Mendelian Inheritance and Its Exceptions
Authors
INTRODUCTION
Mendel's Laws and Principles of Inheritance
Our basic laws of inheritance were derived from a simple series of experiments with garden peas more than a century ago. Gregor Mendel crossed various pure lines of garden peas and, by following their hybrid progeny, observed that traits are inherited as alternate states of independent units of inheritance or genes (which Mendel called “factors”), and that these units come in pairs. Each unit of inheritance can have alternate states (alleles) that segregate at meiosis, with each gamete receiving only one allele (the principle of segregation, Mendel's first law); different alleles assort independently in the gametes (the principle of independent assortment, Mendel's second law). Different alleles can exert different phenotypic effects; broadly speaking, most genes are either dominant or recessive. For instance, if the phenotypes produced by the combinations AA and AB are the same, then A is dominant to B (or conversely, B is recessive to A). The effects of allele B in this case are apparent only in the homozygous state (BB). When neither allele exerts a stronger effect, both are considered codominant, and the offspring may show the phenotypic features of both alleles, as is the case in individuals with type AB blood, who have features of blood groups AA and BB. If the offspring have an intermediate phenotype, such as moderate height in an individual born to a very tall and a very short parent, the alleles are considered semidominant.1, 2
As advances in genetics have confirmed and illuminated the mechanisms underlying Mendel's observations, we have also discovered the need to adapt and modify his principles. Exceptions to Mendel's laws of inheritance are described later in this chapter.
The Molecular Basis of Genetic Inheritance
Our genome is the blueprint for all cellular structures and activities and is stored in the nucleus of every cell. It is made up of tightly wound strands of deoxyribonucleic acid (DNA) organized, in humans, into 23 pairs of chromosomes: 22 autosome pairs (numbered 1–22) and one sex chromosome pair (XX in females and XY in males). The basic form of a DNA molecule is that of a twisted ladder or double helix. Each strand of the helix is a linear arrangement of repeating units called nucleotides that consist of one sugar, one phosphate, and a nitrogen-containing molecule called a base. There are four possible bases—adenine (A), guanine (G), cytosine (C), and thymine (T)—and it is the order of these bases along the sugar-phosphate backbone that makes the DNA sequence. This sequence specifies the genetic instructions required to create a protein and, ultimately, to create an entire organism. In the DNA double helix, the bases are always paired (T with A, and G with C) and often are referred to as base pairs.
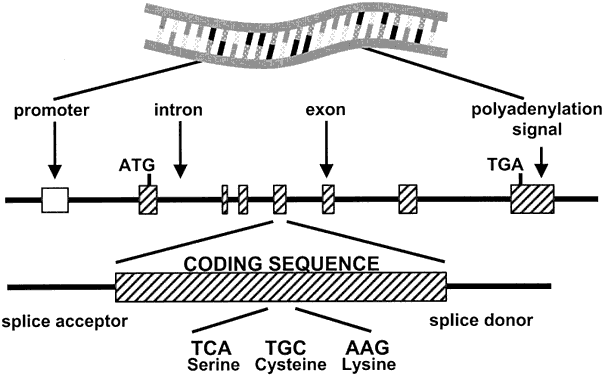
As shown in Fig. 1, genes are composed of exons (coding regions) linked by introns (noncoding regions). Specific triplets of nucleotides encode different amino acids, which are the building blocks of the gene products (proteins). During transcription of the DNA, the introns are removed and the coding exons are spliced together to form a messenger RNA (mRNA) that is an exact mirror image of the successive triplet codons in the exons. The mRNA is exported out of the nucleus into the cytoplasm, where triplet codons are translated into the amino acids of the protein on the ribosomes. Variations in the nucleotide sequence of DNA are common, and when they occur in introns or intervening sequence, they usually are silent. When they affect the coding or regulatory regions of genes, however, they can lead to a change in the gene's function. Copies of specific genes with such a difference in nucleotide sequence are called alleles. In general, each individual carries only two copies of an autosomal gene, and therefore only two alleles, but many different alleles can exist in the population. By definition, nondeleterious alterations, or sequence variants, in alleles occurring in more than 1% of the population are called polymorphisms. When the altered nucleotide responsible for the allelic difference is part of a triplet codon in a coding exon, it can cause a mutation or deleterious change to the amino acid sequence of the resulting protein. In contrast to the normal or wild-type copy, the allele carrying such a change is called the mutant allele. For instance, the mutant allele of the β-globin gene has an A-to-T transition at codon 6, which causes the amino acid glutamine to be replaced by valine at position 6 of the protein (Glu6Val); this one change leads to sickle cell hemoglobin (HbS). Such small-scale changes of only one or a few nucleotides, referred to as point mutations, are a typical cause of Mendelian disorders. When the nucleotide change results in a codon for a different amino acid, as with the sickle cell disease mutation in β-globin, it is called a missense mutation. A nonsense mutation is a nucleotide change resulting in a stop codon (TGA or TAA) that signals the ribosome to stop translating the mRNA and thus truncates the protein. A deletion or insertion of one or two base-pairs (or another number that cannot be divided by three) shifts the reading frame of the mRNA and changes an entire series of amino acids until a stop codon is reached in the novel reading frame (frameshift mutation).1, 2 In general, once a de novo mutation has occurred, it is stably inherited with each mitotic and meiotic cell division, but exceptions to this rule exist (see Somatic Mutations and Dynamic Mutations).
|
TYPES OF MENDELIAN INHERITANCE
To study inheritance patterns in families and uncover possible genetic risk factors and disorders, we recommend constructing a pedigree as a normal part of history-taking for every new patient (Fig. 2). Pedigrees are certainly an essential component of the first prenatal visit but it is also important to seek updated information at follow-up visits to amend the pedigree. With the current pace of advances in human genetics, more and more disease-causing genes will become known, resulting in increased opportunities to offer (prenatal) diagnosis and appropriate interventions.
|
Autosomal-Dominant Inheritance
Autosomal-dominant (AD) disorders are manifested in the heterozygous state. The effect of the mutant allele is so profound that the normal allele cannot compensate, and clinical disease ensues. Fig. 3A illustrates a typical AD pedigree. Note that transmission of the disease appears vertical, with each successive generation being affected. Because only one of the two alleles is abnormal and the alleles segregate at meiosis, an individual has a 50% chance of transmitting the mutation to each offspring. This transmission/inheritance is independent of the sex of the transmitting parent and the offspring: both males and females have an equal chance of being affected. Parents often believe that if they have one child who is affected, the next child is more likely to be healthy; they must be carefully educated to understand that each child independently has a 50% chance of inheriting the mutation.
|
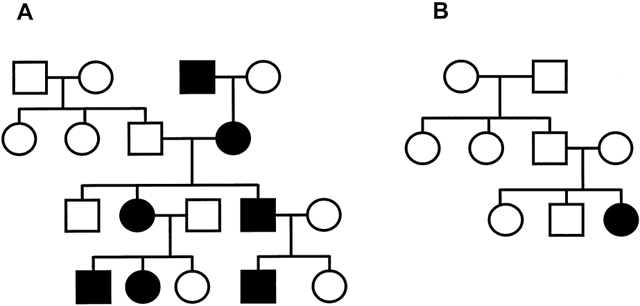
If there is at least one instance of male-to-male transmission, the familial inheritance pattern is most likely to be AD. For some AD diseases, such as achondroplasia, there is a high spontaneous new mutation rate, which can result in an isolated case in a family (see Fig. 3B). Although the siblings of such an isolated case obviously will not transmit the disease, the individual affected with achondroplasia has a 50% risk of transmitting the mutation to his or her child. New mutations are more common in the male germ cells because they undergo a greater number of cell divisions than do female germ cells, and disorders like achondroplasia are often seen with advanced paternal age.
If the homozygous state causes a more severe phenotype, as it does in achondroplasia, then the condition is actually semidominant. Semidominant disorders are often perceived as dominant because of the rarity of the homozygous case. Two people with achondroplasia have a 25% risk of having a son or daughter who is homozygous for the disease-causing mutation in the fibroblast growth factor receptor (FGFR3) gene.
Because of the nature of germ cell proliferation, mutations more commonly arise at later cell divisions, but they can occur in earlier divisions and result in a number of gametes with the same mutation in a phenomenon known as gonadal or germline mosaicism. Type II osteogenesis imperfecta (OI), a lethal skeletal disorder with decreased ossification and severe dwarfism, is a classic example of such a condition. The possibility of germline mosaicism is why an empiric recurrence risk of 6% is quoted to parents with a previously affected child, although more recent research3 has resulted in identification of recessive OI forms.
Penetrance and expressivity
For disorders such as achondroplasia, all heterozygous individuals have an equally severe phenotype. Many AD conditions, however, can have different degrees of severity or variability in the phenotype, even among individuals who possess identical mutations. For example, different members of a family with AD retinitis pigmentosa may have different degrees of visual dysfunction. This variable expressivity can be caused by the influence of environmental conditions and/or the function of genes at other loci, called genetic modifiers. Penetrance, often confused with variable expressivity, indicates the proportion of heterozygous individuals who display any phenotypic manifestations of a given mutation. The concept is used most often clinically when the precise genetic mutation is unknown. Reduced or incomplete penetrance means that a mutation is evident in only a certain percentage of all individuals who carry it. An adult male with the full phenotypic manifestations of Marfan syndrome may have a daughter who is apparently unaffected; yet this daughter produces a grandson who also manifests the disease. In this case, the mother's mutation is said to be nonpenetrant. Penetrance is often associated with a percentage: 80% penetrance means that a given mutation will manifest itself approximately 80% of the time. The other 20% of individuals bearing the mutation will show no evidence of it. Thus, whereas variable expressivity connotes gradations in severity of phenotype, penetrance is an all-or-nothing phenomenon: someone either will or will not manifest clinical disease.
Pleiotropism
Pleiotropism occurs when a single gene exerts multiple and seemingly unrelated effects in different systems. Marfan syndrome, for example, causes skeletal, ocular, and cardiovascular defects. Each separate phenotypic manifestation of an allele can show variable expressivity (mild-to-severe aortic root dilation in Marfan syndrome) and nonpenetrance (not all patients have ectopia lentis).
Loss of function
Loss-of-function mutations usually become manifest only in the recessive state and are apparent only in AD conditions when they affect a protein for which dosage is critical; for example, the protein product might be the rate-limiting step in a metabolic pathway or have a regulatory role. Many AD mutations do not cause loss of function and are deleterious by other mechanisms. When the mutated protein is a structural protein or functions in a multiprotein complex, the mutations can alter the protein conformation and disrupt interactions with other molecules, leading to dysfunction of the entire complex in a dominant-negative manner (e.g., collagen chain mutations in AD osteogenesis imperfecta). Some mutations can cause the protein to acquire a novel behavior, different from that of its wild-type counterpart, and the mutation is referred to as a gain of function mutation. A dominant mutation can also result in the formation of a toxic product that is deleterious to the cell, because the levels of the normally functioning protein increase as the result of a regulatory defect or because the protein has become more resistant to normal degradation.1, 2
Autosomal-Recessive Inheritance
Autosomal-recessive (AR) disorders are clinically apparent only when both alleles at a locus are mutated. When the two alleles carry the same mutation, the individual is a homozygote. When each allele of the gene carries a different mutation, the affected individual is a compound heterozygote. The severity of the phenotype depends on the type of mutation at each of the two alleles. This principle is best illustrated by the mutations in the cystic fibrosis transmembrane conductance regulator gene (CFTR) on chromosome 7. The allele carrying the ΔF508 mutation (deletion of phenylalanine at position 508) is the most common mutant allele in the white population; individuals heterozygous for this mutation are asymptomatic carriers because they have a wild-type allele on the other chromosome 7. Most children born with CF are therefore homozygous for the ΔF508 mutation, but there are more than 100 different mutations in CFTR, and many individuals are compound heterozygotes (e.g. ΔF508/G542X). Furthermore, not all mutations have the same effect on gene function, and compound heterozygotes can display a different or less-severe phenotype: some mutations in CF are associated only with congenital absence of the vas deferens (R117H/ΔF508), whereas others have been found to cause only isolated pancreatic dysfunction or bronchiectasis.
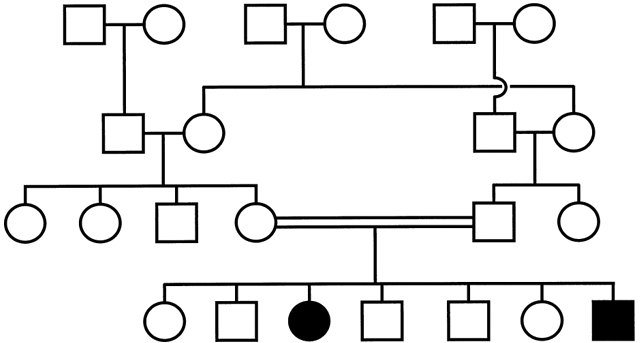
In AR disorders (Fig. 4), both males and females are affected, and the pedigree shows a horizontal pattern of inheritance (i.e. affected siblings). Each parent is an unaffected carrier, and the recurrence risk of the disease for each future child is 25%, while the risk of a child becoming a carrier like the parents is 50%. If the recessive gene is very rare, an affected individual is likely to have been born of a consanguineous mating. In pedigrees with extensive consanguinity, affected individuals can marry heterozygote carriers and have affected children, resulting in a pedigree that resembles that of a dominant condition (pseudodominance).
|
There are many more heterozygotes for a given allele in the population than homozygotes, and the estimation of the chance that a person will be a heterozygote carrier of a recessive disorder is important in counseling situations, such as for siblings of individuals affected with an AR disorder, and their partners. For any disease or trait, an approximation of the frequency of heterozygotes can be obtained by the Hardy-Weinberg (HW) equilibrium equation. In this equation (p2 + 2pq + q2 = 1), p is defined as the frequency of the dominant allele and q as the frequency of the recessive allele for a trait controlled by a pair of alleles. In most practical situations, we know the frequency in the population of the homozygous-recessive individuals or q2. Because the HW equation can also be written as (p + q)2 = 1, we can easily derive that p = 1 - q, and thus the frequency (2pq) of heterozygote individuals. For example, for phenylketonuria (PKU), a common AR disorder, the population incidence (q2) is 1:10,000, leading to a frequency of the recessive allele (q) in the population of 1:100. The allele frequency p is thus 99:100 (or approximately 1) and the number of heterozygote carriers (2pq) in the population is 2 × (1:100) × 1 or 1:50. One can then calculate that a person affected with PKU has a 1 (affected person) × 1:50 (chance the partner is a carrier) × 1:2 (chance that partner passes on the mutant allele) = 1:100. Note that the affected person always transmits a recessive allele in this situation.
X-Linked Inheritance
Because the genes responsible for X-linked (XL) disorders are located on the X chromosome, the risk of inheritance and clinical severity of such diseases differs for males and females. Females have two X chromosomes and can therefore be heterozygous or homozygous for a given XL allele or mutation. Because males are never heterozygous, except for those genes that have a functional Y homologue (see pseudoautosomal region), they invariably manifest the full syndrome when they are hemizygous (e.g. when they inherit the mutant XL disease gene). As is illustrated in the two pedigrees of Fig. 5, the hallmark of XL inheritance is the absence of male-to-male transmission. A heterozygous female has a 50% chance of transmitting the mutation to a son or a daughter, whereas a hemizygous male transmits the mutation to all his daughters but not to his sons.
|
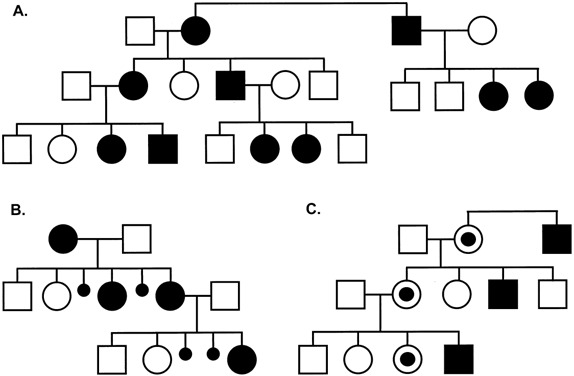
XL inheritance can be dominant or recessive, but the distinction is not always clear. In pedigrees of XL-dominant conditions (see Fig. 5A), both males and females are affected, but the phenotype in males is typically more severe than that of heterozygous females. When the mutation causes loss of function of an essential gene, there may be prenatal male lethality, and no affected males are seen (see Fig. 5B); in this situation, affected women can suffer more pregnancy loss and have fewer liveborn male offspring. With an XL-recessive mutation, heterozygous females are typically unaffected or only mildly affected, whereas hemizygous males have the full phenotype of the condition (see Fig. 5C). Duchenne muscular dystrophy and hemophilia A are typical examples of XL-recessive disorders. Counseling for XL-recessive disorders with an isolated case in a family is difficult, as it may have arisen from a new mutation in the germline of the patient's grandfather or in the germline of his mother. In the latter situation, the recurrence risk would be similar to the population risk, unless there was germline mosaicism. Assuming that there is an equal spontaneous mutation rate in male and female gametes and that affected males do not reproduce, this would occur in approximately one third of the cases, and two thirds of the mothers are carriers. However, for some disorders such as hemophilia A, the new mutation rate in male gametes appears to be higher and the chance that the patient's mother is a carrier is higher than two thirds.
The X chromosome inactivation (XCI) or Lyonization, the irreversible inactivation of one of the two X chromosomes in each somatic cell in females, is at the root of the unique characteristics of XL inheritance. XCI results in silencing of one allele of the majority of genes on the X chromosome in females so that dosage of the products of these genes in males and females is equal. The inactivated X chromosome can be identified as a clump of condensed chromatin, the Barr body. An as-yet incompletely understood counting mechanism ensures that only one X chromosome remains active in each cell (47,XXX females have two Barr bodies per cell). The selection of the active X chromosome is entirely random in females, but once the X is selected in a cell, all its progeny will have the same X active, resulting in a mosaic pattern of cells with either one of the two X chromosomes active in a roughly 50:50 distribution. Skewed XCI results in a higher proportion of cells with one particular X active. Skewed XCI can occur by chance or in monozygotic twinning and may lead to females that express the phenotype of XL-recessive disorders (e.g. Duchenne muscular dystrophy). It also may result from selective survival of cells with one particular X active; for example, in X-autosomal balanced translocations, there is a higher proportion of cells that have inactivated the X chromosome that is not involved in the translocation. This is thought to protect against spreading of the XCI from the translocated X to the autosome and possible inactivation of autosomal genes that are essential for cell survival. The effects of a cell-lethal mutation in XL-dominant disorders may result in preferential survival during development of cells that have the wild-type X active. The demonstration of skewed XCI is often used to support X-linked inheritance in novel disease entities. Skewed XCI does not result from selective pressure on the selection of the X that needs to be inactivated, but rather from lethality of cells with the mutant X active. Some genes on the X chromosome escape XCI, often because they have a Y-linked functional homologue and dosage compensation is not essential. Finally, at the tip of each arm of the X chromosome is a region called the pseudoautosomal region, in which there is a cluster of genes having Y-linked copies. Mutations in these genes can behave as AD or AR, and this inheritance pattern does not preclude that a gene is located on the X chromosome.
Autosomal mutations with sex-limited or sex-modified expression can mimic XL inheritance because only males or only females are affected. Instances of male-to-male transmission, however, would show the true inheritance pattern. The genes involved often have distinct functions in males and females, such as autosomal genes affecting normal development and function of male or female reproductive organs. For example, 21-hydroxylase deficiency caused by AR mutations in the CYP21 gene causes androgen excess (congenital adrenal hyperplasia), which can result in ambiguous genitalia and virilization in females and premature puberty in males. Familial breast and ovarian cancer is another good example of sex-modified expression: even though the increased susceptibility is transmitted as an AD trait, the ovarian cancer phenotype is restricted to females, and males are less likely to have cancer (male breast cancer or cancer of other organs).1, 2
Y-Linked Inheritance
The Y-linked inherited conditions are rare because there are only a small number of Y-linked genes outside the pseudoautosomal region or without a functional X homologue. A pedigree would show only male-to-male transmission, as no other transmission is possible, and one would never see affected females.1, 2 The best-known example of a Y-linked condition is male infertility related to deletions of genes on the Y-chromosome (deleted in azoospermia).4 This condition usually results from new mutations, as complete infertility precludes any transmission, but rare males with some preserved fertility can transmit the mutation.
Genetic Heterogeneity
Mutations in different genes, sometimes transmitted via different inheritance patterns, can cause identical phenotypes. Hereditary deafness is a good example of such genetic heterogeneity; there are numerous loci with AD, AR, and XL inheritance patterns. (A series of different mutations at a single locus is called allelic heterogeneity.) It is not uncommon that two individuals with deafness marry and have children. (In genetic terms, this is referred to as assortative mating.) Counseling in these situations can be challenging, as it is often impossible to predict what the recurrence risk will be, unless the particular gene defect of one of the parents is known. In addition, phenocopies may exist, as in the case of deafness due to congenital rubella, which, in the absence of other stigmata of this acquired disorder, can present as possible hereditary deafness.1, 2
Somatic Mutations
The types of inheritance described so far involve mutations that affect germ cells as well as somatic cells and can be transmitted between generations. Mutations in genes occur at every cycle of cell division and DNA replication, however. Most are corrected by powerful DNA repair mechanisms, but occasionally a mutation escapes correction and becomes permanent in dividing somatic cells. This is the origin of many familial cancer syndromes. In such conditions (e.g. familial breast and ovarian cancer due to mutations in the BRCA1 and BRCA2 genes), the individuals at risk in the families have a germline mutation in one allele that is transmitted in AD fashion and puts them at increased risk of having these cancers develop. In the cells that give rise to the tumor, the second allele is inactivated via a variety of different mechanisms, and at the cellular level the disorder is AR; this is the origin of the two-hit hypothesis first described by Knudson in familial retinoblastoma.1, 2
EXCEPTIONS TO MENDELIAN INHERITANCE
Dynamic Mutations
As noted above, there are exceptions to Mendelian inheritance. One assumption of classic Mendelian genetics is that mutations are stably transmitted (i.e. they are passed unchanged from parent to offspring). The more severe phenotype and earlier onset of disease in each succeeding generation in families with fragile X syndrome (FX) or myotonic dystrophy (DM) were thus quite puzzling. This so-called anticipation was even considered to be the result of ascertainment bias until it was discovered in 1991 that the genes for spinobulbar muscular atrophy and FX contain unstable trinucleotide repeat sequences that can become larger upon germline transmission, and in some cases somatic cell division. DM and a number of AD spinocerebellar ataxias (SCAs) are also caused by unstable trinucleotide repeats or dynamic mutations. There currently are 13 autosomal and two XL inherited diseases known to be caused by triplet repeat expansions (Table 1), as well as a number of cytogenetic abnormalities or fragile sites that are mostly not associated with disease.5 The trinucleotide repeat sequences are CGG, CAG, CTG, GAA, or CGG, and their normal size ranges and disease-causing expansions are given (see Table 1). The triplet repeat may be of an intermediate (premutation) size that does not itself cause disease but is prone to expand to a full mutation during transmission to the next generation, leading to disease in the offspring. The likelihood and degree of expansion may depend on the sex of the transmitting parent for many of the disorders: expansions are more likely during maternal transmission in FX, DM, and FA and during paternal transmission in Huntington's disease and most SCAs. The triplet repeats are in the 5'UTR, 3'UTR, or coding regions of the respective genes except in the case of the AR Friedreich's ataxia and SCA8. The repeats have different, incompletely understood effects on the function of the mutated proteins. In FX, the expanded repeat in the 5'UTR leads to decreased transcription and absent protein while the premutation causes an RNA-mediated pathology. In many CAG repeat disorders, where the mutation is in the coding region of the gene, there is a combinaiton of loss of function of the normal protein and the mutant protein becomes deleterious to the cell, likely because of an altered conformation.6, 7
Table 1. Disorders caused by triplet repeat amplifications
Disorder | Inheritance | Gene | Wild type size | Mutant size | Parental bias |
Fragile X syndrome (FRAXA); Fragile X tremor ataxia syndrome (FXTAS) | XL | FMR1 in Xq27.3 (FRAXA) | (CGC) 6–60 | (CGC) 60–200 (premutation; causes FXTAS) (CGG) 230–1000 (full mutation) | Maternal |
Fragile XE mental retardation (FRAXE mental retardation) | XL | FMR2 in Xq28 (FRAXE) | (GCC) 4–39 | (CGG) 130–150 (premutation)(CGG) 200–900 (full) | Not determined |
Friedreich's ataxia (FRDA) | AR | X25 in 9q13-21.1 | (GAA) 6–32 | (GAA) 80 (premutation) (GAA) 200–1700 (full) | Maternal |
Myotonic dystrophy (DM1) | AD | DMPK in 19q13 | (CTG) 5–37 | (CTG) 50–10,000 | Maternal |
| Myotonic dystrophy (DM2) | AD | ZNF9 in 3q13.3-q24 | (CCTG) 10–26 | (CCTG) 75–11,000 | - |
Spinobulbar muscular atrophy (SBMA or Kennedy's disease) | AD | AR in Xq13-21 | (CAG) 9–36 | (CAG) 38–62 | - |
Huntington's disease (HD) | AD | IT15 in 4p16.3 | (CAG) 6–34 | (CAG) 36–121 | Paternal |
Dentatorubropallidoluysian atrophy (DRPLA) | AD | DRPLA (B37) in 12p31.31 | (CAG) 7–34 | (CAG) 49–88 | Paternal |
Spinocerebellar ataxia type 1 | AD | SCA1 in 6p23 | (CAG) 6–44 | (CAG) 39–82 | Paternal |
Spinocerebellar ataxia type 2 | AD | SCA2 in 12q24.1 | (CAG) 15–24 | (CAG) 32–200 | Paternal |
Spinocerebellar ataxia type 3 Machado-Joseph disease | AD | SCA3 (MJD1) in 14q32.1 | (CAG) 13–36 | (CAG) 61–84 | Paternal |
Spinocerebellar ataxia type 6 Episodic ataxia type 2 | AD | CACNA1A in 19p13 | (CAG) 4–19 | (CAG) 10–33 | - |
Spinocerebellar ataxia type 7 | AD | SCA7 in 3p12-13 | (CAG) 5–34 | (CAG) 37–306 | Paternal |
Spinocerebellar ataxia type 8 | AD | SCA8 RNA in 13q21 | (CTA/CTG) 16–34 | (CTA/CTG) >74 | Maternal |
| Spinocerebellar ataxia type 10 | AD | ATXN10 in 22q13 | (ATTCT) 10–20 | (ATTCT) 500–4,500 | ? |
| Spinocerebellar ataxia type 12 | AD | PPP2R2B in 5q31-q33 | (CAG) 7–28 | (CAG) 55–78 | - |
| Huntington disease-like 2 | AD | HDL2 (JPH3) in 16q24.3 | (CTG) 7–28 | (CTG) 66–78 | - |
| Spinocerebellar ataxia type 17 | AD | TBP in 6q27 | (CAG) 25–42 | (CAG) 47–63 | - |
Two entities in this group of disorders are particularly noteworthy: FX syndrome and myotonic dystrophy.
FX syndrome, one of the most common forms of mental retardation in males (incidence 1/4000), is characterized by moderate-to-severe mental retardation with hyperactivity, autistic features, and facial dysmorphism (e.g. broad jaw, large ears). The phenotype is caused by an expanded CGG repeat (230–1000 repeats) in the 5'UTR of the FX mental retardation 1 (FMR1) gene on chromosome Xq27.3. A repeat size above 200 leads to the full syndrome and is associated with a folate-sensitive fragile site on chromosome metaphase spreads. This characteristic led to the name of the syndrome and was used to confirm the diagnosis before the availability of more sensitive molecular testing (repeat-size detection by Southern analysis).
Fig. 6 illustrates the XL inheritance and additional features that are typical of FX pedigrees. Approximately 30% of female carriers of full mutations have mild-to-severe mental retardation. There are also unaffected transmitting males in FX families who can transmit a premutation to their phenotypically normal daughters, who themselves can have affected sons with full mutations. This dependence of the expression of the FX phenotype on the position in the pedigree is called the Sherman paradox and was based on the observation that the likelihood for mental impairment was higher in the offspring of intellectually normal daughters of transmitting males than in the transmitting males' siblings. It has recently been discovered that some males with the premutation have a previously unrecognized condition, Fragile X Tremor-Ataxia Syndrome (FXTAS).7, 8
|
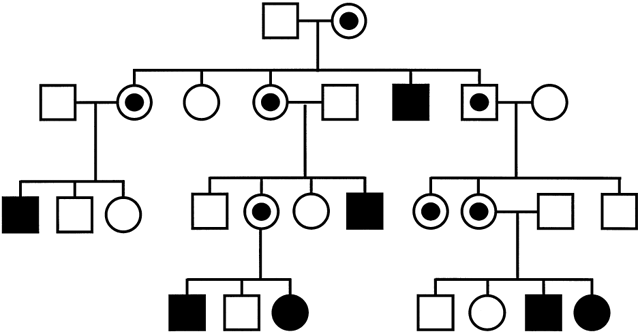
The molecular diagnostic test for the CGC repeat expansion in FX is highly reliable and should be offered to all patients with a family history of FX or mental retardation of unknown cause. Prenatal diagnosis can detect affected males in asymptomatic full or premutation carriers. Female fetuses with a full mutation allele have up to a 50% chance of having mental impairment. Female carriers of premutations are at increased risk of premature ovarian failure when they inherit the mutation from their father.6, 9, 10
Myotonic dystrophy (DM1) is one of the most common inherited muscular disorders and is characterized by myotonia, muscle weakness and atrophy, cardiac conduction system anomalies, frontal balding, cataracts, endocrine abnormalities, and mild mental impairment. It is caused by a trinucleotide CTG repeat expansion in the 3'UTR of the myotonic dystrophy protein kinase gene. There is clear anticipation, with earlier onset and more severe disease in later generations. When the mutation is transmitted through the maternal germline, massive CTG expansions (more than 1000 repeats) can occur, leading to a congenital form of the disease that causes severe hypotonia with facial diplegia, neonatal respiratory insufficiency, and mental retardation. Women carrying an affected fetus are at risk of polyhydramnios and preterm labor. Because of the muscle weakness and cardiac conduction system anomalies, tocolysis with magnesium sulfate can cause life-threatening respiratory depression and arrhythmias.6, 9, 11
Genomic Imprinting
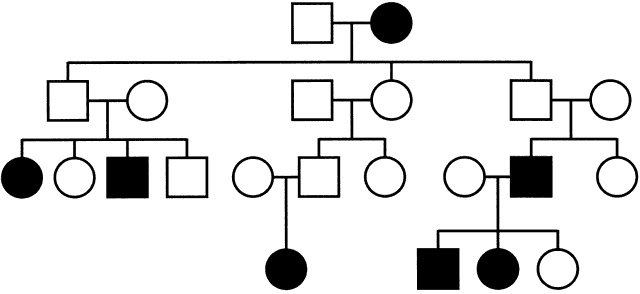
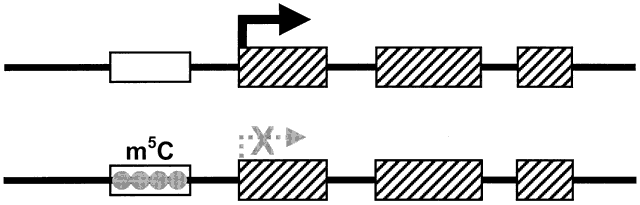
A central assumption of Mendel's laws of inheritance is that genes originating from maternal and paternal genomes are equally expressed in the offspring. In some disorders, however, such as the Beckwith-Wiedemann syndrome, the Prader-Willi syndrome (PWS), and the Angelman syndrome (AS), the sex of the transmitting parent plays a role in the expression of the phenotype in his or her affected children. This led to the discovery that for some genes, only the allele inherited from a particular parent is expressed in the offspring. A gene that is expressed only from the paternally inherited chromosome is maternally imprinted (the maternal allele is inactivated); a gene that is expressed only from the maternally inherited chromosome is paternally imprinted (the paternal allele is inactivated). When such a gene is mutated, only individuals in whom the mutated allele is expressed will have the disease, whereas individuals whose mutated copy is imprinted will be healthy. Fig. 7 shows a characteristic pedigree for a maternally imprinted disorder: whenever the mutation passes through the maternal germline, the offspring are nonmanifesting carriers. Her sons will have affected children, whereas her daughters will again silently transmit the mutation to the next generation. Overall, there should be an equal number of affected individuals and nonmanifesting carriers in each generation. Such genes, therefore, must carry a reversible tag or imprinting mark capable of switching when it passes through the germline of either sex. In general, once this mark has switched and is transmitted to the offspring, it remains stable and is propagated in all further cell divisions, but some instances of tissue-specific imprinting exist. This requires that the previous parental imprinting mark can be erased and that the imprinting mark, when present, leads to silencing of the transcription of the imprinted gene. The molecular mechanism for this phenomenon is complex, involving different modifications of DNA and accessory proteins, such as histones. However, methylation of cytosine residues at specific regulatory regions often associated with the promoters of such genes (Fig. 8), is often present. It has indeed been shown that genes in imprinted loci in the mouse and human genome have methylation of cytosine-guanine rich regions (CpG islands)'imprinting control regions', commonly at their promoters on one of their two alleles and that, with some exceptions, the methylated allele is the silenced one.12, 13, 14, 15
|
|
The number of genes discovered to be imprinted is growing.15 Most, but not all, are grouped together in specific regions of the genome (imprinting clusters) that usually contain both maternally and paternally imprinted genes interspersed with genes that are biallelically expressed. This strongly suggests that a common mechanism regulates parental-specific expression of these groups of genes.13
The functional role of imprinting in mammals is unclear, but the favored explanation is Haig's parental tug-of-war hypothesis. This hypothesis stems from the observation that most paternally expressed genes are growth-promoting, whereas maternally expressed genes restrict growth. Among polygamous species, females must preserve their resources for multiple litters from different partners and so benefit from smaller progeny. Males, conversely, benefit from having larger (and thus more resilient) offspring.12, 13, 14, 15
Uniparental Disomy
Imprinting of genes is responsible for disease phenotypes seen in uniparental disomy (UPD). This is the inheritance in the offspring of both homologues of a chromosome pair from the same parent (maternal UPD when both homologues are inherited from the mother, and paternal UPD when both homologues are inherited from the father). Different genetic mechanisms can lead to UPD. Chromosome nondisjunction leading to a trisomic conceptus is sometimes followed by loss of a chromosome or 'trisomy rescue' and is the most commonly observed mechanism. When the rescue is incomplete, confined placental mosaicism (i.e. the presence of both disomic and trisomic cell lines in the same placenta) can result; this is occasionally associated with fetal mosaicism and, in theory, carries a 30% chance of UPD for the involved chromosome if the chromosome from the parent that only contributes one of the three is lost (Fig. 9). Alternative mechanisms, such as chromosome duplication in monosomy and gamete complementation (diploid gamete from one parent is fertilized by a gamete lacking the same chromosome), can occur.1, 2, 16 UPD exerts no noticeable consequences when it involves chromosomes without imprinting effects. If the chromosomes contain imprinted genes, however, UPD can result in overexpression or underexpression of these genes. For example, up to 30% of cases of AS result from paternal UPD.
|

Imprinting Disorders
The best-known examples of imprinting disorders are PWS and AS and exemplify well the contribution of the various mechanisms that can bring out phenotypes associated with imprinting.1, 2, 12 These two very different developmental disorders result in most instances from identical interstitial cytogenetic deletions on chromosome 15q11–13. In AS, it is the maternal chromosome that has the deletion, whereas in PWS, it is the paternal chromosome. AS is characterized by severe mental retardation, seizures, absence of speech, ataxic gait, hand flapping, inappropriate laughter, and protruding tongue. Seventy percent of patients with AS have a maternal deletion, whereas only 3–5% have UPD15 with maternal deficiency, and 4–6% of patients have point mutations in the UBE3A gene encoding E6-AP ubiquitin protein ligase. This gene is biallelically expressed in most tissues but is paternally imprinted in specific brain regions, illustrating that genetic imprinting can be tissue-specific.17 PWS children have mild-to-moderate mental retardation, hypotonia with growth delay followed by hyperphagia and obesity, hypogonadism, and short stature. It is now known that 65% of PWS results from paternal deletions, whereas almost all the remaining cases of PWS are due to maternal UPD. Another well-characterized imprinting disorder is Beckwith-wiedemann syndrome (BWS), resulting from abnormal imprinting of the chromosome 11p15 imprinted gene cluster containing IGF2, H19 and p57KIP2 genes, or from mutations in p57KIP2. Some recent observations have raised the concern that artificial reproductive technologies may be associated with altered imprinting status of this locus resulting in BWS.18, 19 Whether and how ART increases the absolute risk of this and possibly other imprinting disorders is still under investigation and has been debated.20, 21
Imprinting in Development and Human Reproduction
Triploid conceptuses and the genomes of hydatidiform moles and ovarian teratomas illustrate well the differences between maternal and paternal genomes. The majority of complete hydatidiform moles have a normal karyotype (46,XX), but the whole genome is of paternal origin. This leads to excessive and abnormal placental development and lack of fetal development. Ovarian teratomas are thought to result from the parthenogenetic activation of unfertilized oocytes, and their genome is thus entirely maternally derived. They can contain a variety of more-or-less differentiated tissues but never contain trophoblast. When the extra haploid set of chromosomes of a triploid conceptus is paternally derived, it results in partial hydatidiform mole and reduced fetal development. The converse case, when the extra haploid set is maternal in origin, results in small, underdeveloped placentas.14
Other Epigenetic Inheritance: DNA Methylation and Human Disease
Imprinting is a form of epigenetic gene regulation 'upon' the actual sequence of DNA. The importance of epigenetic gene regulation as a contributor to human genetic disease has become more apparent in the last 5–10 years. As discussed above, in general, methylation of DNA at cytosine in CpG-rich regions upstream of genes can control gene expression, usually making genes transcriptionally inactive, whereas when these sequences are unmethylated, downstream genes are transcriptionally active. Properly regulated DNA methylation and other epigenetic changes have distinct functions at critical stages of development and in essential biologic pathways. It has now been demonstrated that abnormal DNA methylation at specific genes can contribute to human health and disease, including normal aging, cancer, developmental origin of adult-onset disorders. For example, disturbed DNA methylation patterns at oncogenes, tumor suppressor genes, or other genes are associated with many common human cancers, such as breast cancer, colon cancer, and lung cancer.22, 23, 24
Gene-Environment Interactions
Environmental exposure to nutritional, chemical and physical factors can alter the DNA methylation patterns, leading to imprinting disorders and congenital birth defects. Deficiencies of four essential dietary sources of methyl groups (choline, methionine, vitamin B 12 and folic acid) have been associated with an increased risk for a number of diseases.25 It has further been shown in animal experiments that nutritional supplements to the mother can alter gene expression in her offspring without altering the genes themselves.26 Increasing nutritional supplementation of folic acid to pregnant women reduces the risk of spina bifida and other neural tube defects in the offspring. Environmental chemicals, such as certain metals and other xenobiotics may have a probable role in abnormal methylation processes.27 The toxic effects exhibited by metals such as Ni, As, and Cd, as well as by Zn deficiency, may lead to cancer, atherosclerosis, birth defects, neurological disturbances, and pancreatic lesions.
Structural Genomic Variation and Copy Number Variants
Balanced structural genomic variation (inversions, translocations) and unbalanced structural variation, i.e copy number variants (CNVs), are larger-scale genomic changes that are inheritable in a Mendelian pattern. They deserve mention here as it has become exquisitely clear in the last few years that they are an important component of normal human genetic variation, and that they contribute significantly to human inherited and sporadic genetic disease. CNVs (deletions or duplications of larger segments of DNA from 1 kilobase to several megabases) are far more common in the human genome than was previously anticipated. Well over 12,000 CNVs have now been catalogued in publicly accessible databases, such as the Database of Genomic variants (http://projects.tcag.ca/variation/).28 The de novo mutation rate for CNVs is relatively high, about 1.7 x 10-6 to 1.2 x 10-4, and CNVs can therefore be an important contributor to sporadic de novo genetic disease.29 Yet, once present in an individual's genome, they are usually transmitted to the next generation according to Mendelian laws of inheritance. Although CNVs can be associated with clinically important phenotypes, such as autism, developmental delay, birth defects and schizophrenia,30, 31 not all CNVs are clinically significant. For some of the newly discovered CNVs, deciphering their associated clinical phenotypes can be challenging for medical geneticists.
Mitochondrial Inheritance
Cells contain, on average, 103 to 104 mitochondria, each having two to 10 copies of a 16,569 base-pair long double-stranded circular DNA molecule. The mitochondrial genome has 37 genes that encode for a number of proteins of the oxidative phosphorylation chain, for 22 tRNA molecules and rRNAs. All other proteins are encoded in the nuclear genome and imported into the mitochondria. The replication and transcription of mitochondria depend on nuclear-encoded enzymes and transcription factors.32, 33
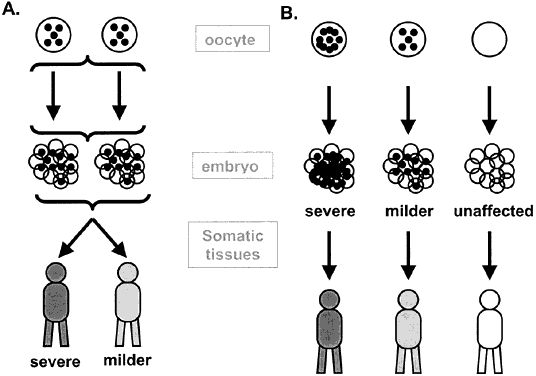
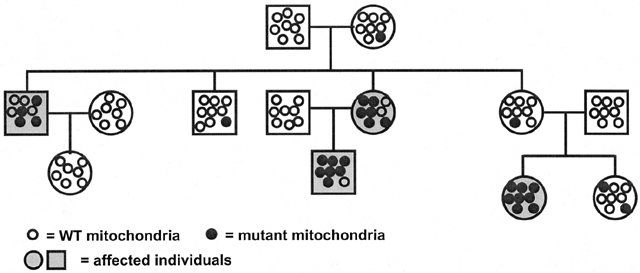
Virtually all (99.99%) mitochondrial DNA is inherited from the mother. The oocyte has a large number (106) of mitochondria, whereas most of the already-depleted supply of mitochondria in the fertilizing sperm is eliminated during the first cell divisions.34 It has been shown that even after intracytoplasmic sperm injection, few if any paternal mitochondria are retained in the developing embryo. Mitochondrial disorders follow two patterns of inheritance: mutations in nuclear-encoded genes follow traditional patterns (AD, AR, XL), whereas mutations in mitochondrial genes follow typical maternal inheritance. Mitochondria divide and replicate their DNA asynchronously from the meiotic and mitotic cell divisions, resulting in a variable number in each cell. In general, all mitochondria of an individual have the same genome (homoplasmy). Mutations in mitochondrial genes can affect all (homoplasmic mutations) or a subset of the mitochondria in each cell (heteroplasmic mutations); the latter leads to variations in the phenotype between individuals within the same family. During oogenesis and/or in the earliest cell divisions after fertilization, a subset of the mitochondria present in the oocyte is selectively amplified, leading to a 'mitochondrial bottleneck' (Fig. 10). The fraction of mutated mitochondria in this amplified population results in different levels of heteroplasmy in the offspring. The pedigree of a typical mitochondrial disorder is given in Fig. 11 and illustrates the absence of paternal transmission, with equal maternal transmission to affected sons and daughters.32, 33
|
|
Mitochondrial disorders are subdivided into three classes. Class I disorders (e.g. Leigh syndrome) are caused by mutations in nuclear genes and have traditional Mendelian patterns of inheritance. Class II disorders result from point mutations in mitochondrial genes; are usually maternally inherited; and often cause muscular, neurologic, and eye phenotypes (e.g. myoclonic epilepsy with lactic acidosis and stroke; neurogenic weakness, ataxia, retinitis pigmentosa, myoclonic epilepsy with ragged red fibers). Class III mutations are mostly sporadic-occurring mitochondrial deletions and duplications that can cause a spectrum of diseases (e.g. diabetes and deafness, Pearson syndrome, Kearns-Sayre syndrome). The accumulation of free oxygen radicals in mitochondria recently has been proposed to play a role in aging and possibly in neurodegenerative disorders such as Parkinson's disease, Alzheimer's disease, and Friedreich's ataxia.32, 33
Moledular Diagnosis and Prenatal Detection of Mitochondrial Disorders
Specific point mutations for many class II mitochondrial diseases are known and detectable, but the sensitivity of the test depends on the degree of heteroplasmy and the specific tissue examined. This makes prenatal genetic counseling and molecular testing in these conditions very challenging, as the mitochondrial content of amniocytes or chorion villi is often not representative of the organs that are commonly affected (e.g. brain, muscle, heart). Interested patients should be informed about these shortcomings and referred to specialized genetic counselors.35, 36
ACKNOWLEDGMENT
This chapter is a revision of an update to a previous version by R. Neil Schimke and Charles R. King.
REFERENCES
Beaudet AL, Scriver CR, Sly WS et al: Genetics, biochemistry, and molecular basis of variant human phenotypes. In Scriver CR, Beaudet AL, Sly WS, Valle D (eds): The Metabolic and Molecular Bases of Inherited Disease, pp 72–79. Vol 1, 7th ed. New York, McGraw Hill, 1995 |
|
Mueller RF, Cook J: Mendelian inheritance. In Rimoin DL, Connor JM, Pyeritz RE (eds): Emery and Rimoin's Principles and Practice of Medical Genetics, pp 87–102. Vol 1, 3rd ed. New York, Churchill Livingstone, 1997 |
|
Morello R, Bertin TK, Chen Y et al: CRTAP is required for prolyl 3- hydroxylation and mutations cause recessiveosteogenesis imperfecta. Cell 127(2):291-304, 2006 |
|
Stuppia L, Calabrese G, Franchi PG et al: Widening of a Y-chromosome interval-6 deletion transmitted from a father to his infertile son accounts for an oligozoospermia critical region distal to the RBM1 and DAZ genes (letter). Am J Hum Genet 59: 1393–1395, 1996 |
|
Debacker K, Kooy RF: Fragile sites and human disease. Hum Mol Genet 16 Spec No. 2:R150-8, 2007 |
|
Wilmot GR, Warren ST: A new mutational basis for disease. In Wells RD, Warren ST (eds): Genetic Instabilities and Hereditary Neurological Diseases, pp 3–12. San Diego, Academic Press, 1998 |
|
Lutz RE: Trinucleotide repeat disorders. Semin Pediatr Neurol 14(1):26-33, 2007 |
|
Jacquemont S, Hagerman RJ, Leehey MA et al : Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA 291: 460–9, 2004b |
|
Zoghbi HY, Orr HT: The spinocerebellar ataxias: In Scriver CR, Beaudet AL, Sly WS, Valle D (eds): The Metabolic and Molecular Bases of Inherited Disease, 8th ed. New York, McGraw Hill, 1995 |
|
Jin P, Warren ST: Understanding the molecular basis of fragile X syndrome. Hum Molec Genet 9: 901–908, 2000 |
|
Geifman-Holtzman O, Fay K: Prenatal diagnosis of congenital myotonic dystrophy and counseling of the pregnant mother: Case report and literature review. Am J Med Genet 78: 250–253, 1998 |
|
Falls JG, Pulford DJ, Wylie AA et al: Genomic imprinting: Implications for human disease. Am J Pathol 154: 635–647, 1999 |
|
Tilghman SM: The sins of the fathers and mothers: Genomic imprinting in mammalian development. Cell 96: 185–193, 1999 |
|
Latham KE: Epigenetic modification and imprinting of the mammalian genome during development. Curr Top Dev Biol 43: 1–49, 1999 |
|
Shaffer LG, McCaskill C, Adkins K et al: Systematic search for uniparental disomy in early fetal losses: The results and a review of the literature. Am J Med Genet 79: 366–372, 1998 |
|
Morison IM, Reeve AE: A catalogue of imprinted genes and parent-of-origin effects in humans and animals. Hum Mol Genet 7: 1599–1609, 1998 |
|
Jiang Y, Lev-Lehman E, Bressler J et al: Genetics of Angelman syndrome. Am J Hum Genet 65: 1–6, 1999 |
|
Maher ER, Brueton LA, Bowdin SC et al : Beckwith-Wiedemann syndrome and assisted reproduction technology (ART). J Med Genet 40: 62–64, 2003 |
|
DeBaun MR, Niemitz EL, Feinberg AP : Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am J Hum Genet 72: 156–160, 2003 |
|
Horsthemke B, Ludwig M : Assited reproduction: the epigenetic perspective. Hum Reprod Update 11:473–480, 2005 |
|
Doornbos ME, Maas SM, McDonnell J et al : Infertility, assisted reproduction technologies and imprinting disturbances: a Dutch study. Hum Reprod 22:2476–2480, 2007 |
|
Widschwendter M, Jones PA : DNA methylation and breast carcinogenesis. Oncogene 21: 5462-5482, 2002 |
|
Giovannucci E, Ogino S : DNA methylation, field effects, and colorectal cancer. J Natl Cancer Inst 97: 1317–1319, 2005 |
|
Tsou JA, Hagen JA, Carpenter CL et al : DNA methylation analysis: a powerful new tool for lung cancer diagnosis.Oncogene 21: 5450-5461, 2002 |
|
Poirier LA : The effects of diet, genetics and chemicals on toxicity and aberrant DNA methylation: an introduction. J Nutr 132:2336S–9S, 2002 |
|
Cooney CA, Dave AA, Wolff GL : Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. J Nutr 132:2393S–2400S, 2002 |
|
Poirier LA, Vlasova TI : The prospective role of abnormal methyl metabolism in cadmium toxicity. Environ Health Perspect 110(Suppl 5): 793–795, 2002 |
|
Iafrate AJ, Feuk L, Rivera MN et al : Detection of large-scale variation in the human genome. Nat Genet 36(9):949-51, 2004 |
|
Lupski JR : Genomic rearrangements and sporadic disease.Nat Genet 39(7 Suppl):S43-7, 2007 |
|
Sebat J, Lakshmi B, Malhotra D et al : Strong association of de novo copy number mutations with autism. Science 316(5823):445-9, 2007 |
|
Walsh T, McClellan JM, McCarthy SE et al : Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320(5875):539-43, 2008 |
|
Lightowlers RN, Chinnery PF, Turnbull DM et al: Mammalian mitochondrial genetics: heredity, heteroplasmy and disease. Trends Genet 13: 450–455, 1997 |
|
Poulton J, Marchington DR: Prospects for DNA-based prenatal diagnosis of mitochondrial disorders. Prenat Diagn 16: 1247–1256, 1996 |
|
Chinnery PF, Howell N, Lightowlers RN et al: Genetic counseling and prenatal diagnosis for mtDNA disease. Am J Hum Genet 63: 1908–1911, 1998 |
|
Marchington DR, Macaulay V, Hartshorne GM et al: Evidence from human oocytes for a genetic bottleneck in an mtDNA disease. Am J Hum Genet 63: 769–775, 1998 |
|
Danan C, Sternberg D, Van Steirteghem A et al: Evaluation of parental mitochondrial inheritance in neonates born after intracytoplasmic sperm injection. Am J Hum Genet 65: 463–473, 1999 |