This chapter should be cited as follows:
Cutts BA, Glob Libr Women's Med
ISSN: 1756-2228; DOI 10.3843/GLOWM.415613
The Continuous Textbook of Women’s Medicine Series – Obstetrics Module
Volume 8
Maternal medical health and disorders in pregnancy
Volume Editor:
Dr Kenneth K Chen, Alpert Medical School of Brown University, USA
Originating Editor: Professor Sandra Lowe

Chapter
Maternal Hemoglobinopathies and Pregnancy
First published: December 2021
Study Assessment Option
By answering four multiple-choice questions (randomly selected) after studying this chapter, readers can qualify for Continuing Professional Development points plus a Study Completion Certificate from GLOWM.
See end of chapter for details.

INTRODUCTION
Normal adult red blood cells contain three types of hemoglobin (Hb) with the main component being hemoglobin A (HbA). Hemoglobin A consists of four main globin chains (two beta (β) and two alpha (α) chains: β2α2). There are two genes for the two β-globin chains (one gene for each β-globin chain) and four genes for the two α-globin chains (two genes for each α-globin chain). Genes for all four globin chains are located on chromosomes 11 and 16 and are inherited – half are maternal and half are paternal. The other two types of hemoglobin are minor fractions containing fetal Hb (HbF) and hemoglobin A2 (HbA2). The switch from fetal Hb (HbF) to adult Hb (HbA) occurs at approximately 3–6 months after birth.1
Hemoglobinopathies encompass the term for all globin gene abnormalities that result in a structurally abnormal hemoglobin (such as sickle cell anemia) or unbalanced globin gene production (such as thalassemia). For the scope of this chapter, we focus on sickle cell anemia, thalassemias and associated syndromes.
Hemoglobinopathies are common inherited disorders of red blood cells affecting approximately 7% of the world’s population (see Figure 1). They are usually autosomal recessive diseases that are the result of gene encoding abnormalities to the globin genes that can be either qualitative – due to globin gene mutation, or quantitative – due to globin gene deletions. The type and size of gene mutations and/or deletions to the hemoglobin gene will impact upon the hemoglobin’s oxygen carrying capacity. In their severest form (major forms) hemoglobinopathies result in red cells with markedly reduced oxygen carrying capacity that carry significant morbidity which is exacerbated by pregnancy.2
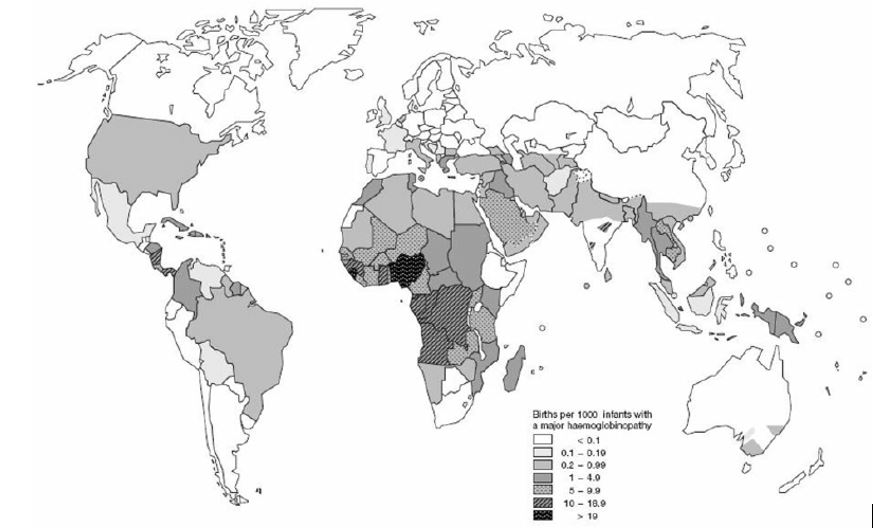
1
Global distribution of hemoglobin disorders in terms of births of affected infants per 1000 births. Adapted from Angastiniotis et al., 1995.3
Hemoglobinopathies impact on pregnancy in two main ways. First, screening for inheritance of abnormal globin genes needs to be considered in the potential offspring. Second, if women have sickle cell anemia or a thalassemia major syndrome, the physiological stress of pregnancy can worsen anemia requiring closer monitoring and potentially intervention to ensure both maternal and fetal safety. Prenatal screening and complications that may arise in pregnancy should be explored in pre-pregnancy counseling.4
CLASSIFICATION
Traditionally hemoglobinopathies are classified as either minor or major with intermedia forms in-between. Minor forms do not usually cause a clinical phenotype, whereas major forms usually present as clinically significant disease. In recent years there has been acknowledgment that the severity of a patient’s hemoglobinopathy exists on a spectrum based on dependence of regular blood transfusion for patient survival. For instance patients with β-thalassemia major are usually transfusion dependent versus β-thalassemia minor patients who are usually transfusion independent. This classification helps both clinicians and potential parents to understand what a pregnancy ‘affected’ by thalassemia means.
SCREENING
Antenatal screening should identify couples at risk of having a child with sickle cell disease or a thalassemia major syndrome. This allows counseling and reproductive choice of continuing or terminating an affected pregnancy. Screening results need to be obtained in a timely manner to allow adequate counseling and fetal testing if requested. Termination is generally considered more acceptable when a diagnosis is made within the first trimester. Diagnosis is by chorionic villous sampling (CVS) between 10 and 12 weeks' gestation, or amniocentesis if the woman is at 15 weeks or greater in gestation with fetal loss rates from both interventions of approximately 0.5–1%.5 Using maternal blood to acquire fetal cell free DNA has enabled non-invasive testing for fetal chromosomal abnormalities by determining chromosomal number and length. It is not able to differentiate specific gene deletions or mutations within chromosomes for autosomal recessive disorders where 50% of the fetal genome is identical to that of the mother.5 Pre-implantation genetic diagnosis can be offered to couples at risk of having a child affected by major hemoglobinopathies; however, this requires couples to undergo assisted reproductive technology to attain unaffected embryos, pregnancy is not certain following this and in most countries it is very expensive for couples to access.5
If resources are not available to perform antenatal screening, neonatal screening programs can be undertaken. At-risk couples with potentially affected neonates are screened and identified to enable early intervention and early management optimization of the affected child.
Screening programs need to first flag at-risk couples. In areas of high prevalence, screening should be universal. Where prevalence is lower, screening should identify risk based on ethnicity, abnormal blood test findings and/or family history. Blood tests done for screening can vary, but should consist of a full blood count and blood film including either a measurement of mean cell volume (MCV) and/or mean cell hemoglobin (MCH) with a predetermined lower limit of cut-off. Patients with normal Hb levels but reduced MCV or MCH levels in the absence of iron deficiency may have a form of thalassemia. Hemoglobin electrophoresis (HbEp) is also performed to measure levels of hemoglobins including HbA, HbA2, HbF, HbS as well as other abnormal hemoglobin variants. Sickle cell trait can only be excluded if HbEp is normal. The above process requires a laboratory with protocols and quality measures in place to allow testing and accurate interpretation of the above.2
COUNSELING FOR HEMOGLOBINOPATHIES
Couples should be counseled about disease morbidity and mortality in sickle cell disease and thalassemia major syndromes that are transfusion dependent.
Sickle cell anemia
In sickle cell anemia (SCA) the higher amounts of HbF continue until approximately 4–6 months of age. With the decline of HbF levels, HbS levels increase with resultant formation of insoluble polymers that cause the typical sickle cell shape of red blood cells that is characteristic of sickle cell anemia. These high numbers of sickle cells can result in significant morbidity and mortality in the first 5 years of life predominantly due to splenic sequestration and complications from hyposplenism such as pneumococcal septicemia. However, early identification of children with SCA has led to successful programs of vaccination and antibiotic prophylaxis and has helped significantly reduce morbidity.
Other long-term complications that occur from SCA are a result of polymerization of HbS at low oxygen tension that eventually damage red cell membrane proteins and cause the typical sickle cell shape of red blood cells in sickle cell anemia. Due to the abnormal shape of the red cells, chronic intravascular and extravascular hemolysis occurs. Red cells have a reduced lifespan and when the red cells hemolyse, free hemoglobin is released resulting in endothelial activation and vasoconstriction which increases the risk of sickle cells becoming caught within small blood vessels. These complications manifest as:
- Chronic hemolysis;
- Vaso-occlusive crisis (sickle cell crisis) occurring in joints, pulmonary vasculature (acute chest syndrome) and other blood vessels;
- Thrombosis (arterial or venous6
The early implementation of hydroxycarbamide (hydroxyurea) in children with sickle cell anemia has been shown to increase levels of HbF and reduce vaso-occlusive crises in children with reduced morbidity and mortality.7 Ongoing work is being undertaken to improve global access to hydroxycarbamide for sickle cell anemia.
Sickle cell anemia refers to a homozygous state where the child has inherited HbS from both parents (HbSS). HbS can also be co-inherited with other abnormal Hb resulting in a compound heterozygous state. These can also manifest as sickle cell anemia and couples should receive counseling about a possible sickle cell anemia phenotype with the below combinations with HbS:
- Hb SC;
- Hb Sβ thalassemia;
- HbSD Punjab;
- Hb SO Arab;
- HbSE;
- Hb SLepore.2
Carrying a single HbS sickle cell gene (sickle cell trait) does not cause significant disease and is only noteworthy to trigger partner testing to determine the existence of HbS or any of the above abnormal hemoglobinopathies.
β-Thalassemia major
Early identification, intervention and optimizing long-term management of blood transfusion in β-thalassemia major patients has significantly improved quality of life. Life expectancy can be normal when there is access to the above care. In β-thalassemia major HbF levels reduce in the months following birth resulting in failure of globin chain production. This manifests clinically as:
- Severe anemia;
- Hemolysis;
- Increased extramedullary hematopoiesis.
Severe anemia requires commencement of regular blood transfusions and iron chelation therapy for long-term survival. Iron chelation is required to reduce the complications from iron overload that result from chronic transfusion. Specifically complications that arise from unmanaged iron overload include:
- Cardiac failure and arrhythmias;
- Hepatic failure;
- Pancreatic dysfunction with impaired glucose tolerance or diabetes mellitus;
- Pituitary dysfunction with hypogonadism;
- Joint arthropathy;
- Hyperpigmentation of skin;
- Short stature.
As with sickle cell trait, thalassemia minor (transfusion-independent thalassemia) generally do not manifest with a clinical phenotype and are only a concern in pregnancy to allow partner testing to determine potential risk of thalassemia major syndromes in a newborn. Whilst a homozygous state of β+/0 thalassemia major usually causes the most profound disruption to globin chain production, other compound heterozygous states that can result in a similar transfusion-dependent phenotype include Hbβ/E.8
α-Thalassemia major
α-Thalassemia has the highest prevalence worldwide. The α-globin genes are inherited in pairs (αα) with one pair inherited from each parent (αα/αα). One or two gene deletions or mutations in the α genes result in a thalassemia minor syndrome (α thalassemia trait) that is not clinically significant and is often first detected in pregnancy screening. Full blood count results usually show a normal hemoglobin level with a mildly reduced MCV and/or MCH.
HbH disease (–/-α)
When three or more α genes are deleted or mutated, HbH disease occurs. The markedly reduced rate of α-chain synthesis presents as a microcytic hypochromic anemia. Excess unmatched β-globin chains result in β-tetramers forming which produces HbH. HbH has a high oxygen affinity, is soluble and prone to oxidation and instability. It precipitates and causes damage to circulating red blood cells with resultant red cell membrane rigidity, fragmentation and hemolysis. Deletional HbH disease usually presents with mild anemia (Hb 90–110 g/L), a reduced MCV (50–65fl) and is more likely to have an intermedia phenotype requiring transfusion during times of physiological stress such as pregnancy or in a perioperative setting. Individuals with deletional HbH disease can deteriorate over time acquiring transfusion dependence. Deletional HbH disease is more common in Asian or African ethnicities. Non-deletional forms tend to have more severe disease with a higher likelihood of transfusion dependence compared to those with deletional forms. The majority of patients with HbH disease develop complications from ineffective erythropoiesis and extramedullary hematopoiesis including mild iron overload, hepatosplenomegaly and gall stones.9,10
Bart’s hydrops
Bart’s hydrops is the absence of all four α chains and is incompatible with life usually results in fetal demise at the end of the second or mid-third trimester. It results from unopposed γ chains forming tetramers called hemoglobin Bart’s. HbBart’s causes markedly impaired tissue oxygenation and as a result the fetus presents with signs of severe anemia, associated cardiac failure and extramedullary hematopoiesis presenting as marked edema, jaundice, ascites and hepatosplenomegaly. It is a common cause of stillbirth and maternal morbidity in South-East Asia due to the higher prevalence of 2 gene α deletion (–SE/aa).11
If both parents have a 2 gene α deletion they have a 25% chance of having a child with Bart’s hydrops and should be offered antenatal screening and termination of an affected pregnancy. Ongoing pregnancy can result in maternal mirror syndrome where the maternal condition “mirrors” the fetal hydropic condition presenting as rapid maternal weight gain and shortness of breath due to severe generalized edema and pulmonary edema. Unmanaged mirror syndrome can result in maternal death.4
FERTILITY AND CONTRACEPTION IN HEMOGLOBINOPATHIES
Women with hemoglobinopathies can have difficulties with fertility and conception due to delayed sexual development. In women with sickle cell anemia ovarian sickling with ischemia and reperfusion injury can occur. Women with a transfusion-dependent hemoglobinopathy and associated iron overload may suffer from hypogonadotropic hypogonadism. Optimization of iron chelation from childhood can help reduce infertility as once fertility is impaired, it is not routinely salvaged with hormone replacement therapy.12
If there is clinical or biochemical evidence of hormonal derangement and/or sub/infertility, referral for further assessment and management is appropriate. Women with central hypogonadism may require ovulation induction which needs to be done at a center of expertise to minimize the risk of ovarian hyperstimulation syndrome and multiple pregnancies. In the fertility setting, pre-pregnancy counseling and partner screening to determine the risk of a child affected by thalassemia major or sickle cell anemia can be discussed and investigated. Postpartum women should be counseled about contraception to avoid early recurrent pregnancy. Progesterone-only contraception should be offered as first-line contraception.13
SICKLE CELL ANEMIA IN PREGNANCY
Pre-conception
Ideally women with sickle cell anemia (SCA) have planned pregnancies with a pre-conceptual discussion around maternal and fetal implications of SCA on pregnancy. If women are presenting for the first time, confirmatory testing for sickle cell anemia should occur. This is also the ideal time to organize partner testing. Where there is a known risk of having a child affected by sickle cell anemia or the maternal risk of carrying a pregnancy is great, other options for having a child can be explored including use of unaffected donor sperm or ovum, pre-implantation genetic diagnosis or surrogacy. In the context of in-vitro fertilization being used, single embryo transfers are recommended to reduce further risk that would result from a multiple pregnancy.4 Immunization status for pneumococcus, influenza and meningococcus should be sought and vaccines updated where appropriate. Pregnancy should be avoided for at least 6 weeks following administration of live vaccines.
Complications in pregnancy
A systematic review and meta-analysis of observational studies quantified the association between sickle cell anemia and adverse pregnancy outcomes. Twenty-one studies with 26, 349 women with sickle cell anemia and associated hemoglobinopathies (HbSS, HbS/β and HbSC) were included. The following maternal and fetal complications were observed:
- Increased maternal mortality – in developed countries rates are approximately <1% compared to underdeveloped countries where rates can be as high as 10% due to lack of antenatal care.
- Increased perinatal mortality – in developed countries rates are approximately 5% with rates being higher in underdeveloped countries.
- Miscarriage rates are increased by approximately 20%.
- Premature delivery is increased with average gestations being between 34 and 38 weeks' gestation. Mothers with HbSS are twice as likely to deliver prematurely compared to HbSC.
- Fetal intrauterine growth restriction is common with approximately 20% of babies being below the 10th centile likely due to vaso-occlusion within the utero-placental circulation resulting in increased placental infarction and abruption.
- Hypertension including both gestational hypertension and pre-eclampsia are increased occurring in approximately one-third of all pregnancies.
- Thrombotic risk is increased in pregnancy and sickle cell anemia can cause persistent platelet and clotting activation. Women should be viewed as high risk for venous thromboembolic disease.
- Infections can be increased due to hyposplenism, but also due to altered complement activation. Urinary tract infections are more common and can result in pyelonephritis. Pneumonia and bony infections also have a higher incidence. All infections increase the risk of painful crises.14,15
Women should commence 5 mg folic acid daily prior to trying to conceive and a discussion of current medications should occur.16 Specifically women on hydroxyurea (HU) should have a discussion involving a multidisciplinary team about the advantages and disadvantages of cessation or continuation in pregnancy. Animal studies have shown potential teratogenic effects with HU taken in higher equivalent quantities; however, where HU has been continued throughout pregnancy, major congenital abnormalities in babies have not been seen. The largest cohort of patients n = 31 resulted in 24 live births with three babies displaying minor anomalies including hip dysplasia, unilateral renal dilation and pilonidal sinus. Fetal growth restriction, fetal demise and preterm delivery were also increased; however, given it was an observational study, it is not known if these complications were secondary to SCA or HU.17 In the majority of women HU cessation is advised prior to trying to conceive; however, if it is thought that the severity of SCA complications outweigh the potential risk of HU exposure, then continuation of HU in pregnancy may be warranted. In this setting women require anomaly ultrasonography at a tertiary facility. The use of HU in adolescence is becoming more prevalent with potential implications also for male fertility. A prospective study of SCA males showed increased spermatogenesis abnormalities with HU exposure, although effects on fertility were not studied.18 Case reports have shown abnormalities are partially reversible with 3–6 months of HU cessation. Based on this, male partners with SCA should be counseled in regards to potential cryopreservation of sperm prior to commencing HU, or, if possible, cessation for 3–6 months prior to trying to conceive.19 Drug cessation at least 3 weeks prior to delivery is advised to reduce drug accumulation in the fetus, associated cytopenias and complications including anemia, infections and bleeding.20
Penicillin prophylaxis due to associated hyposlenism is safe to continue and advised.21 Limited studies examining use of iron chelating agents such as desferrioxamine suggest maternal use for iron overload has not been associated with an increased risk of congenital malformations or adverse pregnancy outcomes.22 It may increase the risk of iron deficiency anemia in the newborn.23 If iron chelating agents are considered vital treatment in pregnancy, close monitoring of maternal and fetal well-being is recommended (see below). Women on angiotensin converting enzyme (ACE) inhibitors or angiotensin receptor blockers for management of hypertension or proteinuria can safely cease these once pregnant.24 In hypertension, women should be transitioned to more suitable medications such as methyldopa or other medications recommended by the International Society for the Study of Hypertensive disorders in Pregnancy (ISSHP), prior to trying to conceive.25 Aspirin 150 mg nocte should be commenced early in the first trimester to ameliorate risk of early onset pre-eclampsia.26
Monitoring – maternal and fetal
The booking visit should occur in the first trimester, ideally before 12 weeks' gestation. At the booking visit a discussion of pregnancy and associated potential complications should occur. If pre-pregnancy counseling and assessment has not occurred, it should happen at this visit including baseline testing as summarized below and a review of current medications. Early involvement with a hematologist and obstetrician with expertise in managing hemoglobinopathies should be sought and consideration should be given to the institution where the patient will have antenatal care and birth. Women will have improved pregnancy outcomes with early involvement of a multidisciplinary team with expertise managing sickle cell anemia.27
A clinical history including frequency of crises, management of crises, transfusion history, organ function and previous pregnancies and outcomes should be undertaken. A plan should be documented that addresses how to initially manage a crisis including intravenous fluid regimens and analgesia, who should be contacted, what investigations should be performed and thresholds for considering blood transfusion. Women should be counseled and educated about signs and symptoms of infection and the need for early presentation if they are unwell. An ultrasound should also be undertaken at this stage to check for an ongoing viable pregnancy.
As well as regular prenatal investigations, the following testing should be undertaken, if not already performed preconceptually, to determine severity of sickle cell anemia and baseline function of organs including:
- Blood pressure;
- Transthoracic echocardiogram to assess cardiac function and to look for evidence of pulmonary hypertension;
- Visual acuity and retinal screening;
- Renal function including urinalysis, biochemistry panel, proteinuria and urine culture;
- Hemoglobin and HbS levels;
- Ferritin levels;
- Baseline pulse oximetry;
- Red blood cell serology and phenotyping to identify any potential red cell alloimmunization. If antibodies are identified, partners should undergo red cell phenotyping to determine potential risk of hemolytic disease of the newborn;
- Partner testing for hemoglobinopathy;
- Immunization status for pneumococcus, influenza and meningococcus should be sought and vaccines updated where appropriate.28
Ongoing management
Women should continue on folate supplementation. Women should be educated to have a low threshold to present to hospital if they have limb, abdominal or chest pain especially in the last trimester where the majority of sickle cell crises occur. There should be a clear medical alert documented in patient notes indicating the diagnosis of sickle cell anemia. There should also be a clear treatment plan in patient notes and ideally carried by the patient as well for action to be taken if a crisis is suspected including medical contact for advice, initial IV fluid usage, analgesia and threshold for blood transfusion or exchange transfusion. Nonsteroidal anti-inflammatories should be avoided before 12 weeks and should not be given after 32 weeks due to premature closure of the patent ductus arteriosus in the fetus.29
Opioid-based analgesia can be given, but requires close monitoring ideally with guidance from anesthetics or a pain team. If the patient is admitted or additional risk factors are present, thromboprophylaxis with low molecular weight heparin should also be initiated to reduce risk of thrombosis. Episodes of hyperemesis should prompt admission for aggressive antiemetic and intravenous fluid hydration to avoid a sickle cell crisis.28
Women should be seen regularly with checks of fetal growth from 20 weeks' gestation. If possible, ultrasonography can be used to assess placental function as well as fetal growth.30 A full blood count should be done monthly to assess Hb and platelet levels as well monthly midstream urine culture to screen and pre-empt urinary tract infections in pregnancy as they can precipitate a sickle crisis as well.
Routine prophylactic transfusion has been shown in a systematic review to reduce maternal mortality, vaso-occlusive episodes, pulmonary complications, neonatal mortality and pre-term birth.31 Regular transfusion is not without risk including alloimmunization, isoimmunization, transmission of infection and iron overload. Benefit to women who have not previously received prophylactic transfusion has not been proven in randomized controlled trials and is not routinely recommended.28
In women who routinely receive exchange transfusion or transfusion outside of pregnancy, consideration should be given to continue these regimens with the patient’s regular hematologist. There is no definite threshold of hemoglobin for transfusion in sickle cell anemia in pregnancy and advice should be sought from a hematologist. Below are listed reasonable indications for blood transfusion:
- Hb ≤60 g/L;
- Hb fall ≥20 g/L;
- Women with previous serious medical, obstetric or fetal complications;
- Women on a regular transfusion regimen prior to pregnancy for prevention of stroke or other sickle cell complications should have their regular transfusion schedule maintained;
- Twin pregnancies;
- Acute chest syndrome or acute stroke – exchange transfusion is advised.
The risk of red cell isoimmunization occurs in approximately 10–20% of women with sickle cell disease.4 It can cause hemolytic disease of the newborn and delayed hemolytic transfusion reactions in the mother. The most common red cell antibodies are to C, E and Kell antigens and blood should be phenotyped to match maternal C, E and Kell antigen status. Blood should be leucocyte depleted and CMV negative to reduce risk of infective transmission through transfusion.32
Labor and delivery
Delivery need not be pre-empted, and timing and type of delivery should be based on obstetric indications, although planned delivery may be necessary in women who are regularly transfused or have regular exchange transfusion so that they can labor when their Hb is optimized. Sickle cell anemia does not preclude vaginal birth or vaginal birth after cesarean section.33 Home birth is not advised.
Women need to be kept warm and well hydrated with intravenous fluids throughout labor and postpartum. Monitor strict fluid input and urine output. Check full blood estimation/count (FBE), blood group and antibody screen on presentation. Continuous cardiotocography monitoring is advised because of an increased risk of fetal distress, stillbirth, placental abruption and compromised placental reserve.34 Epidural analgesia is reasonable to use if desired. Labor and rupture of membranes should not be prolonged to minimize risk of complications. If cesarean section is the required mode of delivery, anesthesia should be with neuroaxial rather than general anesthetic to reduce risk of vasocclusive crises.35 Women should receive thromboprophylaxis with low molecular weight heparin for 6 weeks postpartum and should wear graduated compression stockings. Women with known avascular necrosis of femoral heads or hip replacements should be counseled about birthing positions that will be suitable given limitations of hip movement.
A pediatrician should be alerted if maternal opioids have been administered regularly and for substantial duration for pain in the pregnancy to monitor for clinical evidence of neonatal abstinence syndrome.36 Postpartum women should have their observations and pain monitored regularly for the first 24 hours. Good hydration and oxygenation should be maintained in the postpartum period and early mobilization is advised. If hydroxyurea is to be restarted, women should be counseled in regards to breastfeeding. A single case report showed neonatal cytopenia; however, the Hydroxyurea Exposure in Lactation: a Pharmocokinetics Study (HELPS) showed breastfeeding mothers will transfer only a small amount of hydroxyurea to their infants, so lactation during hydroxyurea therapy should not be absolutely contraindicated.37,38 Women should have routine postnatal and neonatal follow-up as well as checking in with their regular hematologist.
Management of sickle cell crisis in pregnancy
Sickle cell crises usually occur in the third trimester with approximately one-third or more of women with HbSS and HbSC experiencing a crises in pregnancy. Often it is complications from sickle cell disease that precipitate labor and delivery rather than delivery itself causing complications. However, the process of labor can be a precipitant due to the increased risk of dehydration, infection and acidosis.
Pregnant women who are thought to be having a sickle cell crisis need to be admitted to hospital; the obstetric ward is preferable in the final trimester to monitor for obstetric complications including labor. The treating hematology and obstetric teams need to be informed with multidisciplinary involvement of ongoing care. Women need to warmed and rested and oxygenation status should be monitored via oxygen saturations. If women are hypoxic, supplemental oxygen is required (aim to keep SaO2 >94%). Intravenous hydration should be commenced and 3–4 liters of fluid should be given over a 24 hour period initially with close monitoring of fluid balance. Pain relief can initially be with milder analgesics such as paracetamol but opiate-based pain relief is often necessary. If available, anesthetic referral may help in optimizing analgesia in women who have a previous history of significant opioid usage due to frequent crises. Regular pain control assessment using a linear scale and assessment of sedation and conscious levels are required and should be undertaken as part of regular observations. Investigation for cause and evidence of hemolysis, e.g. sepsis, dehydration should be sought including FBE, reticulocytes, urea/electrolytes/creatinine (UEC), blood group and antibody screen, blood cultures, urine culture and chest X-ray. Antibiotic therapy should be reserved for patients presenting with fever and a clinical history consistent with infection, e.g. if a women presents with a crisis and has fever and dysuria, management for Gram negative organisms that cause a urinary tract infection would be appropriate. Discussion with a hematologist for either transfusion or exchange transfusion is appropriate. Low molecular weight heparin thromboprophylaxis should be given.
Chest physiotherapy with incentive spirometry can help in acute chest syndrome.39 Chest X-ray changes in acute chest syndrome are characterized by pulmonary infiltrates, chest pain, shortness of breath and fever. Despite the pathology being vascular in origin, it can be misdiagnosed as either infection or pulmonary edema and responds to oxygen therapy and transfusion. ‘Top-up’ transfusion aiming for a Hb level of 90–100 g/L is reasonable with milder crises, or anemia of Hb ≤60 g/L. In a severe chest crisis (SaO2 reduction >5% baseline level, deteriorating clinical status or progression to multi-lobe involvement), exchange transfusion is appropriate. Performing early limited manual exchange transfusion is preferable compared to waiting for experienced staff and equipment to run an automated exchange.40
THALASSEMIA IN PREGNANCY
Women with α and β minor forms and those with transfusion-independent HbH disease usually can be managed as a normal pregnancy. Normal physiological changes of pregnancy can worsen anemia and the risk of iron deficiency anemia can be increased. Oral iron supplementation should be given if ferritin levels are low (<30 ng/ml).
Women with β-thalassemia major forms or transfusion-dependent intermedia forms are clinically significant in pregnancy and require multidisciplinary management. Favorable pregnancy outcomes are documented in women with thalassemia major.41
Pre-pregnancy
Women with thalassemia major require assessment prior to falling pregnant to optimize current management and plan for changes in management during pregnancy. Assessment of the following should occur:
- Cardiac function – transthoracic echo, ECG, T2 MRI quantification of cardiac iron if possible;
- Hepatic function – liver function, liver and gall bladder ultrasonography and T2 MRI iron quantification if possible;
- Endocrine function for assessment of glucose tolerance with diabetes optimization if present, thyroid function, vitamin D levels and bone density with DEXA (dual-energy X-ray absorptiometry) scan;
- Iron overload status and chelation regimen – if well-controlled chelation therapy may be ceased while trying to naturally conceive;
- Fertility – including menstrual history, hormonal assays, pelvic ultrasonography;
- Rubella, HIV, hepatitis B and C, syphillis serology;
- Smoking cessation;
- Medication regimen.4
Women should commence high dose folic acid 5 mg daily orally to reduce risk of spina bifida and reduce the severity of hemolytic anemia. If women are on ACE-inhibitors or angiotensin receptor blockers for control of hypertension, these should be changed to a ‘pregnancy-safe’ option such as methyldopa. Iron chelation therapy ideally should be ceased in all women in pregnancy, but reports of safe iron chelation in pregnancy have been reported. If the benefits of iron chelation therapy outweigh the risks (usually due to excessive cardiac iron and impaired cardiac function), this should be a decision that occurs between the obstetrician and hematologist. Restarting desferrioxamine during the second and third trimester may be reasonable as seen in a two different case series of 32 and 31 women, respectively, who were chelated in the second and third trimester with favorable fetal outcomes documented.42,43
Women should be counseled in regards to potential pregnancy complications including:
- Impaired fertility due to hypogonadotropic hypogonadism from pituitary hemosiderosis;44
- Partner screening and antenatal testing if desired;
- Higher cardiac outputs induced by pregnancy can result in decompensation and mortality if there is known severe cardiac dysfunction secondary to excessive iron deposition within the myocardium from chronic transfusion. Women with ferritin levels >2500 ug/L are at higher risk of cardiac complications and should delay pregnancy until iron chelation therapy has reduced ferritin levels to an acceptable level.45
- Increased transfusion requirements;
- Increased risk of gestational diabetes due to pre-existing insulin resistance from pancreatic iron overload;
- Increased risk of osteoporosis due to higher rates of pre-existing osteoporosis and osteopenia;
- Acceleration of pre-existing retinopathy and nephropathy if women have pre-existing associated diabetes mellitus;
- High incidence of operative delivery.
Management in pregnancy
Early pregnancy
Women should have an early booking appointment with early commencement of multidisciplinary care to optimize patient and fetal management. Assessment of FBE should be performed at least monthly. Pregnant women need to be aware that pregnancy increases oxygen carrying demands with a 30% increase in red cell mass and a doubling of plasma volume to ensure optimal oxygenation to the developing fetus. Hemoglobin levels are usually maximally diluted in the second trimester and iron demands increase with each trimester increasing the risk of anemia as pregnancy progresses. As a result Hb levels can decrease and transfusion requirements can increase. Women with HbH disease who have previously not required transfusion may require it in pregnancy. Hb threshold for transfusion in pregnancy in women with thalassemia major is usually unchanged compared to non-pregnant thresholds of 100 g/L. This has also been thought to ensure appropriate fetal growth.46 In HbH disease where there is not a prior history of transfusion dependence this threshold is usually <80 g/L – in conjunction with symptomatic anemia. A report on 129 pregnancies in 72 women with thalassemia major and intermedia syndromes (β-thalassemia major n = 28 (42.4%), HbH n = 6 (9.1%) and HbH-Constant Spring) n = 1 (1.5%) over 50% of pregnancies resulted in live births and 88% of live births occurred at full term. Approximately 40% of women received iron chelation therapy and many were transfused throughout pregnancy.41
Renal and liver function (UECs and LFTs) should be performed at booking to check baseline renal and hepatic function. Blood group and antibody screen should be done at booking and as per routine antenatal care and pre-transfusion. Alloimmunization to blood antigens can result in increased risk of hemolytic disease of the fetus and newborn and make cross-matching compatible blood difficult when required.
Cardiac function and iron levels should be assessed. Whilst T2 MRI is the investigation of choice to assess cardiac iron status, it is not readily available in many countries. In women with severely reduced left ventricular function, expansion of plasma volume, and raised cardiac output can precipitate cardiac failure in pregnancy. Women identified as having moderately reduced left ventricular function should be monitored with serial echocardiography in each trimester and regularly reviewed by cardiologists with expertise in pregnancy where possible. Women with known pregestational diabetes mellitus and/or thyroid dysfunction need early endocrinology review and management as per standard recommendations for these conditions in pregnancy. Screening for gestational diabetes should be performed in women with pre-existing impaired glucose tolerance.44
Medications
High-dose folic acid of 5 mg daily should be continued throughout pregnancy. Penicillin prophylaxis can be continued if there is a past history of splenectomy or hyposplenism. Women should not be on ACE-inhibitors or bisphosphonates and consideration should be given to cease iron chelating agents during pregnancy. Despite increasing iron demands in pregnancy, due to increased physiological demands in pregnancy, transfusion requirements also increase with a resultant increase in serum ferritin.44 Vitamin D and calcium therapy should be commenced if there is known reduced bone density prior to pregnancy.
Ongoing management
Women should be advised not to smoke and to take regular exercise (brisk walking for approximately 20 minutes a day is sufficient). Pre-existing back pain will likely be exacerbated in pregnancy. Physiotherapy, compression and analgesia can help relieve symptoms. Thrombosis risk is increased in women with a past history of splenectomy. Thromboprophylaxis should be offered to these women and any woman with thalassemia major whilst they are an inpatient or have additional risk factors.47
Labor and delivery
If there are no contraindications, delivery should be based on obstetric indications. Appropriateness of vaginal delivery should be considered in women with reduced cardiac function and pelvic bony abnormalities especially in women with small stature due to thalassemia major with a normally grown fetus.44
Breastfeeding
Women with hemoglobinopathies should be encouraged to breastfeed when there is no contraindication. Penicillin prophylaxis can be safely continued in breastfeeding. Women on iron chelation therapy prior to pregnancy should be encouraged to resume this postpartum. Desferrioxamine has good safety data in breastfeeding. Deferiprone and deferasirox should not be used until cessation of breastfeeding.44 Whilst there iare few data on the majority of ACE inhibitors in breastfeeding, enalapril is excreted into breast milk in small amounts and has not been found to be harmful in breastfed infants and is considered safe to use.48 Women on HU pre-pregnancy should not breast feed if this is to be restarted. Bisphosphonate therapy should not be commenced until breastfeeding has ceased due to likely excretion into breastmilk and potential adverse bone remodeling effects in the infant. Vitamin D and calcium supplementation can be continued safely.
CONCLUSION
Whilst women with hemoglobinopathies are challenging to manage in pregnancy, with pre-pregnancy optimization and good multidisciplinary care during pregnancy and postpartum, good maternal and neonatal outcomes can be achieved.
PRACTICE RECOMMENDATIONS
- Women with hemoglobinopathies should receive pre-pregnancy counseling including baseline investigations and medication optimization prior to conception.
- Women with hemoglobinopathies should have planned pregnancies to allow early monitoring and intervention if required.
- Women with hemoglobinopathies that have previously been regularly transfused or received exchange transfusion for control of complications should have this continued throughout pregnancy.
- Acute sickle cell crises should be investigated for etiology. Management requires IV hydration, analgesia, thromboprophylaxis and consideration for transfusion.
- Transfusion-dependent thalassemia major requires multidisciplinary management in pregnancy.
- Women with hemoglobinopathies can labor spontaneously and have vaginal deliveries.
- Women with hemoglobinopathies should be encouraged to breastfeed once current medication is reviewed.
CONFLICTS OF INTEREST
The author(s) of this chapter declare that they have no interests that conflict with the contents of the chapter.
Feedback
Publishers’ note: We are constantly trying to update and enhance chapters in this Series. So if you have any constructive comments about this chapter please provide them to us by selecting the "Your Feedback" link in the left-hand column.
REFERENCES
Hoffbrand AV, Moss PAH, Pettit JE. Essential haematology. 5th Singapore: Blackwell Publishing Ltd, 2007. | |
Bain BJ. Haemoglobinopathy Diagnosis. 2nd Malaysia: Blackwell Publishing Ltd, 2010. | |
Angastiniotis M, Modell B, Englezos P, et al. Prevention and control of haemoglobinopathies.Bull WHO 1995;73:375-386 | |
Pavord S, Hunt B. The Obstetric Hematology Manual. United States of America: Cambridge University Press, 2010. | |
Vrettou C, Kakourou G, Mamas T. Prenatal and preimplantation diagnosis of hemoglobinopathies. Int J Lab Hem 2018;40(suppl.1):74–82. | |
Ware RE, de montalembert M, Tshilolo L, et al. Sickle cell disease. Lancet 2017:390;311–23. | |
Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anemia: a multi-center, randomized controlled trial (BABY HUG). Lancet 2011;377:1663–72. | |
Olivieri NF. The beta-thalassemia’s. N Engl J Med 1999;341:99–109. | |
Lal A, Goldrich ML, Haines DA, et al. Heterogeneity of hemoglobin H disease in childhood. N Engl J Med 2011:364;710–8. | |
Sheeran C, Weekes K, Shaw J, et al. Complications of HbH disease in adulthood. Br J Haematol 2014:167;136–9. | |
Fucharoen S, Winichagoon P. Haemoglobinopathies in Southeast Asia. Indian J Med Red 2011;134:498–506. | |
Albu AI, Albu D. Hypogonadism in female patients with beta thalassemia major. Thalassemia and other hemolytic anemias. London (UK): IntechOpen, 59–71, 2018. | |
Manchikanti GA, Grimes DA, Lopez LM, et al. Steroid hormones for contraception in women with sickle cell disease. Cochrane Database Syst Rev 2007;(2):CD0006261. | |
Oteng-Ntim E, Meeks D, Seed PT, et al. Adverse maternal and perinatal outcomes in pregnant women with sickle cell disease: a systematic and meta-analysis. Blood 2015:125;3316–25. | |
Boafor TK, Olayemi E, Galadanci N, et al. Pregnancy outcomes in women with sickle-cell disease in low and high income countries: a systematic review ad meta-analysis. BJOG 2016:123;691. | |
ACOG Committee on Obstetrics. ACOG Practice Bulletin No. 78: Hemoglobinopathies in pregnancy. Obstets Gynecol 2007;109:229. Reaffrimed 2018. | |
Gellen-Dautremer J, Le Jeune S, Receveur M-C, et al. Hydroxyurea exposure throughout pregnancy in Pateints with Sickle-Cell Disease: 4 case reports from Europena Non-Interventional, Multicentric, Prospective Escort-HU Study. Blood 2019;134(Supplement 1):1–27. | |
Berthaut I, Bachir D, Kotti S, et al. Adverse effect of Hydroxyurea on spermatogenesis in patients with sickle cell anemia after 6 bmonths of treatment. Blood 2017;130(21):2354–6. | |
Elalfy MS, Ebeld FSE, Elgamry IR, et al. Hazardous Influence of Hydroxyurea on Spermatogenesis in Thalassemia Intermedia Patients: An Egyptian Cohort Study. Journal of Drug Metabolism and Toxicology 2018;9(2):237–41 | |
Amant F, Han SN, Gziri MM, et al. Chemotherapy during pregnancy. Curr Opin Oncol 2012:24;580–6. | |
Working party of the British Committee for Standards in Haematology Clinical Haematology Task Force. Guidelines for the prevention and treatment of infection in patients with an absent spleen. BMJ 1996;312:430–4. | |
Diamantides MD, Neokleous N, Agapidou A, et al. Iron chelation therapy of transfusion-dependent Beta-thalassemia during pregnancy in the era of novel drugs: is deferasirox toxic? Internation Journal of Hematology 2016;103:537–44. | |
Pearson HA. Iron Studies in infants born to an iron overloaded mother with beta-thalssemia major: possible effects of maternal desferrioxamine therapy. J Pediatr Hematol Oncol 2007;29(3):160–2. | |
Bateman BT, Elisabetta P, Desai RJ, et al. Angiotensin-Converting Enzyme Inhibitors and the Risk of Congenital Malformations. Obstet Gynecol 2017;129(1):174–84. | |
Friedman JM. ACE-inhibitors and congenital anomalies. N Engl J Med 2006;354:2498–500. | |
Duley L H-SD, Meher S, King JF. Antiplatelet agents for preventing pre-eclampsia and its complications. Cochrane Database of Systematic Reviews 2007. | |
Asare EV, Olayemi E, Boafor T, et al. Implemetation of multidiscplinary care reduces maternal mortality in women with sickle cell disease living in low-resource setting. Am J Hematol 2017;92(9):872–8. | |
Royal College of Obstetricians and Gynaecologists. Green-top Guideline No. 61. London: RCOG, 2011. | |
Koren G, Florescu A, Costei AM, et al. Nonsteroidal antiinflammatory drugs during third trimester and the risk of premature closure of the ductus arteriosus: a meta-analysis. Ann Pharmacother 2006;40(5):824–9. | |
Royal College of Obstetricians and Gynaecologists. The investigation and management of the small-for-gestational age fetus. Green-top Guideline No. 31. London: RCOG, 2002. | |
Malinowski AK, Shehata N, D’Souza R, et al. Prophylactic transfusion for pregnant women with sickle cell disease: a systematic review and meta-analysis. Blood 2015;126(21):2424–35. | |
Patient Blood Management Guidelines: Module 5 Obstetric and Maternity. National Blood Authroity 2015. | |
Jain D, Colah R, Lodha P. Sickle cell Disease and Pregnancy. Mediterr J Hematol Infect Dis 2019;11(1):e2019040. | |
Anyaegbunam A, Morel MI, Merkatz IR. Antepartum fetal surveillance tests during sickle cell crisis. Am J Obstet Gynecol 1991;165:1081–3. | |
Bakri M, Ismail EA, Ghanem G, et al. Spinal versus general anesthesia for Ceserean section in patients with sickle cell anemia. Korean Journal of Anesthesiology 2015;68(5):469–75. | |
Shirel T, Hubler CP, Shah R, et al. Maternal opioid dose is associated with neonatal abstinence syndrome in children born to mothers with sickle cell disease. Am J Hematol 2016;91(4):416–9. | |
Sylvester RK, Lobell M, Teresi ME, et al. Excretion of Hydorxyurea into milk. Cancer 1987;60(9):2177–8. | |
Ware R, Marahatta A, Ware J, et al. Hydroxyurea Exposure in Lactation – a Pharmockinetics Study (HELPS). Blood 2018;132(suppl_1):3677. | |
Bellet PS, Kalinyak KA, Shukla R, et al. Incentive spirometry to prevent acute pulmonary complications in sickle cell diseases. New England Journal of Medicine 1995;333:699–703. | |
Sickle cell disease: when and how to transfuse. In: Howard J. (ed.) Haematology Am Soc Hematol Educ Program 2016;(1):625–31. | |
Thompson AA, Kim HY, Singer ST, et al. Thalassemia Clinical Research Network. Pregnancy outcomes in women with thalassemia in North America and the United Kingdom. Am J Hematol 2013;88(9):771–3. | |
Kumar RM, Rizk DE, Khuranna A. Beta-thalassemia major and successful pregnancy. J Reprod Med 1997;42(5):294–8. | |
Pregnancy in Thalassemia and Sickle Cell Disease: The Experience of an Italian Thalassemia Center. In: Sorrentino F, Maffei L, Caprari P, et al. (eds.) Front Mol Biosc 2020;7(16):1–6. | |
Origa R, Comitini F, Pregnancy in Thalassemia. Mediterr J Hematol Infect Dis 2019;11(1):e2019019. | |
Olivieri NF, Nathan DG, MacMillan JH, et al. Survival in medically treated patients with homozygous beta-thalassemia. New England Journal of Medicine 1994;331:574–8. | |
Origa R, Piga A, Quarta G, et al. Pregnancy and β-thalassemia: an Italian multicenter experience. 2010;95:376–81. | |
Pregnancy in Thalassemia and Sickle Cell Disease: The Experience of an Italian Thalassemia Center. In: Sorrentino F, Maffei L, Caprari P, et al. (eds.) Front Mol Biosc 2020;7(16):1–6. | |
Beardmore KS, Morris JM, Gallery ED. Excretion of antihypertensive medication into human breast milk: a systematic review. Hypertens Pregnancy 2002;21(1):85–95. |
Online Study Assessment Option
All readers who are qualified doctors or allied medical professionals can automatically receive 2 Continuing Professional Development points plus a Study Completion Certificate from GLOWM for successfully answering four multiple-choice questions (randomly selected) based on the study of this chapter. Medical students can receive the Study Completion Certificate only.
(To find out more about the Continuing Professional Development awards programme CLICK HERE)