This chapter should be cited as follows:
Nichol A, Hayes K, Glob Libr Women's Med
ISSN: 1756-2228; DOI 10.3843/GLOWM.413433
The Continuous Textbook of Women’s Medicine Series – Obstetrics Module
Volume 4
Fetal development and maternal adaptation
Volume Editor: Professor Asma Khalil, The Royal College of Obstetricians and Gynaecologists, London, UK; Fetal Medicine Unit, Department of Obstetrics and Gynaecology, St George’s University Hospitals NHS Foundation Trust, London, UK

Chapter
Obstetric and Fetal Pharmacology
First published: February 2021
Study Assessment Option
By answering four multiple-choice questions (randomly selected) after studying this chapter, readers can qualify for Continuing Professional Development points plus a Study Completion Certificate from GLOWM.
See end of chapter for details.

INTRODUCTION
Around the world 140 million women give birth each year, many of whom are exposed to medications during the pregnancy.1 A General Practice Research Database showed that approximately two-thirds of women in the UK had been prescribed at least one medication in pregnancy.2 Drug use in pregnancy occurs in four main areas:
- Accidental – it is estimated that around half of all pregnancies are unplanned, so drug exposure may occur before pregnancy is diagnosed.
- Chronic illness – for example in the management of long-term illnesses such as asthma, depression and epilepsy.
- Minor illness – such as urinary tract infection and lower respiratory tract infection.
- Pregnancy-related illness – for example gestational diabetes and pre-eclampsia.
When prescribing any medication in both the pregnant and non-pregnant patient, the risks and benefits must be evaluated by the clinician and the patient. In pregnancy this process may be more complicated if the mother and baby have conflicting needs. A small number of medications such as methotrexate and retinoids are known teratogens which are absolutely contraindicated in pregnancy. However, in some instances the use of known teratogens in pregnancy may be justified by the severity of the maternal condition, for example in the use of warfarin in women with metallic heart valves, or the use of anti-epileptic drugs. The background rate of congenital malformations is around 2%, and known teratogens can have variable effects which may depend on the timing, dose and duration of use. There is often a lack of good quality clinical trial data surrounding the use of medications in pregnancy,3 and in these instances it can be helpful to have a basic understanding of the underlying principles of pharmacology and teratogenesis.4,5,6,7 These are dealt with in the first two sections of this chapter. There then follows an overview of important and commonly used drugs in pregnancy.
PHARMACOLOGY: AN INTRODUCTION
Prescribing medications in pregnancy differs from prescribing in the non-pregnant patient, partly because the effects on both the mother and fetus need to be considered, and also because the physiological changes of pregnancy can affect the pharmacology of medications. Pharmacokinetics relates to the effect of the body on the drug, including how pregnancy may change drug absorption, distribution and metabolism. Pharmacodynamics, on the other hand, involves the effect of the drug on the body, including not only mode of action and pharmacological effect, but also the effect on the fetus including teratogenesis. Each of these will be considered in turn.
PHARMACOKINETICS: WHAT THE BODY DOES TO THE DRUG
This section drug includes:
- Absorption;
- Metabolism;
- Distribution;
- Clearance;
- Placental transfer;
- Excretion into breast milk.
Absorption
Drugs are administered by a variety of routes – oral, intravenous, intramuscular, etc. The vast majority of drugs are given orally, whereby a tablet starts to be broken down by chewing, oral and gastric enzymes, and the effects of gastric pH (acidic). The drug is absorbed across the highly permeable mucosa of the gastrointestinal tract into capillaries which drain into the hepatic portal vein. Although gastric pH increases in pregnancy, gastric emptying is delayed and intestinal motility is reduced, these physiological changes of pregnancy have no measurable effect on drug bioavailability.8
The effects of pregnancy on the absorption of drugs given by alternative routes are difficult to quantify. Drugs given intravenously enter the systemic circulation directly and therefore absorption is unchanged in pregnancy. Intramuscular preparations may theoretically be eluted from the muscle into the circulation more quickly as a result of the increased circulating volume and hyperdynamic circulation, whilst fat stores in pregnancy tend to increase which may in turn have an impact on lipid-soluble drugs given subcutaneously. However, it is unlikely that these effects are significant, and as such dose or route-of-administration adjustments are rarely needed in pregnancy as a consequence of altered absorption.
Metabolism
Once an orally administered drug has been absorbed into the hepatic portal vein it then passes directly to the liver, allowing the body an opportunity to neutralize toxins (not only from drugs, but also from poisons and spoilt foods, for example), before entering the systemic circulation.
Drug metabolism by the liver has two phases:
Phase I – oxidation, reduction or hydrolysis reactions which alter the drug, usually by exposing or adding a chemically active site;
Phase II – conjugation reactions which combine the drug with a more polar water-soluble compound in order to aid elimination.
Phase I metabolism reactions can convert an active drug to an inactive one (as is the case for the majority of medications), prolong the duration of action of a drug by converting an active drug to a different active metabolite (e.g., codeine is metabolized to morphine), or can convert an inactive drug (usually called a prodrug) to an active form (e.g., aciclovir is converted to its active metabolite aciclovir triphosphate).
The predominant phase I reaction is oxidation of the drug by ‘mixed function oxidase’ enzymes, a key example of which is the cytochrome P450 superfamily of enzymes. These enzymes are coded for by over 50 genes, so variations in genotype explain why different people may metabolize particular drugs to differing extents. Additionally, phase I enzymes may be expressed to varying degrees at different stages of pregnancy, with the consequence that gestational age can have a direct impact on drug metabolism. Significantly, the mixed function oxidases also form the basis of many drug interactions. These ‘mixed function’ enzymes are designed to protect the body from whatever toxic (or potentially toxic) compounds come their way – they are not specific for a particular drug. Exposure to a drug (or toxin) upregulates expression of the enzymes, and the greater the expression the quicker a drug is metabolized. Consequently, exposure to one drug can result in faster than expected metabolism of a different drug. Thus, long-term use of a drug which upregulates mixed function oxidase expression – an ‘enzyme inducer’ – can cause faster metabolism and, therefore, decreased efficacy of a different drug. Common examples of enzyme inducers are shown in Table 1.9 By way of contrast, enzyme inhibitors block the function of cytochrome P450 and other mixed function oxidases, giving rise to reduced metabolism of other drugs with an associated risk of toxicity, particularly if there is a narrow therapeutic window.
1
Common enzyme inducers and inhibitors.
Enzyme Inducers: ↑cytochrome P450 → ↑drug metabolism → ↓efficacy | Enzyme inhibitors: ↓cytochrome P450 → ↓drug metabolism → ↑toxicity |
Phenytoin | Grapefruit juice |
Carbamazepine | Erythromycin |
Rifampicin | Clarithromycin |
Ethanol | Ciprofloxacin |
Phase II reactions involve conjugation of the phase I metabolite with another compound which increases water solubility and therefore aids elimination, usually by the kidney. Drugs may be conjugated with:
- Glucuronate;
- Glutathione;
- Sulfate;
- Acetic acid.
The increased hepatic blood flow found in pregnancy and variable effects of genotype and gestational age on enzyme activity, along with the severity of the pathological process being treated, which is often dynamic in pregnancy, all mean that the dose of a drug often needs to be titrated against the clinical effect, rather than according to a specific drug level. Exceptions to this may be if the therapeutic window is narrow and the risks associated with toxicity are high (e.g., the risk of maternal and fetal nephro- and ototoxicity with gentamicin), if poor drug compliance is suspected because of a lack of clinical response (as occasionally seen with anti-epileptic drugs in pregnancy), or if overdose is suspected (e.g., in the case of respiratory arrest in a pre-eclamptic patient on magnesium sulfate when incorrect dosing may be suspected).4,8
Distribution
Distribution involves the movement – and sequestration – of the drug around the body. This depends on a number of factors, including:
- Capillary permeability;
- The concentration of the drug;
- The presence of membrane transporters, including active transporters which can move some drugs against their concentration gradient;
- The solubility of the drug – lipophilic (non-polarized) drugs cross the lipid bilayer of cell membranes far more more easily than water soluble (polarized) drugs;
- Binding of drugs to plasma proteins.
Additionally, water-soluble drugs tend to be distributed through compartments with a high water content such as plasma and amniotic fluid, whilst lipophilic drugs are sequestered throughout the body’s fat stores. This phenomenon is quantified in the ‘volume of distribution’ (Vd), which is defined as ‘the volume in which an amount of drug would need to be distributed to produce a particular concentration in the blood’, or put more simply is the amount of drug in the whole body compared with the amount in the blood. Thus, lipophilic drugs are mainly stored in tissues and, therefore, have a high Vd, whilst water-soluble or polarized drugs are often highly bound to carrier proteins in the blood, and therefore have a low Vd.
During pregnancy there is an increase in circulating blood volume which would be expected to dilute circulating drugs. However, to a certain extent the effect of this is offset by a decrease in the concentration of plasma binding proteins such as albumin. The reduction in binding proteins means that a greater proportion of any given drug is unbound (or free), i.e., in its active state, meaning that whilst the drug is less concentrated, a greater proportion is in its active form, the two effects offsetting one another.8
During pregnancy drugs are also distributed across the placenta. Chorionic villi on the fetal side of the circulation are surrounded by a single layer of trophoblastic cells, the walls of which are largely composed of a lipid bilayer, as shown in Figure 1. These chorionic villi are bathed in pools of maternal blood (intervillous spaces). Thus, the lipid bilayer of trophoblastic cell walls is the main barrier between maternal and fetal circulation within the placenta. As such, small (<500 Da) lipophilic drugs cross the placenta easily, whilst large polarized drugs are excluded. As the vast majority of drugs are small, most drugs cross the placenta to some extent. The exceptions to this are the ‘HIT’ trio, that is heparin (low molecular weight heparin has a molecular weight of 3000–5000 Da), insulin (molecular weight 5000–6000 Da) and tubocurarine (a skeletal muscle relaxant, molecular weight 600 Da).5 Some degree of active transport also takes place across the placenta (e.g., methotrexate is actively transported by the multidrug resistant proteins 1–3 (MDR 1–3), along with other transport processes such as phagocytosis and pinocytosis, although the clinical significance of this, if any, is not yet understood.7 It should also be noted that the placenta itself contains enzymes involved in metabolism. For example, only 10% of oral prednisolone reaches the fetus, as a result of placental metabolism.
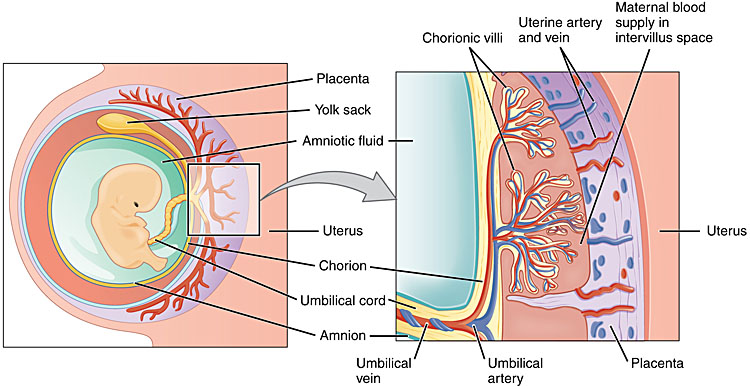
1
Placental structure. Taken from https://opentextbc.ca/anatomyandphysiology/chapter/28–2-embryonic-development/. Under license: https://creativecommons.org/licenses/by/4.0/legalcode.
Overall, the physiological changes of pregnancy tend to balance one another out in terms of drug distribution around the body; however, in the rare cases when monitoring of drug levels is required, it is vital to try and monitor the level of the free or active drug.5,8
Elimination
Elimination is clearance of the drug from the body. This is done mainly by the kidneys, and to a much lesser extent via bowel (drug which has not been absorbed by the gut and that which is excreted in bile) and lungs (e.g., volatile anesthetics). Additionally, in breastfeeding mothers some drug is lost in the breast milk.
Drugs arriving at the kidney in the afferent arterioles are first filtered through the glomerulus and Bowman’s capsule. Unbound drug easily passes from blood to renal tubular fluid, (unless the molecular weight is over 50,000 Da), so almost all drugs pass freely into the tubular fluid. Anion and cation transporters located in the convoluted tubules then actively transport acidic and basic drugs, respectively. Finally, some lipid-soluble drugs are reabsorbed by passive diffusion in the renal medulla.
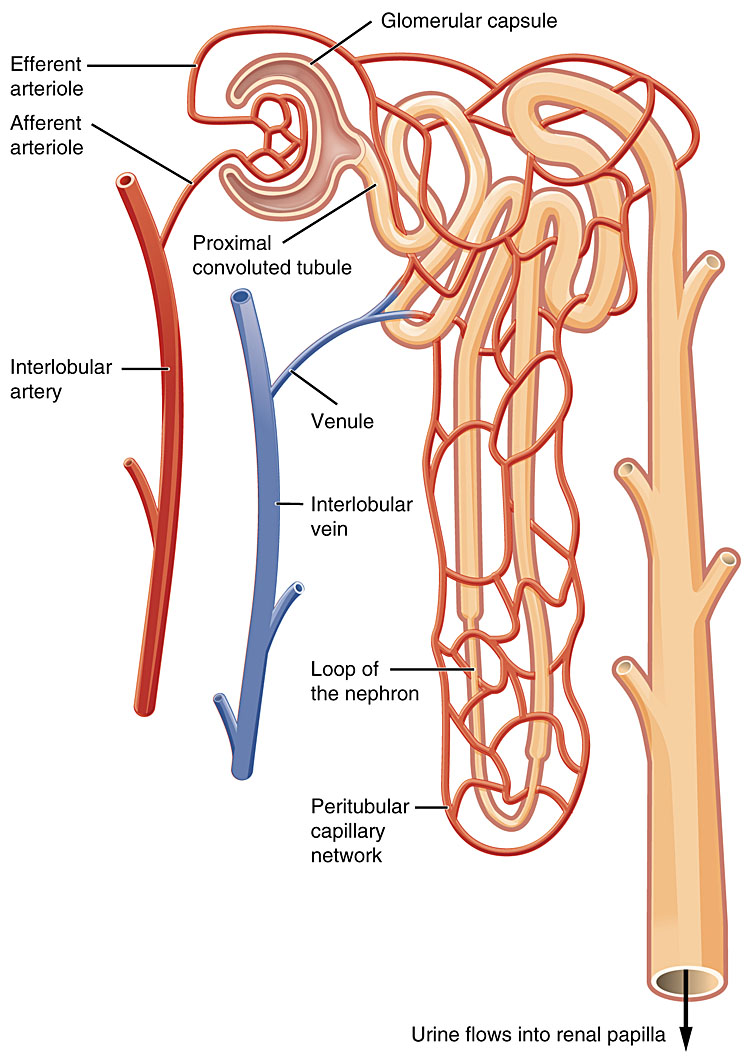
2
Structure of the nephron. Taken from https://opentextbc.ca/anatomyandphysiology/chapter/25–3-gross-anatomy-of-the-kidney/. Under license: https://creativecommons.org/licenses/by/4.0/legalcode.
During pregnancy the renal blood flow increases, giving rise to a 50% increase in GFR (glomerular filtration rate). This leads to increased clearance of renally excreted drugs such as low molecular weight heparin (LMWH) and β-lactam antibiotics such as penicillin. The increased renal excretion of antibiotics like cephalosporins can be helpful in the treatment of urinary tract infections, as more drug becomes concentrated within the urinary tract itself.
The transfer of drugs into breast milk is determined by the drug’s:
- Molecular size;
- Lipophilic properties and polarity (degree of ionization);
- Plasma protein binding.
With the exception of very highly protein-bound drugs, most drugs pass into the breast milk in measurable levels. However, the effect on the infant depends in turn on:
- Maternal plasma levels of the drug;
- Volume of milk ingested – the hindmilk is higher in fat and therefore contains a greater concentration of lipophilic drugs;
- Timing of feed in relation to timing of maternal dose;
- Infant metabolism, which in turn depends on the age of infant.
Whilst many drugs are thought to be safe when breastfeeding, others can cause direct neonatal toxicity (e.g., lithium), cause a risk of hypersensitivity in the infant (e.g., penicillin), may cause sedation which affects the infant’s ability to suckle (e.g., phenobarbitone) or may directly suppress lactation (e.g., cabergoline). It is important to conduct a risk–benefit analysis for that individual, and as always to use the lowest effective dose.
Generally speaking, the physiological changes of pregnancy mean that there is increased circulating volume, increased third spaces (amniotic fluid and peripheral edema), and increased fat stores and renal blood flow, which has the overall effect of reducing plasma levels of many drugs, which is to some degree offset by reduced levels of carrier proteins. Overall, however, when prescribing in pregnancy increased, rather than decreased, levels of some drugs may be required.8
PHARMACODYNAMICS: WHAT THE DRUG DOES TO THE BODY
Drugs have four main modes of action:4,5
- On receptors, e.g., β-blockers;
- On enzymes, e.g., angiotensin converting enzyme (ACE)-inhibitors;
- On membrane channels, e.g., calcium-channel blockers;
- On metabolic processes, e.g., antibiotics which interfere with protein synthesis.
The mode of action of a drug and choosing the right drug for the job is no different in pregnancy to outside of pregnancy, with the exception that the effects on the fetus also need to be considered. In particular, because of the fetal risks including teratogenesis, the risk–benefit equation may be different, and the pool of drugs to choose from may be smaller.
TERATOGENESIS AND FETAL TOXICITY
Teratogenesis is defined as “the process by which congenital malformations are produced in an embryo or fetus”. An example of this was the thalidomide scandal of the 1960s, when thousands of babies were born with phocomelia (shortened or absent limbs) as a result of thalidomide administration as a treatment for hyperemesis gravidarum, despite animal studies having shown no adverse effects of thalidomide.
Evidence for the safety of drugs in pregnancy and breastfeeding is often lacking, as pregnant women are often excluded from clinical trials (unless the drug is specifically targeted at pregnant women), and data come instead from animal studies, ‘off-license’ prescribing, and post-marketing surveillance such as the FDA’s MedWatch scheme in America and the MHRA’s Yellow Card scheme in the UK.3 Previously the FDA categorized the safety of all medications with an ‘A’ to ‘X’ classification in their ‘Pregnancy Risk Categories’. However, this was overly simplistic and in 2015 was replaced with a more descriptive system.10
The background rate of congenital anomalies is around 2%, and teratogenic effects of a drug can be unpredictable. Even with known teratogens such as warfarin, only a minority of infants exposed in utero suffer teratogenic effects.
2
Timing of drug exposure and fetal toxicity.
Timing of exposure | Potential fetal effects |
Pre-conception | Stored in maternal tissues → teratogenicity |
Pre-implantation | ‘All-or-nothing’ effect |
Embryonic | Teratogenicity |
Fetal | Impaired growth and neurodevelopment |
Peripartum | Idiosyncratic neonatal effects and withdrawal |
The timing of exposure to a particular drug is crucial to the teratogenic effect of a particular drug, as summarized in Table 2.5,7 Pre-conception exposure to some drugs can be teratogenic even if the drug is stopped by the time of conception. For example, acitretin, a retinoid used for the treatment of severe psoriasis, is heavily sequestered in body fat, from where it continues to be eluted even after the drug has been stopped. It is highly teratogenic, causing pregnancy loss and major structural and neurodevelopmental abnormalities. Women of reproductive age who are taking acitretin should continue to use effective contraception for 3 years after stopping the drug, and both men and women should not donate blood for 2 years after stopping it. Methotrexate is a folate antagonist which causes miscarriage and major structural abnormalities in the fetus. Both men and women should continue to use effective contraception for 3 months after stopping the drug, because of intracellular accumulation in women and because of effects on spermatogenesis in men.11
Up to 4 weeks from the last menstrual period (LMP) is known as the pre-implantation period, when the morula differentiates into a blastocyst which retains a totipotent inner cell mass. This then implants into the endometrium around 6 days post-fertilization. Exposure to teratogens at this pre-implantation stage tends to have an ‘all-or-nothing’ effect, with either a failure of implantation and miscarriage, or complete recovery as individually damaged cells are replaced by totipotent stem cells.
The ensuing embryonic period of development, up to 11 weeks' gestation, is the main period of organogenesis and thus greatest susceptibility to teratogens with the highest risk of malformation. Exposure to teratogens at different stages results in different anomalies, reflecting the period of maximum sensitivity for each organ system:
Pre-day 20: | limb defects | |
Day 20: | anencephaly | |
Day 34: | transposition of great vessels | |
Day 36: | cleft lip | |
Day 42: | ventriculo-septal defects and syndactyly |
During the fetal period (11 weeks' gestation to term) occasional structural defects can arise as the genitalia, palate and brain continue to form, but drug exposure during this time may more commonly affect fetal growth and neurodevelopment. Whilst fetal growth restriction can be monitored antenatally, neurodevelopmental problems are not manifest until later in the infant’s life, when there may be delay in meeting developmental milestones, attention deficit disorders and cognitive impairment (lower IQ). Proving an association between such neurodevelopmental outcomes and intrapartum teratogen exposure is much more difficult than for structural anomalies.
Drug use in the third trimester can also have intrapartum and short-term neonatal consequences. For example, use of anticoagulants near term carries a risk of placental and maternal hemorrhage, maternally administered systemic chloramphenicol causes neonatal ‘gray baby syndrome’, whilst the neonate may suffer withdrawal symptoms following long-term exposure to SSRIs (selective serotonin reuptake inhibitors) in utero.
Important and commonly used drugs in pregnancy
The following section contains information on the use of some important and commonly used drugs in pregnancy.4,5,6,8,9,11 The list is not exhaustive, and it should be remembered that the safety profile of drugs in pregnancy may change as new information comes to light. Further information can be obtained from:
- British National Formulary (BNF): www.bnf.org
- UK Teratology Information Service (UKTIS): www.uktis.org
- US Food and Drug Administration (FDA): www.fda.gov/drugs
Analgesics
Paracetamol is an analgesic, antipyretic and weak anti-inflammatory whose exact mode of action is not well understood. It is metabolized by cytochrome P450 to a toxic metabolite (NAPQI) which is normally conjugated with glutathione for renal elimination. However, if the elimination pathway is saturated (e.g., in paracetamol overdose) NAPQI accumulates leading to hepatocellular necrosis. However, provided the recommended dose is not exceeded (including weight adjustment if the patient is <50 kg), paracetamol is not known to have any adverse fetal effects in pregnancy or when breastfeeding.
Non-steroidal anti-inflammatory drugs (NSAIDs) such as ibuprofen are non-selective cyclooxygenase (COX) inhibitors. COX-2 is involved in the production of prostaglandins which mediate pain and inflammation, so inhibition of COX-2 is responsible for the therapeutic effects of NSAIDs. COX-1, meanwhile, dilates afferent renal arterioles and helps preserve integrity of the gastric mucosa, which accounts for the renal and gastrointestinal side-effects of NSAIDs. During pregnancy, NSAIDs readily cross the placenta but they are generally considered not to be teratogenic. However, although studies are somewhat conflicting, NSAIDs are widely avoided in pregnancy, particularly during the third trimester, as they have an effect on the fetal kidney giving rise to oligohydramnios, and may cause premature closure of the ductus arteriosus. When breastfeeding NSAIDs are generally considered to be safe as the amount of drug reaching the baby is too small to be harmful, but care should be taken to avoid NSAIDs in the immediate postnatal period in mothers with severe pre-eclampsia because of the risk of acute kidney injury.
Opioids stimulate μ-receptors in the central nervous system, which has the effect of reducing neuronal excitability and therefore the transmission of pain impulses, which, in turn, decreases the sympathetic stress response. Additionally, they cause an obtunded response to hypoxia and hypercapnia, leading to respiratory depression. Opioids cross the placenta by passive diffusion, and whilst studies on their use in pregnancy are limited, there are no known major teratogenic effects. One exception to this is tramadol (a synthetic codeine analog with additional serotonin and noradrenalin reuptake inhibition effects), which has shown embryotoxicity in animal studies and therefore carries a manufacturer’s warning to avoid tramadol during pregnancy. There is also a concern with all opioids that both long-term use and use around the time of delivery may cause neonatal respiratory depression along with neonatal withdrawal symptoms following delivery. Babies at risk should be monitored following delivery. Generally the amount of opioids found in breast milk is thought to be too small to be harmful, with the exception of codeine and fentanyl. Codeine is contraindicated in breastfeeding mothers as it is metabolized by the maternal liver to morphine, but different mothers differ in their metabolic capacity, putting the neonate at an unpredictable risk of morphine overdose. Fentanyl is a synthetic opioid 100 times more potent than morphine. If a mother requires fentanyl for chronic or severe pain, she should avoid breastfeeding during treatment and for between 1 and 5 days afterwards, depending on the route of administration.
Anti-asthmatics
Uncontrolled asthma in pregnancy is associated with a range of maternal and fetal complications, including gestational hypertension and pre-eclampsia, fetal growth restriction and preterm birth, increased cesarean section rate and perinatal mortality. By contrast, anti-asthmatic drugs are generally safe in pregnancy, and in women with well-controlled asthma there is little or no increased risk of adverse outcomes. The risks of uncontrolled asthma to both mother and fetus are far greater than the risk of medications.8,12 Not only should the full range for anti-asthmatic agents be used in pregnancy as required, but pregnancy should, in fact, be seen as an opportunity to optimize the patient’s long-term asthma management.
Short-acting β2-agonists such as salbutamol relax bronchial smooth muscle, increasing airflow to the lungs. Only a tiny amount of inhaled β2-agonist reaches the maternal systemic circulation, and there is good evidence that they are not associated with major congenital malformations or adverse perinatal outcomes. Although the data for long-acting β2-agonists such as salmeterol are slightly more limited, they also appear to be safe for use in pregnancy. As is the case outside of pregnancy, long-acting β2-agonists should always be used in combination with an inhaled corticosteroid (ideally as a combination product such as seretide) because of the risk of asthma-related deaths associated with isolated long-acting β2-agonist use.
Inhaled corticosteroids such as beclomethasone are delivered directly to the target organ where they modify gene transcription in the nucleus, upregulating the production of anti-inflammatory proteins and downregulating the production of pro-inflammatory proteins such as cytokines and interleukins. There is minimal systemic absorption and inhaled corticosteroids have been shown not to cause any increase in the rate of major congenital malformation or adverse perinatal outcomes.
Oral corticosteroids such as prednisolone tend to be used as short courses in acute exacerbations of asthma. As well as modifying the immune response to inflammation they also have a metabolic effect, promoting glucose production from muscle and fat stores, and a mineralocorticoid effect in which they stimulate sodium and water retention by the renal tubule, in exchange for potassium excretion. Data on oral steroid use in pregnancy shows that while they are generally safe there is a possible association with cleft lip and palate.8 However, much of these data are not specific to steroid use in asthma, and as such the pregnancies included in the studies may have had much longer-term exposure to steroids than the short courses usually used in asthma. If a mother with asthma is unwell enough to require oral steroids, then it is felt that the benefit far outweighs any potential risks. Prednisolone is usually the oral steroid of choice as it is actively metabolized by the placenta so only around 10% actually reaches the fetus. If the mother has received more than 7.5 mg prednisolone per day for 2 weeks or more prior to delivery then she should receive parenteral hydrocortisone cover in labor (100 mg every 6–8 hours).
There has been no significant association between theophyllines (such as aminophylline), leukotriene-receptor antagonsists (such as monetlukast), and sodium cromoglicate and major congenital abnormalities or adverse perinatal outcomes. However, theophylline levels should be monitored as theophylline is highly protein bound, so the fall in binding proteins seen in pregnancy leads to a higher proportion of free drug.
Prostaglandidn E2 (e.g., dinoprost) can be safely used for induction of labor in women with asthma, but prostaglandin F2α (e.g., carboprost) can cause bronchospasm and should therefore be avoided in the management of uterine atony.8
All anti-asthmatic agents are currently thought to be safe when breastfeeding, with only small amounts of the maternal dose being excreted into the breast milk.12
Antibiotics
Antibiotics are a large and varied group of drugs with a range of actions. They are among the most frequently prescribed medications in pregnancy.3 Typically antibiotics are only used for a short period of time, but it is worth knowing which antibiotics are thought to be safe, which have proven teratogenic effects, and which carry some risk but may be used if there is no safer alternative and the benefit outweighs the risk.5 The following overview is by no means exhaustive, and as with prescribing any medication in pregnancy, safety should be checked from a reliable source such as the British National Formulary (BNF).11 As always, the lowest effective dose should be used for as short a time as possible.
Antibiotics which are likely to be safe in pregnancy include penicillins and macrolides. Penicillins, including amoxicillin, benzylpenicillin and benzathine penicillin work by weakening the bacterial cell wall, which allows water to permeate and results in bacterial swelling and death. Penicillins do cross the placenta, which is exploited in the treatment of syphilis and for the prevention of early onset group B streptococcus. They are also considered safe in breastfeeding. Macrolides, such as erythromycin, are generally considered safe in pregnancy and breastfeeding, but it should be noted that the manufacturer’s information for clarithromycin and azithromycin recommend against their use unless the benefit outweighs the risk or there is no suitable alternative. It should be remembered that macrolides inhibit the cytochrome P450 enzymes, impairing the metabolism of anti-epileptics and other drugs metabolized by cytochrome P450.
Tetracyclines, such as doxycycline, should always be avoided in pregnancy. Tetracyclines have a bacteriostatic effect by inhibiting bacterial protein synthesis, which allows the host immune system time to kill and remove the bacteria. However, in pregnancy, they have been shown in animal studies to affect bone growth in the first trimester, and in humans in the second and third trimesters they cause permanent discoloration of the infant’s teeth. Furthermore, large parenteral doses also carry a risk of maternal hepatotoxicity. Tetracyclines are found in breast milk and in the UK it is recommended that they should be avoided whilst breastfeeding, whilst the American Academy of Pediatrics consider short courses of tetracyclines safe when breastfeeding as the calcium in maternal milk probably chelates the drug, thus preventing the tooth discoloration.13
Many other antibiotics require a careful risk–benefit analysis before use in pregnancy. In particular, aminoglycosides such as gentamicin and amikacin are extremely useful in the management of severe sepsis, particularly when the source of infection is unknown. However, they also carry a risk of both maternal and fetal ototoxicity (via damage to the vestibulocochlear nerve) and nephrotoxicity (via accumulation in renal tubular epithelial cells). Additionally, they can also interact with magnesium sulfate causing rapid onset respiratory arrest, so extra care should be taken in the management of severe pre-eclampsia. The therapeutic window is narrow, so the dose depends on weight (booking weight in pregnancy and ideal weight in obese patients) and on renal function. Drug levels always need to be checked if repeated doses are required. Thus in pregnancy the use of aminoglycosides is usually restricted to cases of severe sepsis, usually as a second-line or add-on agent, when the benefit is thought to clearly outweigh the risk.
Trimethoprim (and co-trimoxazole which is a combination of sulphamethoxazole and trimethoprim) should generally be avoided in pregnancy as it is a folate antagonist and therefore teratogenic in the first trimester. Co-trimoxazole carries an additional risk of neonatal hemolysis when used in the third trimester, and kernicterus in jaundiced and G6PD-deficient infants when used in breastfeeding. Short-term trimethoprim use is generally considered safe when breastfeeding.
Nitrofurantoin is metabolized by bacteria to an active metabolite which causes bacterial DNA damage, and reaches therapeutic concentrations in the urine because of renal excretion. It is contraindicated towards term because it carries a risk of neonatal hemolysis, but is generally safe when breastfeeding except for G6PD-deficient infants in whom there is a risk of hemolytic anemia.
There are limited data on the use of quinolones such as ciprofloxacin in pregnancy, but animal studies have shown an association with arthropathy, and there are usually safer alternatives available so their use in pregnancy tends to be avoided. Similarly, they should be avoided when breastfeeding, because of the potential risk of arthropathy in growing infants and a possible association with neonatal Clostridium difficile infection.
In general, systemic chloramphenicol use is reserved for life-threatening infections such as Haemophilius influenzae and typhoid fever. When possible its use should be avoided in late pregnancy because of the potential risk of ‘gray baby syndrome’ in which the neonatal liver is unable to adequately conjugate the drug for excretion, resulting in vomiting, poor-feeding, loose stools and ashen-gray skin. The manufacturer also advises avoiding its use in breastfeeding. There are no known data regarding the use of topical chloramphenicol for eye infections.
Anticoagulants
Heparin inhibits thrombin and factor Xa, which are key components of the final common coagulation cascade (see Figure 3), thus impairing clot formation. Low molecular weight heparin (LMWH) preferentially inhibits factor Xa, and is usually preferred as it has a more predictable effect.8

3
Mechanism of blood clotting. Taken from https://opentextbc.ca/anatomyandphysiology/chapter/18–5-hemostasis/. Under license: https://creativecommons.org/licenses/by/4.0/legalcode.
As heparin is a large molecule (3000–5000 Da) it does not cross the placenta and is therefore safe for use in pregnancy and breastfeeding. If it is used therapeutically in pregnancy (rather than prophylactically), then monitoring of anti-Xa levels may be required, under the guidance of a hematologist. Additionally, platelet levels should also be monitored with prolonged use because of the risk of heparin-induced thrombocytopenia (HIT).
Warfarin interferes with the vitamin K activation of clotting factors II, VII, IX and X, and is metabolized by cytochrome P450. It crosses the placenta and is teratogenic in the first trimester, particularly around 6–10 weeks' gestation. However, effects can be unpredictable, with around 10% of exposed pregnancies being affected. Potential risks in the first trimester include calcification (‘stippling’) of the fetal bone epiphyses and nasal and limb hypoplasia, which results in short limbs and digits. Use later in pregnancy has been associated with eye anomalies and central nervous system defects, along with bleeding risks to the fetus, placenta or mother. Intracranial bleeding may cause fetal mental retardation, placental bleeding may cause growth restriction, preterm delivery and stillbirth, whilst the mother is at risk of major obstetric hemorrhage. Broadly speaking warfarin is regarded as contraindicated in pregnancy, although this does depend on the indication for warfarin. All women of reproductive age should be aware of the risks of warfarin in pregnancy, and as far as possible women on warfarin should have preconception counseling to plan pregnancy and anticoagulation. For some women it may be appropriate to use only LMWH, whilst for others it may be appropriate to conceive on warfarin then use LMWH between 6 and 12 weeks before reverting to warfarin. However, for women with metal heart valves LMWH does not sufficiently anticoagulate, so generally it is advised to continue warfarin throughout the pregnancy. The woman herself must be involved in all of decision-making, along with specialist advice from joint obstetric and maternal physicians. This specialist advice should also include a comprehensive delivery plan.8
There are no adequate data regarding the use of direct oral anticoagulants (DOACs) in pregnancy. Rivaroxaban and apixaban both directly inhibit factor Xa, whilst dapigatran directly inhibits thrombin. Animal studies show that these compounds cross the placenta, and so far data regarding their teratogenic potential use in humans is largely complicated by confounding factors. As such, the International Society on Thrombosis and Haemostasis currently recommends against pregnancy in women taking DOACs, and if a woman does become pregnant whilst taking a DOAC, then this should be discontinued and changed to LMWH at the first opportunity. In addition to the potential teratogenic risk, DOACs are also presumed to carry ongoing risks throughout the pregnancy of fetal intracranial bleeding, placental bleeding and maternal hemorrhage.14
Antidiabetic agents
Metformin is an oral antidiabetic agent which increases tissue sensitivity to insulin (and therefore only works if there are some residual functioning pancreatic islet cells) and reduces gluconeogenesis. It is excreted unchanged by the kidney, and although its use in pregnancy is off-license in the UK, there is strong evidence for its safety. Accordingly, NICE (the UK’s National Institute for Health and Care Excellence) has stated that the likely benefits from improved blood glucose control outweigh the potential for harm.15 Metformin is also safe when breastfeeding for women with pre-existing diabetes. (Patients with gestational diabetes should stop their anti-diabetic agents after delivery.)
Insulin stimulates glucose uptake into tissues from the circulation, stimulates glycogen, lipid and protein synthesis, and drives potassium ions into cells in the same manner as endogenously produced insulin. Its use in pregnancy (and breastfeeding) is safe, although dose requirements are likely to alter during pregnancy, sometimes falling in the first trimester and usually increasing in the second and third trimesters. Dose requirements need frequent reviews by an experienced diabetologist throughout pregnancy. Dose adjustments are also required in the context of intercurrent illness and antenatal corticosteroid administration. Furthermore, women taking insulin must also be warned about the risks of hypoglycemia and decreased hypo-awareness, particularly in the first trimester and when breastfeeding.
Sulphonylureas are oral hypoglycaemic agents which stimulate endogenous insulin release from pancreatic islet cells. Gliclazide crosses the placenta, and carries a possibility of neonatal hypoglycemia and should therefore be avoided in pregnancy and breastfeeding. Glibenclamide does not cross the placenta, and is safe when breastfeeding, but NICE recommends that all oral hypoglycemic agents other than metformin should ideally be stopped preconception as there is a paucity of data regarding use in the first trimester, but that glibenclamide could be considered in women with gestational diabetes who either cannot tolerate metformin or who cannot achieve adequate blood sugar control with metformin but who decline insulin.15
Pioglitazone, a thiazolidinedione, is an insulin sensitizer which is contraindicated in pregnancy and breastfeeding because of antenatal toxicity in animal studies. Its presence in breast milk has also been demonstrated in animal studies.15
Antidepressants and antipsychotics
Selective serotonin re-uptake inhibitors (SSRIs), such as paroxetine, fluoxetine, sertraline and citalopram, work by inhibiting the re-uptake of serotonin by pre-synaptic cells, increasing the amount of serotonin available post-synaptically. They are often used as first-line agents in the management of depression because of their relatively good safety profile, and are also used for panic disorder and obsessive compulsive disorder. General advice regarding the use of SSRIs in pregnancy and breastfeeding is that they should only be used if the benefit outweighs the risk.8,16 Studies on their safety in pregnancy have shown conflicting results, but there is increasing evidence of an association between both paroxetine and fluoxetine use, and fetal cardiac malformations. There are currently insufficient data to draw any clear conclusions as to whether other SSRIs – or indeed SNRIs (serotonin and noradrenaline re-uptake inhibitors, such as venlafaxine) – pose similar risks. Concerns about the risk of persistent pulmonary hypertension of the newborn have been raised in relation to SSRI use beyond 20 weeks, although more data are needed to clarify this rare but potentially serious association. SSRI use towards term also carries a risk of neonatal withdrawal, and neonates who have been exposed in utero should be monitored for feeding problems, irritability, respiratory distress, hypoglycemia and other signs of withdrawal. All of these potential risks should be balanced against the importance of maternal mental health well-being during pregnancy and the postnatal period, and the possibility of relapse when stopping the drug in a patient who is otherwise stable should also be considered, discussed with the woman, and kept under review.
Tricyclic antidepressants, such as amitriptyline, work by inhibiting reuptake of serotonin and noradrenaline, and are usually used for depression, anxiety and neuropathic pain. There is a long history of use in pregnancy and there is no clear evidence of major congenital anomaly. Use towards term should prompt neonatal observation for withdrawal effects, as with SSRIs.
Benzodiazepines such as diazepam enhance the activity of the inhibitory neurotransmitter GABA (gamma aminobutyric acid), and are used in the management of anxiety and status epilepticus. They were previously thought to carry a risk of orofacial and cardiac malformations, although more recent studies have not confirmed this association. Benzodiazepines can cause dependency and if a plan is made to stop the drug then this should be done very gradually. New prescriptions should be at the lowest effective dose for the shortest period of time. Neonates exposed near term are at risk of withdrawal and should be monitored for respiratory depression. Benzodiazepines should not be withheld in status epilepticus.
Antipsychotics are typically divided into first generation or ‘typical’ antipsychotics (such as haloperidol, chlorpromazine) and second generation or ‘atypical’ antipsychotics (such as quetiapine and olanzapine). They are used in the management of schizophrenia, bipolar disorder (particularly during episodes of mania) and for acute psychomotor agitation. Data on their use in early pregnancy are limited and often complicated by confounding factors. Use in the third trimester should prompt neonatal monitoring for extrapyramidal effects and withdrawal symptoms such as agitation, altered muscle tone, tremor, drowsiness, difficulty in feeding and respiratory distress, and delivery should be planned in a unit which is able to monitor and treat these effects. There is some evidence from animal studies to suggest that antipsychotics may adversely affect the developing neonatal nervous system, and as such it is generally not advised to breastfeed if maternal antipsychotics are required.
Lithium is a mood stabilizer used in the manic phase of bipolar disorder, whose mode of action is not fully understood but appears to influence nerve excitation and synaptic transmission. Lithium exposure in pregnancy may have a possible association with Ebstein’s cardiac anomaly, although the absolute risk (if any) is thought to be low. More significantly, lithium has a narrow therapeutic window, with risk of maternal undertreatment if levels are too low and both maternal and fetal toxicity if levels are too high. NICE recommends that lithium levels are monitored every 4 weeks until 36 weeks, then weekly until delivery, then rechecked intrapartum. Additionally, the neonate is at risk of lithium toxicity, so neonatal lithium levels should also be checked shortly after delivery. It is also important to ensure the woman knows lithium levels may be high in breast milk, with a risk of toxicity for the baby.16
Antiepileptic drugs
Antiepileptic drugs (AEDs) are used in 1 in every 200 pregnancies in the UK and are associated with significant teratogenic effects.17,18 First trimester use carries a risk of major and minor structural abnormalities:
Major abnormalities | Minor abnormalities |
Neural tube defects | Dysmorphic features |
Congenital heart defects | Hypoplastic digits and nails |
Orofacial defects |
The risk of teratogenicity increases with the number of AEDs used, and is particularly associated with valproate use. Although estimates vary widely it is thought that with a single AED the rate of congenital anomaly is around 5%, but this rate rises with polytherapy. Women with epilepsy who are not on any AEDs have a similar risk of congenital malformations to the general population. Use of AEDs later in pregnancy is associated with a two-fold increase in growth restriction and the Royal College of Obstetricians and Gynaecologists (RCOG) advises that women with epilepsy taking AEDs should have growth scans from 28 weeks onwards.18
Despite the risks associated with AED use in pregnancy, most women with epilepsy will deliver a healthy baby. Furthermore, there are significant risks to the fetus from epilepsy itself, including the risk of hypoxia and trauma during a seizure, along with sudden unexpected death in epilepsy (SUDEP). AEDs should never be stopped abruptly and all pregnant women with epilepsy should be managed in a multidisciplinary setting with specialist neurology advice.17 Women with epilepsy should take folic acid 5 mg for several months prior to conception and throughout the pregnancy, and AEDs should ideally be used in monotherapy, at the lowest effective dose. Dosing should be evaluated clinically rather than by monitoring drug levels, with the exception of lamotrigine the levels of which can fall profoundly in pregnancy, or in situations where poor compliance is suspected. All pregnant women in the UK taking AEDs should be enrolled on the UK Epilepsy and Pregnancy Register (www.epilepsyandpregnancy.co.uk) and neonates should be given vitamin K after delivery. Mothers should usually be encouraged to breastfeed.
Valproate is contraindicated in women of reproductive age in all but very exceptional circumstances, as in addition to carrying a 10% risk of congenital malformation, valproate also has a 30–40% risk of neurodevelopmental disorders (walking and talking later, lower IQ, poor language skills and memory problems), along with a five-fold increase in autism compared with the general population. In 2018 the MHRA (Medicines and Healthcare products Regulatory Agency) launched ‘Prevent’, the Valproate Pregnancy Prevention Programme.19 This states that valproate is absolutely contraindicated in women of childbearing potential for conditions other than epilepsy, and only in epilepsy if there are no suitable alternatives, and if all of the conditions of the program are met. These include the need for highly effective contraception, annual specialist review and an agreement of risk undertaken jointly by the patient and specialist. Reasonable alternative first-line agents include lamotrigine or levetiracetam.
Carbamazepine is a potent hepatic enzyme inducer, which is typically used for focal seizures. It is considered to be one of the safest AEDs for use in pregnancy, carrying a risk of cardiac and facial defects between 2 and 5%. It is safe when breastfeeding.
Phenytoin is also a hepatic enzyme inducer, which is used for both generalized and focal epilepsy, along with the management of status epilepticus. It has a narrow therapeutic window and has significant maternal side-effects with long-term use including skin coarsening, acne and gum hypertrophy, making it a less common first-line agent in young women. Despite this it is relatively safe in pregnancy, although it carries a risk of facial defects (1–2%) and fetal hydantoin syndrome. It is safe when breastfeeding.
Lamotrigine is used for the management of both generalized and focal epilepsy, and is also considered to be amongst the safest AEDs in pregnancy, with a risk of cardiac and facial defects between 2 and 5%. Free drug levels may fall, particularly in the third trimester, so dose adjustment and monitoring of plasma levels may be required. It is safe when breastfeeding.
Levetiracetam is a relatively new drug for focal epilepsy, and appears to have a reasonable safety profile, with a 1–2% risk of cardiac and neural tube defects. However, being relatively new, data are somewhat limited, including in both pregnancy and breastfeeding.
Antihypertensives
Antihypertensives are among the most commonly prescribed medications in pregnancy, not only for pre-existing hypertension, but also for pregnancy-induced hypertension and pre-eclampsia. NICE guidance states that the aim of treatment for women with uncomplicated chronic hypertension is to keep blood pressure below 150/100 mmHg, and below 140/90 mmHg for patients with target organ damage secondary to chronic hypertension.20 The aim of treatment is reduced the risk of cerebral hemorrhage, but care should be taken not to over treat blood pressure as this can reduce placental (and therefore fetal) perfusion.
NICE recommends labetalol, a combined α- and β-blocker as a first-line antihypertensive agent in pregnancy. It is generally considered to be safe in pregnancy, although there remains an as yet unresolved concern about fetal growth restriction. Labetalol can be given orally for chronic hypertension and intravenously in hypertensive emergencies, but should be avoided in patients with asthma. In general, pure β-blockers (such as atenolol) are avoided in pregnancy as data are limited, and there is a possible association with growth restriction and preterm delivery.
Methyldopa is an α-blocker prodrug which is metabolized to norepinephrine. It is recommended by NICE as an alternative first-line agent to labetalol. Clinical trial data on its use in pregnancy are limited, but it has a long history of use in pregnancy without any known adverse fetal outcomes. Methyldopa can cause reduced variability on the cardiotocograph, and can cause maternal depression and deranged liver function tests, along with hemolytic anemia.
Modified release nifedipine is a calcium-channel blocker, also recommended by NICE as an alternative to labetalol. Whilst it is not licensed for use in pregnancy, it is often used to treat hypertension in pregnancy and as a tocolytic in the prevention of preterm labor. It has maternal side-effects of headache, flushing and tachycardia. It should not be given sublingually as this provokes a rapid fall in blood pressure which can result in placental hypoperfusion. The manufacturer advises avoiding the use of nifedipine before 20 weeks of pregnancy, although this should be weighed up against the risk of uncontrolled hypertension.
Hydralazine causes direct relaxation of arteriolar smooth muscle, leading to a selective decrease in vascular resistance in the cerebral, coronary and renal circulation. When given intravenously for hypertensive emergencies, it is thought that placental vessels remain relatively unaffected. It is metabolized by acetylation in the liver, which can give rise to a lupus-like syndrome in women who are slow acetylators.
Angiotensin converting enzyme (ACE) inhibitors, such as ramipril, are contraindicated in pregnancy as they are teratogenic and fetotoxic. Use in the first trimester is associated with cardiovascular and neurological malformations, whilst use later in pregnancy has been associated with fetal renal failure and oligohydramnios, fetal hypotension and stillbirth. If a patient normally takes an ACE-inhibitor pre-pregnancy this should be changed to a safer alternative before conceiving. There is a paucity of data regarding the use of angiotensin II receptor blockers, such losartan, in pregnancy. However, as they are very similar to ACE-inhibitors it is generally recommended to avoid them.
Diuretics such as furosemide are not normally used for the treatment of hypertension in pregnancy as they can further deplete the intravascular volume which is already compromised in pre-eclampsia. However, it is reasonable to use them for other indications such as pulmonary edema, if required.8
PRACTICE RECOMMENDATIONS
- Prescribing any medication in pregnancy should prompt a careful discussion with the woman about the risks and benefits of the medication, of any alternatives, and of the possible risks of not taking the medication.
- When medication is required in pregnancy the lowest effective dose for the shortest possible time should be chosen.
- Increased circulating volume and increased renal clearance means that circulating drug concentrations may fall during pregnancy, so some medications may require an increase in dose.
- Consideration should be given to the first trimester as the period of highest sensitivity to potential teratogens, as this is the key period of organogenesis.
- Whilst many medications do carry risks of teratogenesis and fetal toxicity, these should be balanced against the benefits of good control of chronic conditions such as asthma, epilepsy, diabetes and mental health problems.
- Pregnant patients with chronic medical conditions should be cared for in joint medical and obstetric multidisciplinary clinics.
CONFLICTS OF INTEREST
The author(s) of this chapter declare that they have no interests that conflict with the contents of the chapter.
Feedback
Publishers’ note: We are constantly trying to update and enhance chapters in this Series. So if you have any constructive comments about this chapter please provide them to us by selecting the "Your Feedback" link in the left-hand column.
REFERENCES
Making Childbirth a Positive Experience: World Health Organisation, Feb 2018. https://www.who.int/reproductivehealth/intrapartum-care/en/. | |
Hardy J, Leaderer B, Holford T, et al. Safety of medications prescribed before and during early pregnancy in a cohort of 81,975 mothers from the UK General Practice Research Database. Pharmacoepidemiol Drug Saf 2006;15:555-64. | |
Chan M, Wong I, Sutcliffe A. Prescription drug use in pregnancy: more evidence of safety is needed. The Obstetrician and Gynaecologist 2012;14:87–92. | |
Brown M, Sharma P, Mir F, et al. Clinical Pharmacology. London: Elsevier. | |
Hayes K. Pharmacokinetics, pharmacodynamics and teratogenesis. In: Fiander A, Thilaganathan B (eds.) Your Essential Revision Guide: MRCOG Part One: The Official Companion to the Royal College of Obstetricians and Gynaecologists Revision Course, 2nd edn. England: Cambridge University Press, 393–403. | |
Hayes K. Non-hormonal therapy in obstetrics and gynaecology. In: Fiander A, Thilaganathan B (eds.) Your Essential Revision Guide: MRCOG Part One: The Official Companion to the Royal College of Obstetricians and Gynaecologists Revision Course, 2nd edn. England: Cambridge University Press, 405–11. | |
Tower C. Medication in pregnancy. In: Luesley D, Baker P, Hodder A (eds.) Obstetrics and Gynaecology: An evidence-based text for MRCOG, 2nd edn. England, 192–7. | |
Nelson-Piercy C. Handbook of Obstetric Medicine, 5th edn. Florida: CRC Press. | |
Hitchings A, Lonsdale D, Burrage D, et al. The Top 100 Drugs: Clinical Pharmacology and Practical Prescribing. London: Churchill Livingstone Elsevier. | |
FDA Pregnancy and Lactation Labelling Final Rule https://www.fda.gov/vaccines-blood-biologics/biologics-rules/pregnancy-and-lactation-labeling-final-rule. | |
British National Formulary www.BNF.org. | |
British guideline on the management of asthma: A national clinical guideline. British Thoracic Society and Scottish Intercollegiate Guidelines Network, 2014. | |
LactMed Drugs and Lactation Database: Tetracycline https://www.ncbi.nlm.nih.gov/books/NBK501108/. | |
Cohen H, Arachchillage D, Middledorp S, et al. Management of direct oral anticoagulants in women of childbearing potential: guidance from the SSC of the ISTH. Journal of Thrombosis and Haemostasis 2016;14:1673–6. | |
NICE NG3. Diabetes in Pregnancy: management from preconception to the postnatal period. National Institute for Health and Care Excellence, 2015. | |
NICE CG192. Antenatal and postnatal mental health: clinical management and service guidance. National Institute for Health and Care Excellence, 2014. | |
Bhatia M, Adcock J, Mackillop L. The management of pregnant women with epilepsy: a multidisciplinary collaborative approach to care. The Obstetrician and Gynaecologist 2017;19:279–88. | |
RCOG Green-top Guideline No. 68: Epilepsy in Pregnancy, 2016. | |
Medicines and Healthcare Products Regulatory Agency. Valproate medicines (Epilim, Depakote): Pregnancy Prevention Programme materials online. May 2018. https://www.gov.uk/drug-safety-update/valproate-medicines-epilim-depakote-pregnancy-prevention-progamme-materials-online. | |
NICE CG107. Hypertension in Pregnancy: diagnosis and management. National Institute for Health and Care Excellence, 2010. |
Online Study Assessment Option
All readers who are qualified doctors or allied medical professionals can automatically receive 2 Continuing Professional Development points plus a Study Completion Certificate from GLOWM for successfully answering four multiple-choice questions (randomly selected) based on the study of this chapter. Medical students can receive the Study Completion Certificate only.
(To find out more about the Continuing Professional Development awards program CLICK HERE)