This chapter should be cited as follows:
Lust K, Tellam J, Glob Libr Women's Med
ISSN: 1756-2228; DOI 10.3843/GLOWM.414493
The Continuous Textbook of Women’s Medicine Series – Obstetrics Module
Volume 2
Maternal health and disorders in pregnancy
Volume Editor: Dr Kenneth K Chen, Alpert Medical School of Brown University, USA

Chapter
Adrenal Disorders in Pregnancy
First published: February 2021
Study Assessment Option
By answering four multiple-choice questions (randomly selected) after studying this chapter, readers can qualify for Continuing Professional Development points plus a Study Completion Certificate from GLOWM.
See end of chapter for details.

PHYSIOLOGY OF ADRENAL GLAND FUNCTION IN PREGNANCY
There are two major components to the adrenal glands: the cortex and medulla. The adrenal cortex consists of the outermost zona glomerulosa (ZG) which produces mineralocorticoids (aldosterone), the middle zona fasciculata (ZF) which produces glucocorticoids (cortisol) and the zona reticularis which produces androgens (dehydroepiandrosterone, DHEA, and the sulfated version DHEA-S).1,2 The adrenal medulla produces adrenaline and noradrenaline.
Cortisol
Cortisol is essential for maintaining blood pressure, electrolyte physiology and glycemic control. Ten to fifteen per cent circulates freely in non-pregnant women, with the remainder bound to cortisol binding globulin (CBG) and albumin.3
Both free and bound cortisol are elevated throughout pregnancy and spike at delivery. By the third trimester, cortisol levels have increased by 2–3 fold. Diurnal variation is preserved, characterized by high levels of cortisol in the morning and low levels at night.4
Multiple mechanisms are involved (Figure 1):5
- The placenta is a secondary site for production of adrenocorticotropic hormone (ACTH), corticotropin releasing hormone (CRH) and cortisol.
- Usual negative feedback loops are altered. Unlike hypothalamic CRH, placental CRH increases with cortisol (positive feedback loop) leading to further increases in both CRH and cortisol.
- Raised estrogen levels in pregnancy increase corticosteroid binding globulin (CBG). This causes an increase in total cortisol and a reduction in cortisol clearance. The half-life of cortisol is doubled.
- Raised progesterone levels in pregnancy displace cortisol from CBG, further elevating free cortisol.
- Fetal hypothalamic-pituitary-adrenal (HPA) axis activity begins at approximately 20 weeks' gestation.6 Cortisol can cross the placenta bi-directionally (mother to fetus, and fetus to mother) and is regulated by placental 11-βhydroxysteroid dehydrogenase type 2 (11-βHSD type 2).3,4 Excess maternal steroid exposure has been linked to transient fetal HPA axis suppression, low birth weight, metabolic dysfunction and behavioral alterations in offspring.6
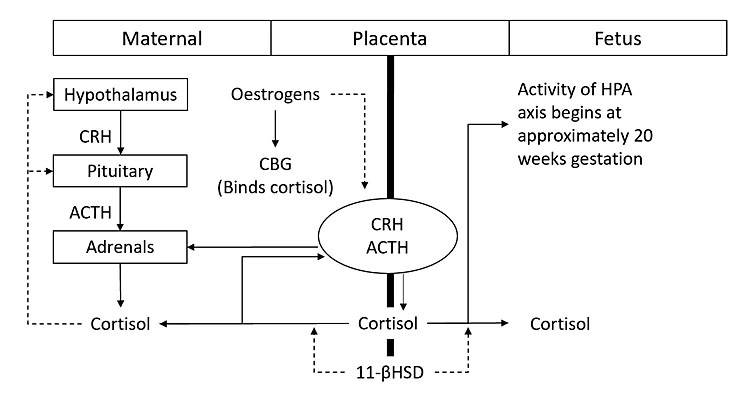
1
Hypothalamic-pituitary-adrenal (HPA) axis in pregnancy. Solid arrows indicate positive feedback and dashed arrows indicate negative feedback. CRH, corticotropin releasing hormone; ACTH, adrenocorticotropic hormone; CBG, corticosteroid binding globulin.5
Renin-angiotensin-aldosterone system (RAAS)
Aldosterone controls salt and water balance in the kidney. Its net effect is sodium retention with resultant water reabsorption to maintain blood pressure. It has a complex regulatory system detailed in Figure 2.
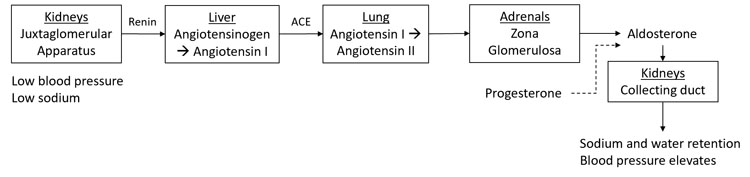
2
Renin-angiotensin-aldosterone system (RAAS) in pregnancy. Solid black arrows indicate positive feedback and dashed black arrows indicate negative feedback. ACE, angiotensin-converting enzyme.5
Upregulation of the RAAS system also occurs in pregnancy. Plasma renin activity (PRA) elevates in early pregnancy and reaches 3–7 fold by the third trimester. This is accompanied by receptor based refractoriness to the effects of angiotensin and hence blood pressure does not rise. Aldosterone follows a similar pattern, peaking at 38 weeks at 5–20 fold. There is ongoing aldosterone responsiveness to usual stimuli (for example posture, blood volume, dietary salt). Progesterone is an aldosterone antagonist and plays a role in regulation of aldosterone activity.5
Dehydroepiandrosterone/dehydroepiandrosterone sulfate (DHEA/DHEA-S)
There is increased androgen secretion by the adrenal cortex in pregnancy. Placental aromatase metabolizes these to estradiol and estrone.7 Although there is greater production of androgens, overall the levels decrease due to the aromatization process.
Adrenaline/noradrenaline
Adrenaline and noradrenaline are essential for maintaining homeostasis in multiple systems, particularly the cardiovascular system. Metabolites known as metanephrines are measured to assess for states of adrenaline/noradrenaline excess and are unchanged in pregnancy.8
ADRENAL INSUFFICIENCY
Adrenal insufficiency (AI) is relatively rare in pregnancy and the exact prevalence of AI in pregnancy is unknown. AI can be divided into primary, secondary or tertiary causes (Table 1).9,10 In primary adrenal insufficiency there is elevated adrenocorticotropic hormone (ACTH) and destruction of the adrenal cortical gland function leading to glucocorticoid, adrenocorticoid and mineralocorticoid deficiency. Secondary AI is due to lack of ACTH stimulation and tertiary is due to a lack of corticotropin releasing hormone (CRH) stimulation.
In developed countries autoimmune adrenalitis (Addison’s disease) is the most common cause of primary AI, whereas in developing countries tuberculosis destruction of the adrenal gland is more common. The prevalence of primary AI in developed countries is reported to be 96–140 per one million population.11,12 Exogenous corticosteroids for the treatment of other systemic diseases (>5 mg/day prednisone or equivalent doses of other corticosteroids for longer than 3 weeks in the past year) increases the risk of AI.10 A recent systematic review of glucocorticoid therapy and subsequent AI in adults demonstrated that AI was present at <0.5 mg prednisolone equivalent dose/day, <4 weeks of exposure, cumulative dose <0.5 g, and following tapered withdrawal of glucocorticoids. The authors advised that clinicians should be vigilant for AI with all degrees of glucocorticoid exposure.13 The ingestion of corticosteroids can lead to ACTH and CRH suppression resulting in adrenal atrophy.
AI was previously associated with increased maternal and fetal morbidity and mortality. Modern glucocorticoid and mineralocorticoid replacement therapy along with improvements in obstetric and neonatal care ensures that the majority of women can expect good maternal and fetal outcomes.9,10
Adrenal insufficiency type | Pathophysiology | Medical disorders |
Primary | Loss of adrenal cortical gland function |
|
Secondary | Dysfunction of pituitary gland with ACTH deficiency |
|
Tertiary | Dysfunction of hypothalamus with CRH deficiency |
|
*Combined secondary and tertiary AI.
Clinical features
AI is usually diagnosed prior to pregnancy; a recent review reporting only 17.7% of cases being diagnosed in pregnancy with equal distribution in all three trimesters.9 Symptoms of AI are nonspecific and can mimic normal physiological symptoms of pregnancy such as fatigue, low blood pressure, nausea, vomiting and linea nigra. AI should be considered in women presenting with excessive fatigue, weight loss, malaise, vomiting, dizziness, hyperpigmentation, abdominal pain, salt craving and or electrolyte disturbance. Symptoms may worsen across the day. Pigmentation from Addison’s involves nonexposed parts of the skin, creases of hands, extensor surfaces, mucous membranes and in scars. Laboratory findings in primary AI may include hyponatremia, hyperkalemia, mild anemia and metabolic acidosis. Hypoglycemia and hypercalcemia may occur in an adrenal crisis. A prior personal or family history of autoimmune disease should raise the clinical suspicion of AI in a woman presenting with symptoms.9,10 Adrenal crises can be associated with hyperemesis gravidarum, infections, surgery and delivery.
Diagnosis
- If AI is suspected, a screening test involves paired early morning ACTH and cortisol. The diagnosis of primary AI is highly likely if the if the cortisol is (<140 nmol/L (5 μg/dL) in combination with an ACTH level more than twofold the upper limit of the reference range.
- The corticotropin stimulation test is the “gold standard” for diagnosis of primary AI. Studies in normal healthy pregnant women have demonstrated higher peak total cortisol responses postcorticotropin stimulation test with 250 μg intravenous corticotropin in the second and third trimester compared to the non-pregnant state.14 Therefore, trimester specific cortisol cut-offs postcorticotropin stimulation of 700 nmol/L (25 μg/dL), 800 nmol/L (29 μg/dL) and 900 nmol/L (32 μg/dL) for the first, second and third trimesters, respectively, returning to the usual cut off postpartum of 500 nmol/L (18 μg/dL) have been suggested.1,2,7,9,10,15 Corticotropin is considered safe for use in pregnancy.16
- Antibody testing should also be undertaken if Addison’s disease is suspected as 21-hydroxylase antibodies may be positive in >90% of patients and 17 hydroxylase antibodies in 30% of patients.12,17
- Ongoing studies continue into the utility of salivary cortisol as an additional investigative tool in AI. At present its use is not established.
- If secondary or tertiary adrenal insufficiency due to a structural lesion is considered; an MRI without contrast is the imaging modality of choice and can be performed safely in pregnancy.
Effects on pregnancy
AI is associated with reduced fertility which is likely contributed to by coexisting medical disorders. A recent review in the UK of maternal and neonatal outcomes of women with Addison’s disease compared to control women over a 9 year period revealed a statistically significant increase in preterm premature rupture of the membranes, premature delivery, cesarean section, wound complications, postpartum infection, transfusion, length of hospital stay greater than 3 days, maternal death and venous thromboembolism in the women with Addison’s disease.12
Effects on fetus
The study by Schneiderman et al. also revealed that impaired fetal growth and congenital anomalies were more common in women with Addison’s disease compared to controls.12
Management
Women with pre-existing AI should undergo preconception planning to optimize therapy and to plan alterations to therapy during pregnancy, to screen for comorbidities, such as other autoimmune or pituitary conditions, and infertility as well as standard pre-pregnancy assessment to optimize conception success and pregnancy outcomes.
Women with AI in pregnancy should be seen at a minimum every trimester by a physician with experience and expertise in this area to ensure optimal outcomes from mother and baby. They should be monitored for clinical symptoms and signs of under or over glucocorticoid replacement. Over replacement in pregnancy may be associated with maternal edema, hypertension, pre-eclampsia, gestational diabetes mellitus and fetal adverse effects including low birth weight, premature delivery, long term effects on the fetal HPA axis and possibly increased risk cardio-metabolic disease in offspring.9,10
In pregnancy, hydrocortisone is the glucocorticoid of choice for replacement therapy as it is an active glucocorticoid, provides physiological cortisol replacement in divided doses and is metabolized by placental 11-βhydroxyl steroid dehydrogenase. The usual dose of hydrocortisone (15–25 mg/day) should be increased by 20–40% from 24 weeks gestation to mimic the physiological increase in free cortisol.9,10,13 Hydrocortisone should be given in two or three divided doses with the highest dose being given in the morning on wakening. In cases of moderate stress, the oral dosage of hydrocortisone should be doubled until recovery (usually 2–3 days).9,10,15 Patients with chronic AI should be advised to be strictly compliant with their medications, particularly when unwell. If they are unable to tolerate oral corticosteroids, they should have injectable corticosteroids available and be able to administer these, such as hydrocortisone 100 mg subcutaneously or intramuscularly.15 Women with AI should have identification on them and a medical alert stating their diagnosis and medication.
Adrenal crisis is an emergency and if suspected should be treated with an immediate parental injection of 100 mg hydrocortisone, appropriate fluid resuscitation and 200 mg hydrocortisone per 24 hours either by continuous infusion or 6 hourly bolus injection.15 An investigation for the cause of the adrenal crisis should be undertaken and appropriate management instituted.
Delivery requires an increase in glucocorticoid replacement which is usually given intravenously. Stjernholm et al. showed that serum cortisol increases 1.5 times in vaginal delivery compared to an elective cesarean section. There was no correlation between the type of analgesia and cortisol level.18 It is recommended hydrocortisone 100 mg intravenously be administered at the onset of active labor followed by a continuous infusion of hydrocortisone 200 mg/24 hours or bolus dose of 50 mg every 6 hours.9,10,15 A recent study by Owa et al. in women taking systemic corticosteroids for autoimmune conditions has demonstrated that low dose corticosteroid supplementation during labor and delivery with 50 mg intravenous hydrocortisone at the onset of labor and every 8 hours until delivery followed by 25 mg every 8 hours for 24 hours was not associated with any differences in maternal or fetal outcomes and could be considered standard of care. They demonstrated a trend towards less complications associated with high dose glucocorticoids.19
Women with primary AI may require an increase in the usual dose of fludrocortisone (usual nonpregnancy dose range 0.05–0.2 mg/d) in the third trimester due to the antimineralocorticoid effect of rising progesterone. Some studies have documented sixfold increases in the dose of fludrocortisone in pregnancy. The dose of fludrocortisone is adjusted based on the presence of postural hypotension or elevated potassium. The dose may need to be reduced in pregnancy in the presence of hypertension or hypokalemia.9,10,15,20 Postpartum the dose should be reduced promptly back to the pre-pregnancy dose as progesterone falls quickly after delivery of the placenta.9,10,15,20
CUSHING’S SYNDROME
Syndromes of pathologic cortisol excess are divided into ACTH dependent and ACTH independent. Causes of ACTH dependent Cushing’s include pituitary adenoma, exogenous ACTH administration and ectopic ACTH. Causes of ACTH independent Cushing’s include exogenous glucocorticoid administration, adrenal adenoma and adrenal carcinoma. The term Cushing’s disease is reserved for those with a pituitary adenoma alone, with the remaining conditions known as Cushing’s syndrome.
In pregnancy, the most common cause of cortisol excess is adrenal in origin found in 50–60% of cases.5 This contrasts with outside pregnancy where cortisol excess is most commonly secondary to a pituitary adenoma (i.e. Cushing’s disease, 70% of cases). Fertility in syndromes of cortisol excess is affected due to cortisol and androgen suppression of gonadotrophs.21 Systemic estrogen and progesterone contraception should be avoided due to an increased risk of thrombosis.21
Clinical features
Differentiating physiologic hypercortisolemia in pregnancy from pathological cortisol excess can be challenging due to similar clinical features. The best discriminating symptom is proximal myopathy as this is not a pregnancy-related symptom.1 Other features seen in Cushing’s syndrome include menstrual irregularities/amenorrhea, weight gain, truncal adiposity, easy bruising, purple abdominal striae (particularly abdominal, axillary and hips), moon facies, buffalo hump, thin skin, hirsutism, behavioral changes, minimal trauma fracture, hypertension, diabetes and thromboembolism.1,5
Diagnosis
Signs and/or symptoms of hypercortisolemia require investigation for pathologic hypercortisolemia (Figure 3):2,3,21,22
- Midnight salivary cortisol. Diurnal variation is preserved in pregnancy, therefore, this is a helpful screening test. Suspect Cushing’s if midnight salivary cortisol is consistently greater than 9 nmol/L in first trimester (0.25 μg/dL), 7.2 nmol/L in second trimester (0.26 μg/dL) and 9.1 nmol/L in third trimester (0.33 μg/dL) on at least two separate occasions.
- Urinary free cortisol. Pregnancy specific range is known and is up to 3-fold non-pregnant values, therefore this test is useful as a screening test in pregnancy. Suspect Cushing’s if urinary free cortisol is greater than 3-fold upper limit of non-pregnant normal values or, if available, greater than pregnancy specific range.
- ACTH. This should be normal in pregnancy, therefore low or low normal ACTH and evidence of pathologic hypercortisolemia is most likely to be Cushing’s syndrome secondary to adrenal adenoma.
- Dexamethasone suppression test (DST). Low dose (1 mg) DST has a high rate of false positive result due to estrogen induced elevated CBG levels. High dose (8 mg) DST may be considered if Cushing’s disease is suspected; however, it may not be required if the diagnosis is Cushing’s syndrome. Therefore, this test is not first line. Cortisol level should suppress by at least 50% in the normal population. Suppression of less than 50% indicates autonomous cortisol secretion. Depression, obesity, chronic disease and some medications (for example, phenytoin) can also cause false positive results.
- Consider abdominal MRI for further investigation for adrenal lesion(s) once pathologic hypercortisolemia is confirmed. Abdominal CT is generally avoided in pregnancy due to high fetal exposure to ionizing radiation. Ultrasound may identify a lesion in 60% of cases outside of pregnancy; the expanding gravid uterus can make maternal adrenal glands more difficult to visualize, hence may not be helpful at later gestation.
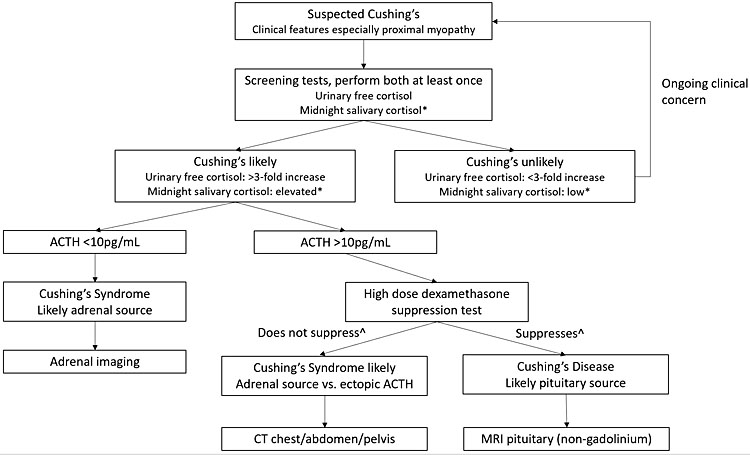
3
Suggested investigation of suspected pathologic hypercortisolemia. *Reference range in pregnancy: first trimester <6.9 nmol/L (<0.25 μg/dL), second trimester <7.2 nmol/L (<0.26 μg/dL) and <9.1 nmol/L (0.33 μg/dL) in third trimester. ^Suppresses is characterized by >50% reduction in cortisol with high dose dexamethasone test, while does not suppress is <50% reduction. ACTH, adrenocorticotropic hormone.
Effects on pregnancy
Patients with treated Cushing’s do well in pregnancy. Untreated Cushing’s, however, is associated with the risks outlined below in Table 2:5,23
2
Complication risk with untreated Cushing’s in pregnancy.
Complication | Risk in pregnancy |
Hypertension | 68% |
Prematurity | 43% |
Diabetes | 25% |
IUGR | 21% |
Pre-eclampsia | 14% |
Stillbirth | 6% |
Spontaneous abortion/intrauterine death | 5% |
Psychiatric disorders | 5% |
Cardiac failure | 3% |
Wound infection | 2% |
Maternal/infant death | 2% |
Management
In a review of 136 pregnancies where treatment was instituted prior to 20 weeks' gestation, live birth increased from 76% to 89%.5 Therefore, it is important to diagnose and treat Cushing’s in pregnancy. Surgery is the management of choice. Trans-sphenoidal resection of pituitary lesions and adrenal surgery to excise adenomas have been successfully performed in pregnancy.5 Prior to 24 weeks in the second trimester is ideal for pituitary surgery. Laparoscopic adrenalectomy appears to be safe and effective until 32 weeks. Third trimester surgery has been reported, however, there is a higher risk of premature delivery.21 After surgical treatment, patients must be started on cortisol replacement due to the risk of AI (see Adrenal insufficiency section for dosing advice).
Medical therapy with metyrapone is an alternative if surgery is contraindicated or if symptoms are severe while surgery is pending. It is important to monitor for severe hypertension as a side-effect due to excess deoxycorticosterone if metyrapone is commenced.5,21,24 Ketoconazole is teratogenic in animal studies, however, there are case reports of its successful use in pregnancy.25,26,27,28 Potential side-effects include IUGR and anti-androgenic effects in the male fetus.5 Cabergoline has been used in some cases successfully.21,24,29 Mitotane is contraindicated and pasireotide has not been explored.21
HYPERALDOSTERONISM
Secondary causes of hypertension are thought to affect 0.24% of pregnancies.30 Primary hyperaldosteronism, otherwise known as Conn’s syndrome, is the most common cause of hyperaldosteronism.31 Other less common conditions of aldosterone excess include idiopathic hyperaldosteronism, glucocorticoid-remediable aldosteronism, also known as familial hyperaldosteronism type 1), familial hyperaldosteronism type 2 (FHII), familial hyperaldosteronism type 3 (FHIII), renin-responsive adenomas, ectopic aldosterone production (from ovaries or kidneys) and syndromes of apparent mineralocorticoid excess.2,31,32
Pathogenesis
Hyperaldosteronism results in sodium and water retention causing hypertension and hypokalemia. It is important to investigate the cause of hyperaldosteronism as management can differ and there are genetic consequences. Primary hyperaldosteronism is characterized by an aldosterone producing adrenal adenoma with resultant high aldosterone levels. For idiopathic hyperaldosteronism, the cause is unknown, however, there may be hyperplasia in one or both adrenal glands. Glucocorticoid-remediable aldosteronism is an autosomal dominant cause of hypertension. This is due to a chimeric CYP11B gene that responds to ACTH rather than angiotensin II. Therefore, aldosterone production is regulated by ACTH instead of the RAAS. Exogenous corticosteroids that suppress ACTH also lower aldosterone levels in this condition. Familial hyperaldosteronism type II (FHII) is a rare autosomal dominant form of primary hyperaldosteronism. Familial hyperaldosteronism type III (FHIII) is caused by a potassium channel mutation. “Apparent mineralocorticoid excess” is a condition characterized by deactivation of 11β-OHSD at the mineralocorticoid receptor (MR). This can be due to genetic or exogenous causes (excess liquorice ingestion). 11β-OHSD converts cortisol (MR activity) to cortisone (no MR activity), thus protecting the MR from inappropriate activation by cortisol. With deactivation of 11β-OHSD, both cortisol and aldosterone act at the MR leading to the syndrome of apparent mineralocorticoid excess (Table 3).2,31,32
3
Pathogenesis and management of hyperaldosteronism.
Syndrome | Pathogenesis | Treatment in pregnancy* |
Primary hyperaldosteronism | Adrenal adenoma | Antihypertensive medications, unilateral adrenalectomy if unresponsive to treatment |
Idiopathic hyperaldosteronism | Unknown, can have hyperplasia of one or both adrenal glands | Antihypertensive medications |
Glucocorticoid-remediable aldosteronism/familial hyperaldosteronism type 1 | Chimeric CYP11B gene responding to ACTH instead of angiotensin II | Glucocorticoids |
Familial hyperaldosteronism type 2 | Autosomal dominant primary hyperaldosteronism | Antihypertensive medications, unilateral adrenalectomy if unresponsive to treatment |
Familial hyperaldosteronism type 3 | Potassium channel mutation | Antihypertensive medications, bilateral adrenalectomy in severe cases unresponsive to treatment |
Apparent mineralocorticoid excess | Deactivation 11β-hydroxysteroid dehydrogenase | Cease liquorice ingestion, antihypertensive medications, corticosteroids to block ACTH if unresponsive to treatment |
*Note: outside pregnancy, the mainstay of treatment is mineralocorticoid receptor blockade (e.g. spironolactone). These are contraindicated in pregnancy due to their potential anti-androgenic effects on a male fetus in particular.
Clinical features
- Hypertension
- Hypokalemia
Diagnosis
It is thought that hyperaldosteronism is underdiagnosed in pregnancy given the paucity of reported cases in the literature.30 A diagnosis of aldosterone excess should be suspected when hypertension is present prior to pregnancy with evidence of prior or current hypokalemia. Patients are usually on multiple antihypertensive agents at high doses and may need significant potassium supplementation. A family pedigree should be performed to assess for a familial cause.
Investigations for hyperaldosteronism:
- Aldosterone excess should be suspected if renin is suppressed or low normal, as pregnancy is a state of physiologic hyperreninemic hyperaldosteronism.2,30 It can be challenging to diagnose.
- Assessment of an aldosterone to renin ratio alone may lead to a false negative result. Also, other factors including medications interfere with the aldosterone to renin ratio which can lead to false positive or false negative results (Table 4).33
- It has been suggested to diagnose hyperaldosteronism in pregnancy if plasma aldosterone is elevated and plasma renin activity is <4 ng/mL/h.34
- If the diagnosis is suspected, imaging is indicated to assess for an adrenal adenoma (MRI or ultrasound).
- Saline or fludrocortisone suppression tests are rarely performed in pregnancy due to fragile hemodynamic status.
- Adrenal vein sampling is not performed due to significant radiation exposure.30
4
Interference with aldosterone and renin levels.
False negative | False positive |
Hypokalemia | Estrogens |
Low salt diet | β-adrenergic blockers |
Concomitant renovascular hypertension | α-methyldopa |
Pregnancy | Clonidine |
Spironolactone | Nonsteroidal anti-inflammatory medications |
Dihydropyridine calcium channel blockers | |
Angiotensin receptor blockers | |
Angiotensin-converting enzyme (ACE) inhibitors | |
Possibly selective serotonin reuptake inhibitors (SSRIs) |
Effect on pregnancy
In a review of 32 pregnancies with hyperaldosteronism, there were significant risks to mother and fetus.30 There were five intrauterine fetal deaths and two neonatal deaths. Intrauterine growth restriction occurred in five pregnancies. There were ten cases of pre-eclampsia, three cases of HELLP and six cases of placental abruption. Early delivery prior to 38 weeks occurred in 51% of pregnancies at a mean of 32 weeks. Virilization occurred in one female neonate. Spironolactone and potassium supplementation were used from 35 weeks' gestation in this case. Maternal risk included pulmonary edema occurring antenatally in one pregnancy and postpartum in two pregnancies. One pregnancy was complicated by postpartum renal failure.
Apparent mineralocorticoid excess can have other deleterious effects on the pregnancy. 11-β-hydroxysteroid dehydrogenase type-2 (11β-OHSD2) is produced by the placenta and has a role in normal development of the fetus. Inactivating mutations have been linked with low birth weight, IUGR and possibly pregnancy failure.31
Management
Genetic counseling should be offered to women with suspected genetic causes of hyperaldosteronism. The aldosterone antagonist spironolactone is not recommended in pregnancy due to its anti-androgenic effects.30 Eplerenone has been used in three pregnancies without complications as has amiloride in 20 pregnancies.30 Management of blood pressure is paramount. Assessment for complications of hypertension should be performed (ECG, echocardiogram, urine protein/creatinine ratio and creatinine). Definitive management is indicated prior to considering pregnancy. Pre-eclampsia risk should be assessed, and aspirin commenced if indicated. Throughout the pregnancy, electrolytes should be tested to watch for hypokalemia or complications from hypertension. Monitor the fetus for IUGR. If hypertension or hypokalemia is uncontrolled, or there is evidence of reduced fetal growth, surgical excision of an adrenal adenoma may be indicated; however, this does carry risk of significant fetal morbidity and mortality.30 Fetal gestation will have an impact on decisions related to surgery to remove the tumor and consultation with all clinicians involved in management to ensure the best outcome for mother and neonate should occur.
PHEOCHROMOCYTOMA
Pheochromocytomas are neuroendocrine chromaffin cell tumors of the adrenal medulla and produce catecholamines; either adrenaline, noradrenaline or dopamine. Paragangliomas arise from the extra adrenal chromaffin cells of either the sympathetic nervous system and produce catecholamines, or the parasympathetic nervous system which do not produce catecholamines.35 Pheochromocytomas account for 80–90% of neuroendocrine chromaffin cell tumors with paragangliomas (extra adrenal pheochromocytomas) accounting for 10–20% of tumors.2,35 Pheochromocytomas occur in 0.2–0.6% of patients with hypertension and in 3–7% of patients with an incidentally discovered adrenal mass.35,36
The prevalence of pheochromocytoma in pregnancy is rare occurring in <0.007% of all pregnancies.35,37,38 Up to 30% of patients with pheochromocytomas in pregnancy are identified as having a pathogenic germ line mutation.35,39 The major hereditary syndromes associated with gene mutation related pheochromocytomas and their clinical features are detailed in a recent review by Lenders et al.35 In most cases (90%), the disease is characterized by a single catecholamine-secreting adrenal adenoma. Involvement of both adrenal glands is more commonly seen in cases with a strong family history of pheochromocytoma. Approximately 10–20% of these tumors are malignant, with a higher incidence among paragangliomas.2 Significant improvements in diagnosis and management of pheochromocytoma, surgical and anesthetic techniques, as well as obstetric and neonatal care has resulted in reduced maternal and fetal mortality from 48% and 55% respectively in the 1960s to 9.8% and 16%, respectively, from a systematic review published in 2015 by Wing et al.35,38 Antenatal diagnosis and treatment is associated with zero maternal deaths.39
Clinical features
Clinical symptoms of pheochromocytoma in pregnancy do not differ from non-pregnant patients. Hypertension in pheochromocytoma can develop at any stage of pregnancy unlike hypertension related to pregnancy which occurs after 20 weeks' gestation. Hypertension is reported to occur in up to 87% of patients and may be paroxysmal or persistent. The presence of orthostatic hypertension in pregnancy without an explanation in the presence of hypertension is suggestive of pheochromocytoma.35,37,38 Symptoms tend to worsen with increasing gestation which may be explained by mechanical factors including growing uterus, fetal movements, uterine contractions, abdominal palpation.35 Other common features include headaches, heat intolerance, palpitations, chest pain, sweating, abdominal pain, nausea, weight loss and nervousness.2,40 Other features which may be present include hyperglycemia or impaired glucose tolerance. Rarer presenting symptoms include cardiomyopathy, myocardial ischemia, arrhythmias, seizures and acute pulmonary edema. Signs of associated syndromes may be present on examination, e.g. café-au-lait spots, freckles and fibromas.35,36,37,41
Maternal and fetal complications
There is an increased incidence of adverse maternal and fetal outcomes when a pheochromocytoma occurs in pregnancy. Table 5 summarizes outcomes from two recent systematic reviews.8,38 A recent UKOSS study on adrenal tumors in pregnancy over a 4 year period also reported pregnancy outcomes from both the UKOSS data and those identified in the literature.40 They reported an increased incidence of preterm delivery of 55% with the mean gestational age being 32.5 weeks.40
Complication | Incidence (n = 128) % |
Hypertension | 87 |
Hypertensive crisis | 24 |
Delivery Vaginal Cesarean Not stated | 23 55 22 |
Maternal mortality | 8.6 |
Fetal mortality | 16 |
Diagnosis
Recent studies report 73–80% of women are diagnosed antenatally.38,39 Survival is improved for both the mother and fetus if the diagnosis is made antenatally therefore a high level of suspicion and aggressive investigation is warranted if the women have suggestive symptoms. In healthy pregnant women, urinary and plasma catecholamine levels are not increased. Eclampsia but not pre-eclampsia may be associated with an increase in plasma catecholamines at the time of the seizure. This rise resolves within a few days.42 Maternal catecholamines cross the placenta minimally due to the presence of noradrenaline transporters and catecholamine metabolizing enzymes in the placenta.
Prior to organizing investigations, it is important to take a detailed history of current pharmacological therapy as some drugs, as detailed in Table 6 may cause falsely elevated test results for plasma and urinary metanephrines.36
6
Medications that may falsely elevate plasma or urine metanephrine results on screening for pheochromocytoma.27
Medication | Plasma | Plasma | Urine | Urine |
Paracetamol | ++ | − | ++ | − |
Labetalol | − | − | ++ | ++ |
Sotalol | − | − | ++ | ++ |
α-methyldopa | ++ | − | ++ | − |
Tricyclic antidepressants | ++ | − | ++ | − |
Buspirone | − | ++ | − | ++ |
Phenoxybenzamine | ++ | − | ++ | − |
Monoamine oxidase inhibitors | ++ | ++ | ++ | ++ |
Sympathomimetics | + | + | + | + |
Cocaine | ++ | + | ++ | + |
Sulfasalazine | ++ | − | ++ | − |
Investigations for pheochromocytoma:
- A recent study has demonstrated that measurement of plasma and urinary free metabolites (metanephrines and 3-methoxytyramine) in the non-pregnant population in patients with a low probability of pheochromocytoma has a similar diagnostic performance, whereas the plasma test is superior to the urine test in patients with a high risk of pheochromocytoma.8,43 Plasma free metabolites are produced within the tumor and circulate continuously while catecholamine release is episodic and variable.35,36
- False positive results may occur due to drugs, hyperglycemia, strenuous exercise raised intracranial pressure and inappropriate sampling. To minimize false positive results which may occur for plasma free metabolites it is recommended that the patient rest supine for at least 20 minutes prior to the blood being taken.35
- The clonidine suppression test is used to discriminate patients with mildly elevated plasma normetanephrines results due to either a false positive result from increased sympathetic activity from a true positive result due to a chromaffin cell tumor. Studies and reviews have suggested that this test is contraindicated in pregnancy due to potential for adverse effects on the fetus.35,36 This is likely secondary to the possibility of maternal hypotension and resultant reduced placental perfusion. There is limited experience with the test in pregnancy.
- As soon as the diagnosis is confirmed, an imaging study should be performed to localize the lesion. The imaging modality recommended in pregnancy is an MRI without gadolinium and/or ultrasound. Depending on gestation the diagnostic utility of ultrasound may be limited by the gravid uterus. The diagnostic sensitivity of an MRI is similar to CT at 90–100%, with specificity of MRI being 70–80%. MIBG scan (123-I metaiodobenzylguanidine) is contraindicated in pregnancy due to the potential for radioactive compounds to cross the placenta.
- Tumor biopsy is contraindicated as this may precipitate a hypertensive crisis.35,37
- Genetic testing and counseling should be undertaken to determine if there are any underlying genetic mutations which require ongoing management for the women or her family.35
Treatment
Treatment recommendations for pheochromocytoma in pregnancy are based on consensus recommendations. Medical therapy is commenced prior to definitive surgical treatment. Medical therapy aims to treat hypertension, prevent paroxysms and correct hypovolemia. Hypertension management in pregnancy is a balance between control of maternal blood pressure while not compromising utero-placental blood flow. α-Adrenergic receptor blockers are commenced which control blood pressure and allow partial restoration of normal blood volume with subsequent β-adrenergic receptor blockade 10–14 days later.35,37,38,42 The two most common α-adrenoceptor blockers used are phenoxybenzamine and doxazosine. The drug which has been used most often is phenoxybenzamine, which is safe in pregnancy and can be started at 10 mg twice daily; the dose can be increased by 10 mg every 2–3 days until blood pressure and symptoms are controlled aiming for a final dose of 1 mg/kg/day. Phenoxybenzamine has prominent side-effects including nasal congestion, dizziness, drowsiness, blurred vision and gastrointestinal upset. It has a half-life of 24 hours with effects continuing for up to a week postcessation. Phenoxybenzamine also crosses the placenta and accumulates in the fetus and may cause neonatal hypotension and respiratory depression.35,38,42,43 Doxazosine has been used increasingly in pregnant patients as it has less maternal side-effects, a lower incidence of reflex tachycardia and postoperative hypotension. It crosses the placenta but with a lower fetal : maternal plasma ratio than phenoxybenzamine and has good neonatal outcomes. Due to its improved profile in pregnancy doxazosin is becoming the preferred α-blocker in pregnancy.2,35,41,44
β-Adrenergic blockade is commenced to prevent catecholamine induced tachyarrhythmias and α-blocker induced reflex tachycardia. β-blockers should not be used alone and should be commenced after appropriate α-blockade. Propranolol (40 mg three times a day) or atenolol (25–50 mg once per day) are the preferred drugs. Women should also be encouraged to increase salt and fluid intake to reduce postural hypotension and postoperative hypotension.35,44 Hypertensive crisis can be managed with calcium channel blockers, e.g. nicardipine or nifedipine, as well as magnesium sulfate which also induces vasodilation and reduces catecholamine release and the sensitivity of α-adrenoceptors to catecholamines.35
Surgery is the definitive treatment for pheochromocytoma. If the diagnosis of pheochromocytoma is made within the first 24 weeks of gestation and there has been adequate α-blockade for at least 2 weeks preoperatively then surgery is recommended before 24 weeks' gestation. If the diagnosis is made after this time, then surgery should be postponed to occur as either a combined cesarean and tumor removal procedure or postpartum. Laparoscopic adrenalectomy for lesions <6 cm is the preferred surgical approach with reports in the literature indicating less pain, less blood loss, fewer hospitalization days and less surgical morbidity.35,36 Patients require long-term follow-up postsurgery to monitor blood pressure and detect recurrence.
In women who are diagnosed with pheochromocytoma which cannot be resected, there are limited data to recommend cesarean section over vaginal delivery. The decision on mode of delivery is dependent on patient preference, parity, previous cesarean section, maternal and fetal condition and success of medical treatment. Oxytocin and uterotonic agents should be used cautiously as they may be associated with tachycardia and hypotension.2 Recent literature has reported successful vaginal delivery with regional anesthesia.38,39,41
Pheochromocytoma in pregnancy is a rare condition associated with high rates of morbidity and mortality in mothers and babies despite significant improvements in medical, surgical, anesthetic, obstetric and neonatal care. Timely diagnosis and instigation of treatment are crucial in improving outcomes. Decisions regarding timing of adrenal surgery and timing and mode of delivery should be made on an individual basis in consultation with the woman and experienced multidisciplinary teams in tertiary centers to ensure optimum outcomes for mother and baby.
ADRENAL INCIDENTALOMAS
Adrenal incidentalomas are unsuspected lesions ≥1 cm in diameter that are detected on imaging studies (excluding studies of patients who are being evaluated for cancer) in subjects that do not have clinical features of adrenal disease.45,46 There are rare case reports of incidentalomas occurring in pregnancy.47
Principles of management of adrenal incidentalomas detected in pregnancy are the same as in the non-pregnant state. This includes investigating for hormone hypersecretion and differentiating malignant from benign lesions via their radiographic imaging characteristics. MRI is the preferred investigative modality due to the lack of radiation exposure. Ultrasound may also be used. Women require referral to a tertiary center with multidisciplinary teams given the rarity of this condition. Management decisions must balance the risks and benefits of surgical removal of incidentalomas in pregnancy. If there is no evidence of hormone excess or malignancy, surveillance in pregnancy may be appropriate.3,48
ADRENOCORTICAL CARCINOMAS
Adrenocortical carcinoma (ACC) is a rare malignancy with an estimated incidence of 1–2 per million population.49 The pathogenesis is not completely understood and diagnosis is associated with poor prognosis with a survival rate at 5 years reported to be <15–35%.49,50,51 Patients usually present with mass effect or hormone excess.49,52
There are limited case reports of ACC in pregnancy.50,53 A series by Abiven-Lepage in 2010 reported 12 pregnancy associated cases.53 In this series they reported worse outcomes when ACC is diagnosed in pregnancy or postpartum than in the non-pregnant populations.
Gestation at diagnosis will impact on management. Termination of pregnancy should be discussed with the woman and her partner. Given the rarity of this condition and the poor prognosis, women with suspected ACC should be seen in tertiary centers with a multidisciplinary team approach.49,51 Early diagnosis and management is essential to optimize outcomes.
CONGENITAL ADRENAL HYPERPLASIA (CAH)
Classic congenital adrenal hyperplasia (CAH) is a rare autosomal recessive disorder affecting 1 in 16,000 births. Jewish people of Eastern European origin have a higher incidence of this disorder. The milder forms are more common with a gene frequency of 1 : 200–1 : 400.54 It is characterized by defective conversion of cholesterol to cortisol/aldosterone due to adrenal enzyme mutation. There is cortisol deficiency with or without aldosterone deficiency depending on the enzyme affected (Figure 4). Precursors are shunted down the androgen pathway with subsequent clinical and biochemical androgen excess. Progesterone excess acting as an antagonist to aldosterone can further exacerbate aldosterone deficiency. Ninety per cent of cases are due to 21-hydroxylase deficiency which causes both cortisol and aldosterone deficiency (salt wasting/classical CAH) or partial deficiency (non-classic or simple virilising CAH). Eight to nine per cent are due to 11-βhydroxylase deficiency which causes both cortisol and aldosterone deficiency. Non-classic CAH results in deoxycortisone excess which has similar activity to aldosterone, therefore salt wasting does not occur.55,56
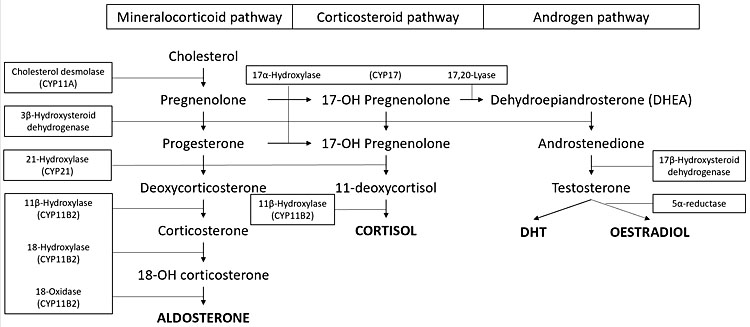
4
Adrenal hormone synthesis.
Clinical and biochemical features54,57
Clinical features vary based on the enzyme affected.
Salt wasting/classic CAH: complete 21-hydroxylase deficiency
Cortisol deficiency results in poor cardiac function, reduced glomerular filtration rate, poor development of adrenal medulla with possible catecholamine deficiency and increased antidiuretic hormone. Aldosterone deficiency results in hyponatremia, hyperkalemia and profound hypovolemia. Androgen excess results in hirsutism, precocious puberty, virilization of external genitalia, in women, psychosexual problems, polycystic ovarian syndrome (PCOS), acne, amenorrhea, delayed menarche and premature menopause.
The female fetus is exposed to excess androgens from week 7 of gestation. This results in enlarged clitoris, partly fused labia majora and a common urogenital sinus (in place of separate urethra/vagina). Ovaries, uterus and fallopian tubes are normally formed. The male fetus may have subtle hyperpigmentation and penile enlargement. At puberty, males can present with painful testicular adrenal-like rest tumors. Untreated/undiagnosed individuals have premature closure of epiphyses with resultant short stature, early pubic hair, ongoing clitoral growth in the female/penile growth in the male (with small testes) and possible precocious puberty.
Non-classic CAH/simple virilizing form: partial 21-hydroxylase deficiency
Presentation is variable and based on level of residual 21-hydroxylase deficiency. Some remain undiagnosed, while others may present with hirsutism at puberty, infertility, menstrual irregularities or hypotension/hypovolemia.
11-βhydroxylase deficiency
Cortisol deficiency and androgen excess as in classic CAH. Aldosterone deficiency and deoxycortisone excess: deoxycortisone has mineralocorticoid activity, therefore there is paradoxical mineralocorticoid excess causing sodium retention and hypertension.
Diagnosis
- 17-hydroxyprogesterone is usually elevated in classic CAH; however, it may not be elevated in the non-classic form.54
- Prenatally, classic CAH may be diagnosed by elevated 11-deoxycortisone in amniotic fluid, chorionic villus biopsy at weeks 10–11 or elevated maternal urinary tetrahydrodeoxycortisol after week 8.56,58
- Postnatally, the gold standard test is a short synacthen test. Synthetic ACTH (0.125–0.25 mg) is injected intravenously – basal and 60 minute 17-hydroxyprogesterone levels are taken. Levels above 10,000 ng/dL suggests classic CAH, while levels 1,500–10,000 ng/dL suggest non-classic CAH.54
Effects on pregnancy
There are multiple possible effects on pregnancy including miscarriage due to inadequate corpus luteum, pre-eclampsia and fetal growth restriction; furthermore, cesarean section may be required due to android pelvis.56
Management
Parents should be screened for CAH if there is a history of a previously affected infant or a known mutation in one parent. Preconception counseling should be offered to all with known diagnosis of CAH or if there has been an affected infant. Pre-eclampsia is more common, therefore appropriate monitoring should occur throughout the pregnancy. Steroid replacement should continue at pre-pregnancy doses. Androgens levels should be monitored; free testosterone is reduced or unchanged in pregnancy, therefore it is a good marker to monitor. Other androgens usually change in pregnancy. If free testosterone levels become elevated, steroid replacement dose should increase. Mineralocorticoid replacement generally does not require a dose change in pregnancy.56
Placental aromatase deactivates maternal androgens, therefore there is some protection against virilization of a female fetus. The male fetus does not have any excess risk. Administration of dexamethasone to reduce androgens in any female fetus is a controversial strategy with a significant risk of treating an unaffected fetus. It is therefore not recommended.56
Neonates with suspected CAH should be commenced on cortisol replacement. Classic CAH requires cortisol and aldosterone replacement, while non-classic CAH may only require cortisol replacement or no treatment at all, depending on the degree of enzymatic dysfunction. Mineralocorticoids should be dosed at 0.1–0.2 mg fludrocortisone daily in infants. Adequacy of dose is assessed by renin levels in conjunction with other clinical features (blood pressure, electrolytes).54
For the non-pregnant patient, nocturnal dexamethasone is sometimes administered to suppress ACTH surge at a dose of 0.5–0.75 mg.59 Dexamethasone is avoided in pregnancy due to transplacental passage and thus fetal exposure. Prednisolone 5–7.5 mg daily is usually effective for pregnant women with CAH treated with steroids outside of pregnancy. Adequacy of steroid treatment in pregnancy is assessed by 17-hydroxyprogesterone and androstenedione levels. 17-hydroxyprogesterone levels should be 100–1000 ng/dL (3–30 nmol/L) and androstenedione levels should be normal.
PRACTICE RECOMMENDATIONS
- All women with preexisting adrenal disorders should undergo preconception counseling.
- Diagnosing adrenal disease in pregnancy can be challenging:
- The fetal-placental unit alters usual maternal hormone feedback loops;
- Pregnancy results in unique maternal reference ranges making diagnosis of disordered adrenal function more difficult;
- Usual workup is not always able to be performed due to (1) physiologic changes in pregnancy thereby invalidating tests and/or (2) unacceptable risks to the mother/fetus;
- Pregnancy-related symptoms may mask an underlying pathologic process.
- Assessment and management of some adrenal conditions require urgent intervention in pregnancy, while others can be deferred until the postpartum period.
- Some adrenal disorders in pregnancy are high risk for adverse maternal and fetal outcome, therefore these patients should be watched closely in a specialist multidisciplinary clinic if available.
CONFLICTS OF INTEREST
The author(s) of this chapter declare that they have no interests that conflict with the contents of the chapter.
Feedback
Publishers’ note: We are constantly trying to update and enhance chapters in this Series. So if you have any constructive comments about this chapter please provide them to us by selecting the "Your Feedback" link in the left-hand column.
REFERENCES
Levin G, Elchalal U, Rottenstreich A. The adrenal cortex: Physiology and diseases in human pregnancy. Eur J Obstet Gynecol Reprod Biol 2019;240:139–43. | |
Manoharan M, Sinha P, Sibtain S. Adrenal disorders in pregnancy, labour and postpartum – an overview. J Obstet Gynaecol 2019:1–10. | |
Kamoun M, Mnif MF, Charfi N, et al. Adrenal diseases during pregnancy: pathophysiology, diagnosis and management strategies. Am J Med Sci 2014;347(1):64–73. | |
Jung C, Ho JT, Torpy DJ, et al. A longitudinal study of plasma and urinary cortisol in pregnancy and postpartum. J Clin Endocrinol Metab 2011;96(5):1533–40. | |
Lekarev O, New MI. Adrenal disease in pregnancy. Best Pract Res Clin Endocrinol Metab 2011;25(6):959–73. | |
Mastorakos G, Ilias I. Maternal and fetal hypothalamic-pituitary-adrenal axes during pregnancy and postpartum. Ann N Y Acad Sci 2003;997:136–49. | |
Kuijper EA, Ket JC, Caanen MR, et al. Reproductive hormone concentrations in pregnancy and neonates: a systematic review. Reprod Biomed Online 2013;27(1):33–63. | |
Orioli L, Debieve F, Donckier J, et al. Pheochromocytoma during pregnancy: Case report and review of recent literature. Ann Endocrinol (Paris). 2017;78(5):480–4. | |
Anand G, Beuschlein F. Management of endocrine disease: Fertility, pregnancy and lactation in women with adrenal insufficiency. Eur J Endocrinol 2018;178(2):R45-R53. | |
Langlois F, Lim DS, Fleseriu M. Update on adrenal insufficiency: diagnosis and management in pregnancy. Current Opinion in Endocrinology, Diabetes and Obesity 2017;24(3):184–92. | |
Hannah-Shmouni F, Stratakis CA. An overview of inborn errors of metabolism manifesting with primary adrenal insufficiency. Rev Endocr Metab Disord 2018;19(1):53–67. | |
Schneiderman M, Czuzoj-Shulman N, Spence AR, et al. Maternal and neonatal outcomes of pregnancies in women with Addison's disease: a population-based cohort study on 7.7 million births. BJOG 2017;124(11):1772–9. | |
Joseph RM, Hunter AL, Ray DW, et al. Systemic glucocorticoid therapy and adrenal insufficiency in adults: A systematic review. Semin Arthritis Rheum 2016;46(1):133–41. | |
Suri D, Moran J, Hibbard JU, et al. Assessment of adrenal reserve in pregnancy: defining the normal response to the adrenocorticotropin stimulation test. J Clin Endocrinol Metab 2006;91(10):3866–72. | |
Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and Treatment of Primary Adrenal Insufficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2016;101(2):364–89. | |
Therapeutic Goods Australia. Prescribing medicines in pregnancy database. 2019. | |
Bratland E, Husebye ES. Cellular immunity and immunopathology in autoimmune Addison's disease. Mol Cell Endocrinol 2011;336(1–2):180–90. | |
Stjernholm YV, Nyberg A, Cardell M, et al. Circulating maternal cortisol levels during vaginal delivery and elective cesarean section. Arch Gynecol Obstet 2016;294(2):267–71. | |
Owa T, Mimura K, Kakigano A, et al. Pregnancy outcomes in women with different doses of corticosteroid supplementation during labor and delivery. J Obstet Gynaecol Res 2017;43(7):1132–8. | |
Quinkler M, Oelkers W, Remde H, et al. Mineralocorticoid substitution and monitoring in primary adrenal insufficiency. Best Pract Res Clin Endocrinol Metab 2015;29(1):17–24. | |
Brue T, Amodru V, Castinetti F. Management of endocrine disease: Management of Cushing's syndrome during pregnancy: solved and unsolved questions. Eur J Endocrinol 2018;178(6):R259-R66. | |
Fan J, Tang J, Fang J, et al. Ultrasound imaging in the diagnosis of benign and suspicious adrenal lesions. Med Sci Monit 2014;20:2132–41. | |
Lindsay JR, Jonklaas J, Oldfield EH, et al. Cushing's syndrome during pregnancy: personal experience and review of the literature. J Clin Endocrinol Metab 2005;90(5):3077–83. | |
Nakhleh A, Saiegh L, Reut M, et al. Cabergoline treatment for recurrent Cushing's disease during pregnancy. Hormones (Athens). 2016;15(3):453–8. | |
Berwaerts J, Verhelst J, Mahler C, et al. Cushing's syndrome in pregnancy treated by ketoconazole: case report and review of the literature. Gynecol Endocrinol 1999;13(3):175–82. | |
Boronat M, Marrero D, Lopez-Plasencia Y, et al. Successful outcome of pregnancy in a patient with Cushing's disease under treatment with ketoconazole during the first trimester of gestation. Gynecol Endocrinol 2011;27(9):675–7. | |
Costenaro F, Rodrigues TC, de Lima PB, et al. A successful case of Cushing's disease pregnancy treated with ketoconazole. Gynecol Endocrinol 2015;31(3):176–8. | |
Zieleniewski W, Michalak R. A successful case of pregnancy in a woman with ACTH-independent Cushing's syndrome treated with ketoconazole and metyrapone. Gynecol Endocrinol 2017;33(5):349–52. | |
Ferriere A, Cortet C, Chanson P, et al. Cabergoline for Cushing's disease: a large retrospective multicenter study. Eur J Endocrinol 2017;176(3):305–14. | |
Morton A. Primary aldosteronism and pregnancy. Pregnancy Hypertens 2015;5(4):259–62. | |
Escher G. Hyperaldosteronism in pregnancy. Ther Adv Cardiovasc Dis 2009;3(2):123–32. | |
Mulatero P, Tizzani D, Viola A, et al. Prevalence and characteristics of familial hyperaldosteronism: the PATOGEN study (Primary Aldosteronism in TOrino-GENetic forms). Hypertension 2011;58(5):797–803. | |
Young Jr. WF, Calhoun DA, Lenders JWM, et al. Screening for endocrine hypertension: an Endocrine Society scientific statement. Endocrine Reviews 2017;38(2):103–22. | |
Malha L, August P. Secondary Hypertension in Pregnancy. Curr Hypertens Rep 2015;17(7):53. | |
Lenders JWM, Langton K, Langenhuijsen JF, et al. Pheochromocytoma and Pregnancy. Endocrinol Metab Clin North Am 2019;48(3):605–17. | |
Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014;99(6):1915–42. | |
Eschler DC, Kogekar N, Pessah-Pollack R. Management of adrenal tumors in pregnancy. Endocrinol Metab Clin North Am 2015;44(2):381–97. | |
Wing LA, Conaglen JV, Meyer-Rochow GY, et al. Paraganglioma in Pregnancy: A Case Series and Review of the Literature. J Clin Endocrinol Metab 2015;100(8):3202–9. | |
Biggar MA, Lennard TW. Systematic review of phaeochromocytoma in pregnancy. Br J Surg 2013;100(2):182–90. | |
Quartermaine G, Lambert K, Rees K, et al. Hormone-secreting adrenal tumours cause severe hypertension and high rates of poor pregnancy outcome; a UK Obstetric Surveillance System study with case control comparisons. BJOG 2018;125(6):719–27. | |
van der Weerd K, van Noord C, Loeve M, et al. Endocrinology in pregnancy: Pheochromocytoma in pregnancy: case series and review of literature. Eur J Endocrinol 2017;177(2):R49-r58. | |
Khatun S, Kanayama N, Hossain B, et al. Increased concentrations of plasma epinephrine and norepinephrine in patients with eclampsia. Eur J Obstet Gynecol Reprod Biol 1996;69(2):61–7. | |
Eisenhofer G, Prejbisz A, Peitzsch M, et al. Biochemical Diagnosis of Chromaffin Cell Tumors in Patients at High and Low Risk of Disease: Plasma versus Urinary Free or Deconjugated O-Methylated Catecholamine Metabolites. Clin Chem 2018;64(11):1646–56. | |
Versmissen J, Koch BC, Roofthooft DW, et al. Doxazosin treatment of phaeochromocytoma during pregnancy: placental transfer and disposition in breast milk. Br J Clin Pharmacol 2016;82(2):568–9. | |
Jason DS, Oltmann SC. Evaluation of an Adrenal Incidentaloma. Surg Clin North Am 2019;99(4):721–9. | |
Ioachimescu AG, Remer EM, Hamrahian AH. Adrenal incidentalomas: a disease of modern technology offering opportunities for improved patient care. Endocrinol Metab Clin North Am 2015;44(2):335–54. | |
Fallo F, Pezzi V, Sonino N, et al. Adrenal incidentaloma in pregnancy: clinical, molecular and immunohistochemical findings. J Endocrinol Invest 2005;28(5):459–63. | |
Garrett RW, Nepute JC, Hayek ME, et al. Adrenal Incidentalomas: Clinical Controversies and Modified Recommendations. AJR Am J Roentgenol 2016;206(6):1170–8. | |
Long SE, Miller BS. Adrenocortical Cancer Treatment. Surg Clin North Am 2019;99(4):759–71. | |
Kotteas E, Ioachim E, Pavlidis N. A pregnant patient with adrenocortical carcinoma: case report. Onkologie 2012;35(9):517–9. | |
Megerle F, Kroiss M, Hahner S, et al. Advanced Adrenocortical Carcinoma – What to do when First-Line Therapy Fails? Exp Clin Endocrinol Diabetes 2019;127(2–03):109–16. | |
Fassnacht M, Dekkers O, Else T, et al. European Society of Endocrinology Clinical Practice Guidelines on the Management of Adrenocortical Carcinoma in Adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2018;179(4):G1-G49. | |
Abiven-Lepage G, Coste J, Tissier F, et al. Adrenocortical carcinoma and pregnancy: clinical and biological features and prognosis. Eur J Endocrinol 2010;163(5):793–800. | |
Speiser PW. Congenital Adrenal Hyperplasia. F1000Res. 2015;4(F1000 Faculty Rev):601. | |
White PC. Steroid 11 beta-hydroxylase deficiency and related disorders. Endocrinol Metab Clin North Am 2001;30(1):61–79, vi. | |
Speiser PW, Arlt W, Auchus RJ, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2018;103(11):4043–4088. | |
Claahsen-van der Grinten HL, Sweep FC, Blickman JG, et al. Prevalence of testicular adrenal rest tumours in male children with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Eur J Endocrinol 2007;157(3):339–44. | |
Rosler A, Weshler N, Leiberman E, et al. 11 Beta-hydroxylase deficiency congenital adrenal hyperplasia: update of prenatal diagnosis. J Clin Endocrinol Metab 1988;66(4):830–8. | |
Krieger DT, Allen W, Rizzo F, et al. Characterization of the normal temporal pattern of plasma corticosteroid levels. J Clin Endocrinol Metab 1971;32(2):266–84. |
Online Study Assessment Option
All readers who are qualified doctors or allied medical professionals can automatically receive 2 Continuing Professional Development points plus a Study Completion Certificate from GLOWM for successfully answering four multiple-choice questions (randomly selected) based on the study of this chapter. Medical students can receive the Study Completion Certificate only.
(To find out more about the Continuing Professional Development awards program CLICK HERE)