s postnatal use in thousands of women, no post-marketing data on its prophylactic effects has been published.
Discussion
It is apparent from the human studies of mAb-Ds that their characteristic activities were determined by the cell line that produced them. Those from human B-cell lines (BRAD-3, BRAD-5) behaved like polyclonal anti-D Ig but were slightly less effective. The HH derived mAb-Ds enhanced D-immunization. The CHO mAb-Ds were slow and erratic in red-cell clearance, which suggests they would not be able to prevent D-immunization. By contrast, the YB2/0 mAb-Ds cleared red cells extremely quickly but with signs of an associated inflammatory response. What could be causing these differences?
The human IgG molecule has two branched oligosaccharide chains, each linked to one of the heavy chains at Asparagine-297 in the central (CH2) domain of the Fc region, near the interaction sites for Fc receptors (Figure 1). The oligosaccharide, or glycan, has a basic structure of N-acetylglucosamine and mannose residues to which other sugars may or may not be attached. Fucose may be present on one or both chains of the oligosaccharides, near the Fc protein. The two branches may independently terminate in N-acetylglucosamine or galactose. The various combinations give 16 different oligosaccharides, or glycoforms. Then sialic acid may cap galactose, giving further heterogeneity (Figure 4).
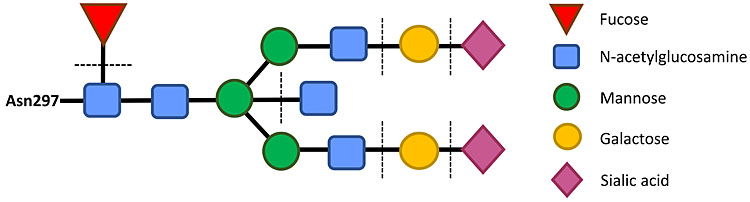
4
Glycosylation of human IgG Fc. The oligosaccharide chain (or glycan) is attached to the IgG Fc at residue Asparagine-297 in the CH2 domain. The two oligosaccharide chains, one on each Fc chain, occupies most of the space between the two Fc regions of the IgG molecule. Other amino acid residues in the CH2 domain of IgG form the binding site for FcγRIIIa. Fucose on IgG glycans blocks this interaction. The dotted lines indicate sugars that may or may not be present and can modulate the effector activity of the IgG. In Figures 1 and 2, the oligosaccharide chains are depicted as brown stars on the Fc.
The sequence and arrangement of sugars in the oligosaccharide are not coded for by DNA, unlike the protein sequence. After the IgG protein is synthesized, the sugars are added by multiple enzymes called glycosyltransferases to form the oligosaccharide chains. These enzymes (also coded for by DNA) are characteristic for each cell line, reflecting their species of origin.65,66
The absence of galactose (and therefore also of sialic acid) on human IgG was first discovered in inflammatory diseases such as rheumatoid arthritis.67 In contrast, galactose was found to be higher on IgG of pregnant women than non-pregnant women.68 Galactose is also increased on alloimmune IgG anti-D.69,70 Fucose was shown to block the interaction between IgG and FcγRIIIa, measured by ADCC assays71 and by x-ray crystallography.72 Therefore, high amounts of fucose would prevent the phagocytosis of anti-D coated red cells by macrophages in the spleen.
Progress in further understanding the biological relevance of glycosylation was held back by the complexity of their structure. Only in the last decade have analytical methods been developed for accurate and quantitative large-scale determination of the glycoforms of IgG. A recent study measured the percentages of 12 glycosylation features on a large sample of mAb-Ds (described above) from the four cell lines used (B-LCL, HH, CHO, YB2/0) and compared them with polyclonal anti-D Ig.43 The percentages of the two most important sugars affecting the function of IgG, galactose and fucose, are compared with other characteristics of polyclonal anti-D Ig and mAb-Ds (Table 1 and Figure 5).
1
Comparison of characteristics of polyclonal and monoclonal anti-Ds that have been tested in studies of RBC clearance or prevention of D-immunization.
Year | Anti-D | Isotype | Cell line | ADCC activity | Auto- | Rate of clearance. Order of | Prevention of immu- | Galac- | Fucose (%) |
Polyclonal | IgG1>3 | B | ++++ | Auto, Allo | ++++ | Yes | 84 | 80 | |
1990 | BRAD-3 | IgG3 | B-LCL | ++ | Auto | +++ Pre-sensitized | 61 | 77 | |
1995 | BRAD-3 | IgG3 | B-LCL | +++ | Allo | +++ mAb, RBC | Yes (3 of 10) | 61 | 77 |
1995 | BRAD-5 | IgG1 | B-LCL | +++ | Allo | +++ mAb, RBC | Yes (1 of 8) | 64 | 81 |
2002 | BRAD-3 + BRAD-5 | IgG1>3 | B-LCL | +++ | Allo | +++ RBC, mAb | Yes (24 of 92), No (2 of 92) | 63 | 80 |
1990 | FOG-1 | IgG1 | HH | + | Auto | + Pre-sensitized | 24 | 93 | |
1999 | AD1+AD3 | IgG1+3 | HH | + | Allo | + RBC, mAb | No (3 of 5) | 38 | 97 |
2000 | G7 | IgG1 | HH | + | Allo | ++ RBC, mAb | No (3 of 9) | 42 | 90 |
2000 | G12 | IgG1 | HH | + | Allo | ++ RBC, mAb | No (5 of 8) | 24 | 93 |
2000 | G12 | IgG1 | HH | + | Allo | +++ mAb, RBC | ? 2 subjects | 24 | 93 |
2002 | Rhoclone | IgG1 | HH | + | Post-natal | Not done. RBC, mAb | ? 110 women | ||
2019 | Rhoclone | IgG1 | HH | + | Post-natal | Not done. RBC, mAb | ? 105 women | ||
2020 | Rhoclone | IgG1 | CHO | + | Post-natal | Not done. RBC, mAb | ? 144 women | ||
2004 | MonoRho | IgG1 | CHO | + | Allo | ++ RBC, mAb | ? 31 subjects | 30 | 92 |
2006 | FOG-1 | IgG1 | YB2/0 | +++++ | Auto | +++++ Pre-sensitized | 15 | 23 | |
2006 | R297 | IgG1 | YB2/0 | +++++ | Auto | +++++ Pre-sensitized | 45 | 33 | |
2021 | Roledumab (R-593) | IgG1 | YB2/0 | Not done | Ante- and post-natal | Not done. RBC, mAb | ? 62 women. Data withheld |
*D-positive RBC were injected into D-positive subjects (autologous studies) or D-negative subjects (allogeneic studies).
†Subjects given BRAD mAbs were challenged with D-positive RBC (see Figure 2) to determine whether prophylaxis was effective or failed. The subjects and women in the other studies were not challenged and their status (failure, protected responder or non-responder is not known (?)) as they had not produced anti-D at the end-point (180 days) of the studies or patient observations.
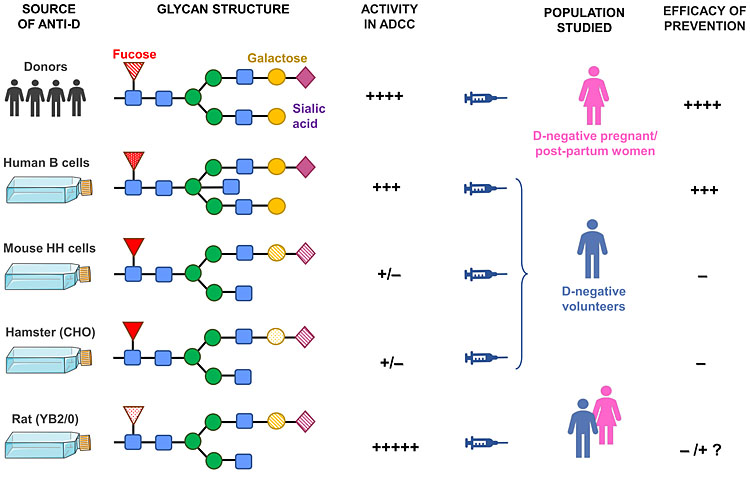
5
Pictorial summary of the effect of glycosylation variants on anti-D function. In order to achieve an effective prevention of hemolytic disease, prophylactic anti-D must have low fucose and high galactose content.
The HH mAb-Ds had low amounts of non-human sugars (α-galactose and N-glycolylneuraminic acid) derived from the mouse cells. These were probably the cause of the rapid development of immune anti-D in the recipients of RBC coated with HH mAb-Ds because foreign sugars are immunogenic and humans have naturally occurring anti-α-galactose. Alpha-galactose on animal cells is the main factor preventing xenotransplantation.
The galactosylation (β-galactose) was high for polyclonal anti-D Ig (84%) and quite high for B-LCL mAb-Ds but was very low for the animal-cell-derived mAb-Ds, HH, CHO and YB2/0, also the PER.C6 mAb-D. The sialylation was therefore very low as well,43 extremely so for FOG-1-YB2/0 (2%); this mAb-D cleared some RBCs in the liver, which has asialoglycoprotein receptors. It has been observed that in rheumatoid arthritis, decreased galactosylation of IgG correlated with elevated interleukin (IL)-6 and C-reactive protein (CRP), markers of systemic inflammation.73 Therefore, the low levels of galactose, and also of sialic acid, would make these mAb-Ds liable to inflammatory responses, as was observed for a CHO and the two YB2/0 mAb-Ds.
The fucosylation of B-LCL mAb-Ds was similar to polyclonal anti-D Ig, higher with HH and CHO mAb-Ds but much lower on YB2/0 mAb-Ds. The high levels of fucose on HH, CHO and PER.C6 mAb-Ds would explain why ADCC and RBC clearance was variable but generally slow. In contrast, the low fucose on YB2/0 mAb-Ds enabled very rapid phagocytosis of RBCs in vivo, but this induced some hemolysis and TNFα, both inflammatory.
In a D-negative recipient, these small inflammatory stimuli might induce stimulation rather than suppression of the anti-D immune response. For pregnant and post-partum women, inflammation accompanying phagocytosis of D-positive RBCs by mAb-Ds must definitely be avoided.
A paradox of pregnancy is that the semi-allogeneic fetus is tolerated by the mother but she is not immunosuppressed. Unique immunological changes are orchestrated by trophoblast cells of the placenta, which release cytokines, hormones and cellular products to the uterus and maternal blood.74 This immune deviation prevents rejection of the fetus in the uterus whilst enhanced systemic antibody-mediated immunity ensures the woman is protected against environmental infections,75,76 so ensuring that both mother and fetus survive. Thus an inflammatory anti-D given to pregnant women who already have heightened antibody responses would be disastrous.
The great success of animal-cell-derived mAbs for cancer treatment and other diseases may be that inflammatory reactions unintentionally caused by cross-species glycosylation will help destroy diseased cells. In contrast, for HDFN prophylaxis, RBCs must be destroyed by anti-D without inflammation caused by incompatible glycosylation.
CONCLUSION
A mAb-D for prophylaxis should be both highly efficiently recognized by FcγRIIIa on effector cells of the immune system and promote rapid clearance of fetal D-positive RBCs transferred by fetomaternal hemorrhage. At the same time, the anti-D must be compatible with the immune system of pregnant women; that means the glycosylation must not be inflammatory. To date, no mAb-D has fulfilled all these requirements, although the two made from human B cell lines were nearly as effective as the current polyclonal anti-Ds in routine use (Figure 5).
It is likely that some form of glycoengineering would be required for a novel effective mAb-D. If that is considered successful in in vitro assays, lengthy clinical trials in hundreds of volunteers must be undertaken with challenge injections of RBCs to ascertain the numbers of responders, non-responders and failures, as described above. Considerable financial investment would be required both for generating the mAb-Ds and performing Phase 1, 2 and 3 trials. Unfortunately, it is unlikely that organizations who have already tested mAb-Ds would be willing to start again.
It is surprising and worrying that a mAb-D that has not been shown to meet all the licensing and pre-marketing testing requirements, is available for prophylaxis in India and other low–income countries. It should be the responsibility of the drug manufacturers to do a well-controlled post-marketing follow-up of any monoclonal anti-D they sell. There is a need for an international licensing authority to ensure that medicines are appropriately tested before they are available in the market for clinical use.
Despite its great success, the era of immunizing, boosting and plasmapheresis of donors for the preparation of polyclonal anti-D Ig should surely come to an end. After 40 years of trial and error with monoclonal antibodies, it is time for the international community to come together now, so that we can see the way forward to ensure an effective and affordable alternative to plasma-derived polyclonal anti-D is available to treat all those mothers who need it.
PRACTICE RECOMMENDATIONS
- These should be listed by the Editors in Guidelines for Rhesus Disease Prevention (Recommended international guidelines and comparisons). However, we suggest the readers follow the BCSH 2014 Guidelines by Qureshi et al.,22 which are the most comprehensive and cover all aspects of prophylaxis.
- Monoclonal anti-D (mAb-D) should not be given as prophylaxis to pregnant or post-partum women because none of them have undergone significant clinical trials showing prevention of D-immunization after a second D-positive pregnancy, nor have they been licensed for clinical use.
ACKNOWLEDGEMENTS
Servier Medical Art images (smart.servier.com) were used in Figures 1, 2 and 5 with modifications.
CONFLICTS OF INTEREST
The author(s) of this chapter declare that they have no interests that conflict with the contents of the chapter.
Feedback
Publishers’ note: We are constantly trying to update and enhance chapters in this Series. So if you have any constructive comments about this chapter please provide them to us by selecting the "Your Feedback" link in the left-hand column.
REFERENCES
Mollison PL. Blood Transfusion in Clinical Medicine, 5th edn. Oxford, UK: Blackwell Scientific Publications, 1972. | |
Woodrow JC, Clarke CA, McConnell RB, et al. Prevention of Rh haemolytic disease: results of the Liverpool “low risk” clinical trial. Brit Med J 1971;ii:610–2. | |
Levine P. Serological factors as possible causes in spontaneous abortions. J Hered 1943;34:71–80. | |
Nevanlinna HR, Vaino T. The influence of mother-child ABO incompatibility on Rh immunisation. Vox Sang 1956;1:26–33. | |
Murray S, Knox, EG, Walker W. Rhesus haemolytic disease of the newborn and the ABO groups. Vox Sang 1965;10:6–31. | |
Finn R. In: Report of the Liverpool Medical Institution. Lancet 1960;i:526. | |
Clarke CA, Donohue WTA, McConnell RB, et al. Further experimental studies on the prevention of Rh haemolytic disease. Brit Med J 1963;I:979–84. | |
Stern K, Goodman HS, Berger M. Experimental isoimmunization to hemoantigens in man. J Immunol 1961;87:189–98. | |
Freda VJ, Gorman JG, Pollack W. Successful prevention of experimental Rh sensitization in man with an anti-Rh gamma2 – globulin antibody preparation: a preliminary report. Transfusion 1964;4:26–32. | |
Freda VJ, Gorman JG, Pollack W. Rh factor: prevention of isoimmunization and clinical trial on mothers. Science 1966;151(3712):828–30. | |
Pollack W, Gorman JG, Freda VJ, et al. Results of clinical trials of RhoGAM in women. Transfusion 1968;8(3):151–3. | |
Woodrow JC. Rh immunization and its prevention. Series Hematology vol III, 1970;3:6–151. | |
Combined Study. Prevention of Rh haemolytic disease: results of the clinical trial. A combined study from centres in England and Baltimore. Brit Med J 1966;2:907–14. | |
Combined Study. Prevention of Rh-haemolytic disease: final results of the “high risk” clinical trial. A combined study from centres in England and Baltimore. Brit Med J 1971;2:607–9. | |
Bowman JM, Chown B, Lewis M, et al. Rh isoimmunization during pregnancy: antenatal prophylaxis. Can Med Assoc J 1978;118(6):623–7. | |
Bowman JM. Controversies in prophylaxis, in Haemolytic Disease of the Newborn. In: Garratty G. (ed.) Amer Assoc Blood Banks. Arlington, VA, 1984;67–85. | |
Tovey LA, Townley A, Stevenson BJ, et al. The Yorkshire anti-D immunoglobulin trial in primigravidae. Lancet 1983;2(8344):244–6. | |
Mollison PL, Hughes-Jones NC, Lindsay M, et al. Suppression of primary Rh immunization by passively-administered antibody. Experiments in volunteers. Vox Sang 1969;16(4):421–39. | |
Pollack W, Ascari WQ, Kochesky RJ, et al. Studies on Rh prophylaxis. I. Relationship between doses of anti-Rh and size of antigenic stimulus. Transfusion 1972;11(6):333–9. | |
WHO (1971) Prevention of Rh sensitization. Technical Report Series 468. | |
Samson D, Mollison PL. Effect on primary Rh immunization of delayed administration of anti-Rh. Immunology 1975;28(2);349–57. | |
Qureshi H, Massey E, Kirwan D, et al. BCSH guideline for the use of anti-D immunoglobulin for the prevention of haemolytic disease of the fetus and newborn. Transfus Med 2014;24(1):8–20. | |
Practice Bulletin No.181: Prevention of Rh D alloimmunization. Obst & Gynecol 2017;130–2–e57–70. | |
National Blood Authority. Prophylactic use of RhD immunoglobulin in pregnancy care 2021. ISBN 978-0-9945576-8-1. | |
Chilcott J, Tappenden P, Lloyd Jones M, et al. The economics of routine antenatal anti-D prophylaxis for pregnant women who are rhesus negative. BJOG 2004;111(9);903–7. | |
Xie X, Fu Q, Bao Z, et al. Clinical value of different anti-D immunoglobulin strategies for preventing Rh haemolytic disease of the fetus and newborn: a network meta-analysis. PLoS One 2020;15(3):e02 30073. | |
Mollison PL, Engelfriet CP, Contreras M. Blood Transfusion in Clinical Medicine 9th edn. Oxford, UK: Blackwell Scientific Publications, 1993. | |
Pilgrim H, Lloyd-Jones M, Rees A. Routine antenatal anti-D prophylaxis for RhD negative women: a systematic review and economic evaluation. Health Technol Assess 2009;13(10):1–126. | |
Mollison PL. Some aspects of Rh haemolytic disease and its prevention, in Haemolytic disease of the newborn. In: Garratty G. (ed.) Amer Assoc Blood Banks, Arlington VA, 1984;1–32. | |
Mollison PL, Engelfriet CP, Contreras M. Blood Transfusion in Clinical Medicine 8th edn. Chapter 6. Oxford, UK: Blackwell Scientific Publications, 1987. | |
Gorick BD, Hughes-Jones NC. Relative functional binding activity of IgG1 and IgG3 anti-D in IgG preparations. Vox Sang 1991;61(4):251–4. | |
McMaster Conference. Prevention of Rh immunization. Vox Sang 1977;36:50–64. | |
Bowman JM. Controversies in Rh prophylaxis. Who needs Rh immune globulin and when should it be given? Am J Obstet Gynecol 1985;151(3):289–94. | |
No authors listed. Statement from the Consensus Conference on anti-D prophylaxis. 7 and 8 April 1997. The Royal College of Physicians of Edinburgh. The Royal College of Obstetricians and Gynaecologists, UK. Vox Sang 1998;74:127–8. | |
European Medicines Agency. Committee for Medicinal Products for Human Use. Guideline on the clinical investigation of human anti-D immunoglobulin for intravenous and/or intramuscular use. CPMP/BPWG/575/99 Rev. 1, London, 2007. | |
Nagelkerke SQ, Bruggeman CW, den Haan JMM, et al. Red pulp macrophages in the human spleen are a distinct cell population with a unique expression of Fc-γ receptors. Blood Adv 2018;2(8):941–53. | |
Borges da Silva H, Fonseca R, Pereira RM, et al. Splenic macrophage subsets and their function during blood-borne infections. Front Immunol 2015;6:480. | |
Kumpel BM. On the immunologic basis of Rh immune globulin (anti-D) prophylaxis. Transfusion 2006;46(9):1652–6. | |
Coopamah MD, Freedman J, Semple JW. Anti-D initially stimulates an Fc-dependent leukocyte oxidative burst and subsequentially suppresses erythrophagocytosis via interleukin-1 receptor agonist. Blood 2003;102(8):2862–7. | |
Thornton JG, Page C, Foote G, et al. Efficacy and long term effects of antenatal prophylaxis with anti-D immunoglobulin. BMJ 1989;298(6689):1671–3. | |
Koelewijn JM, de Haas M, Vrijkotte TG, et al. One single dose of 200 μg of antenatal RhIG halves the risk of anti-D immunization and hemolytic disease of the fetus and newborn in the next pregnancy. Transfusion 2008;48(8):1721–9. | |
Kumpel BM, Elson CJ. Mechanism of anti-D-mediated immune suppression – a paradox awaiting resolution? Trends Immunol 2001;22(1):26–31. | |
Kumpel BM, Saldova R, Koeleman CAM, et al. Anti-D monoclonal antibodies from 23 human and rodent cell lines display diverse IgG Fc-glycosylation profiles that determine their clinical efficacy. Sci Rep 2020;10(1):1464. doi: 10/1038/s41598-019-57393-9. | |
Kumpel BM. Coordinator’s report: An assessment of the functional activity of human Rh monoclonal antibodies after their evaluation by nine laboratories. Transfus Clin Biol 1996;6(3):439–52. | |
Kumpel BM, Jackson DJ. Characterisation and functional activity of human Rh monoclonal antibodies. Transfus Clin Biol 1996;6(3):453–8. | |
Kumpel BM, Beliard R, Brossard Y, et al. Section 1C: Assessment of the functional activity and IgG Fc receptor utilisation of 64 IgG Rh monoclonal antibodies. Coordinator’s report. Transfus Clin Biol 2002;9(1):45–53. | |
Kumpel BM. Efficacy of RhD monoclonal antibodies in clinical trials as replacement therapy for prophylactic anti-D immunoglobulin: more questions than answers. Vox Sang 2007;93(2):99–111. | |
Kumpel BM, Goodrick MJ, Pamphilon DH, et al. Human Rh D monoclonal antibodies (BRAD-3 and BRAD-5) cause accelerated clearance of Rh D+ red blood cells and suppression of Rh D immunization in Rh D- volunteers. Blood 1995;86(5):1701–9. | |
Koskimies S. Human lymphoblastoid cell line producing specific antibody against Rh-antigen D. Scand J Immunol 1980;11(1):73–7. | |
Thomson A, Contreras M, Gorick B, et al. Clearance of Rh D-positive red cells with monoclonal anti-D. Lancet 1990;336(8724):1147–50. | |
Goodrick J, Kumpel B, Pamphilon D, et al. Plasma half-lives and bioavailability of human monoclonal Rh D antibodies BRAD-3 and BRAD-5 following intramuscular injection into Rh D-negative volunteers. Clin Exp Immunol 1994;98:17–20. | |
Kumpel BM. In vivo studies of monoclonal anti-D and the mechanism of immune suppression. Transfus Clin Biol 2002;9(1):9–14. | |
Olovnikova NI, Belkina EV, Drize NI, et al. Fast clearance of Rhesus-positive erythrocytes by monoclonal anti-Rhesus antibodies – insufficient condition for effective prevention of Rhesus sensitization. Bull Exp Biol Med 2000;129(1):77–81 (in Russian). | |
Miescher S, Spycher MO, Amstutz H, et al. A single recombinant anti-RhD IgG prevents RhD immunization: association of RhD-positive red blood cell clearance rate with polymorphisms in the FcγRIIA and FcγRIIIA genes. Blood 2004;103(11):4028–35. | |
Chapman GE, Ballinger JR, Norton MJ, et al. The clearance kinetics of autologous RhD-positive erythrocytes coated ex vivo with novel recombinant and monoclonal anti-D antibodies. Clin Exp Immunol 2007;150(1):30–41. | |
Robak T, Windyga J, Trelinski J, et al. Rozrolimupab, a nixture of 25 recombinant human monoclonal RhD antibodies, in the treatment of primary immune thrombocytopenia. Blood 2012;120(18):3670–6. | |
Armour KL, Parry-Jones DR, Beharry N, et al. Intravascular survival of red cells coated with a mutated human anti-D antibody engineered to lack destructive activity. Blood 2006;107(7):2619–26. | |
Beliard R, Waegemans T, Notelet D, et al. A human anti-D monoclonal antibody selected for enhanced FcγRIII engagement clears RhD+ autologous red cells in human volunteers as efficiently as polyclonal anti-D antibodies. Br J Haematol 2008;141(1):109–19. | |
Yver A, Homery M-C, Fuseau E, et al. Pharmacokinetics and safety of Roledumab, a novel human recombinant monoclonal anti-RhD antibody with an optimized Fc for improved engagement of FCγRIII, in healthy volunteers. Vox Sang 2012;103:213–22. | |
Pharmacokinetics and Safety of Roledumab, in RhD-negative Pregnant Women Carrying an RhD-positive Foetus. Clinicaltrials.gov/ct2/show/NCT02287896. | |
Olovnikova NI, Ershler MA, Grigorieva OV, et al. Impact on N-glycosylation profile of monoclonal anti-D antibodies as a way to control their immunoregulatory and cytotoxic properties. Biochemistry (Moscow) 2012;77(8):925–33. | |
Chauhan AR, Bhattacharyya MS, Turakhia N, et al. Efficacy and safety of monoclonal anti-D immunoglobulin in comparison with polyclonal anti-D immunoglobulin in prevention of Rho Isoimmunization. J Assoc Physicians India 2002;50(Oct):1341–2. | |
Chauhan AR, Nandanwar YS, Ramaiah A, et al. A multicenter, randomized, open-label trial comparing the efficacy and safety of monoclonal anti-Rh (D) immunoglobulin with polyclonal anti-Rh (D) immunoglobulin for the prevention of maternal Rh-isoimmunization. J Obstet Gynecol India 2019;69(5):420–5. | |
Mayekar RV, Paradkar GV, Bhosale AA, et al. Recombinant anti-D for prevention of maternal-foetal Rh(D) alloimmunization: a randomised multi-centre clinical trial. Obstet Gynecol Sci 2020;63(3):315–22. | |
Marth JD, Grewal PL. Mammalian glycosylation in immunity. Nat Rev Immunol 2008;8(11):874–87. | |
Jefferis R. Recombinant proteins and monoclonal antibodies. Adv Biochem Eng Biotechnol 2021;175:281–318. | |
Parekh RB, Dwek RA, Sutton BJ, et al. Association of rheumatoid arthritis and primary osteoarthritis with changes in the glycosylation pattern of total serum IgG. Nature 1985;316(6027):452–7. | |
van de Geijn FE, Wuhrer M, Selman MH, et al. Immunoglobulin G galactosylation and sialylation are associated with pregnancy-induced improvement of rheumatoid arthritis and the postpartum flare: results from a large prospective cohort study. Arthritis Res Ther 2009;11(6):R193 doi: 10/1186/ar2892. | |
Kapur R, Della Valle L, Verhagen OJHM, et al. Prophylactic anti-D preparations display variable decreases in Fc-fucosylation of anti-D. Transfusion 2015;55(3):553–62. | |
Mori F, Salvatore A, Ascione E, et al. Evaluation of prophylactic polyclonal anti-D antibodies: Differences in Fc-glycosylation in commercial products. Vox Sang 2021;1–2 DOI: 10.1111/vox.13213. | |
Shields RL, Lai J, Keck R, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human FcγRIII and antibody-dependent cellular toxicity. J Biol Chem 2001;277(30):26733–40. | |
Ferrara C, Grau S, Jäger C, et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcγRIII and antibodies lacking core fucose. Proc Natl Acad Sci USA 2011;108(31):12669–74. | |
Troelsen LN, Jacobsen S, Abrahams JL, et al. IgG glycosylation changes and MBL2 polymorphisms: association with markers of systemic inflammation and joint destruction in rheumatoid arthritis. J Rheumatol 2012;39(3):463–9. | |
Kumpel BM, Sibley K, Jackson DJ, et al. Ultrastructural localisation of glycoprotein IIIa (GPIIIa, β3 integrin) on placental syncytiotrophoblast microvilli: implications for platelet alloimmunization during pregnancy. Transfusion 2008;48(10):2077–86. | |
Kumpel BM, Manoussaka MS. Placental immunology and maternal alloimmune responses. Vox Sang. 2012;102(1):2–12. | |
PrabhuDas M, Bonney E, Caron K, et al. Immune mechanisms at the maternal-fetal interface\; perspectives and challenges. Nat Immunol 2015;16(4):328–34. |
Online Study Assessment Option
All readers who are qualified doctors or allied medical professionals can automatically receive 2 Continuing Professional Development points plus a Study Completion Certificate from GLOWM for successfully answering four multiple-choice questions (randomly selected) based on the study of this chapter. Medical students can receive the Study Completion Certificate only.
(To find out more about the Continuing Professional Development awards program CLICK HERE)