Neurologic Sequelae of Birth
Authors
INTRODUCTION
Cerebral injury from hypoxemia, whether it is prenatal, intrapartum, or neonatal, is a significant contributor of neurologic morbidity and mortality in neonates. Perinatal asphyxia occurs in approximately 6 per 1000 term live births.1 There is little agreement about what constitutes fetal asphyxia. No single parameter taken in isolation seems to predict the severity of the hypoxic ischemic injury or the long-term neurologic sequelae. Investigators have shown the inadequacy of the Apgar score as a correlate to significant brain insult.2 Fetal acidosis, meconium staining, late decelerations, and respiratory depression are nonspecific markers of neonatal distress. Signs attributed to asphyxia may be due to a neurologically impaired fetus. Fetal acidemia may be induced by parameters other than hypoxemia, and abnormal fetal heart rate patterns may be the result, rather than the cause, of neurologic impairment.
Five principal mechanisms of asphyxia have been described during the antepartum and intrapartum periods: (1) interruption of umbilical circulation, (2) altered placental gas exchange, (3) inadequate maternal perfusion of the placenta, (4) decreased maternal oxygenation, and (5) failure of the infant to transition from fetal to neonatal cardiopulmonary circulation.3 There are many etiologic factors in fetal asphyxia (Table 1). Placental abruption, cord prolapse, and uterine rupture result in an acute, hypoxic-ischemic insult. Partial or intermittent hypoxic-ischemic injury can occur in cases of placental insufficiency. The clinical features of hypoxic-ischemic encephalopathy, however, are nonspecific.
TABLE 1. Clinical Factors Associated With Hypoxic-Ischemic Injury
Maternal hypotension or hypertension
Maternal diabetes
Maternal anemia
Maternal cardiopulmonary disease
Preeclampsia
Intrauterine growth retardation
Umbilical cord compression
Placental abruption, previa, or insufficiency
Altered fetal heart rate or acid-base disturbance
Thick meconium
Infant cardiopulmonary disease
Neurologic signs and symptoms may be observed in the neonate with perinatal asphyxia. It often is difficult to determine the precise timing of insult.4 Whereas an intrapartum event may lead to neurologic signs hours to days later, infants who have sustained a hypoxic insult in utero may have few, if any, clinical abnormalities in the neonatal period. Term infants who suffer an acute asphysic insult resulting in severe long-term neurologic handicaps invariably show a recognizable clinical encephalopathy during the first few days of life.4 A detailed examination will aid in the assessment of severity of hypoxic-ischemic encephalopathy. The spectrum is divided into three classes that have prognostic value.5 Mild encephalopathy is characterized by jitteriness, uninhibited deep tendon reflexes, and hyperalertness. This is not associated with long-term sequelae. Hypotonia, seizures, and lethargy are hallmarks of moderate encephalopathy. The outcome of moderate encephalopathy is more variable. If symptoms persist for longer than 1 week, neurologic sequelae may exist in up to 40%.5 Severe encephalopathy is associated with coma, seizures, brain-stem dysfunction, and increased intracranial pressure. The majority of these infants are affected with long-term sequelae, such as microcephaly, mental retardation, spastic quadriplegia, and seizures.
Radiologic examination helps in the assessment of the extent of cerebral injury. Although ultrasound cannot identify specific patterns of injury, it is a portable tool allowing bedside assessment of germinal matrix and intraventricular hemorrhage. It also can detect changes in echogenicity seen with early periventricular leukomalacia in the premature infant. Computed tomographic (CT) scanning is the investigation of choice in the clinical evaluation of the hypoxic-ischemic brain injury of the term newborn.4 Optimal timing of the scan is debated. It often is obtained during the initial evaluation of the asphyxiated infant, but the maximal extent of tissue damage visualized on CT usually is 2 to 5 days after the insult. CT scanning may identify parasagittal injury, focal ischemia, injury to the basal ganglia, and posterior fossa pathology. Although magnetic resonance imaging is the best technique available for evaluating the anatomic structures of the brain as well as myelination pattern, it is difficult to interpret in the newborn because of the high water content of the brain.6 The brain appears hyperintense on T2-weighted images, making it difficult to distinguish from neuropathic changes due to early ischemic insults.6 An electroencephalogram recording also can aid in the evaluation of an asphyxiated infant. A progression of electroencephalographic patterns correlates with the evolution of the clinical hypoxic-ischemic encephalopathy.4 Often, it shows a discontinuous pattern with marked slowing and reduced amplitude. The low-voltage pattern may deteriorate to an isoelectric tracing that parallels the decline in clinical condition. If there is rapid resolution of the electroencephalogram abnormalities, however, prognostic outcome improves.5
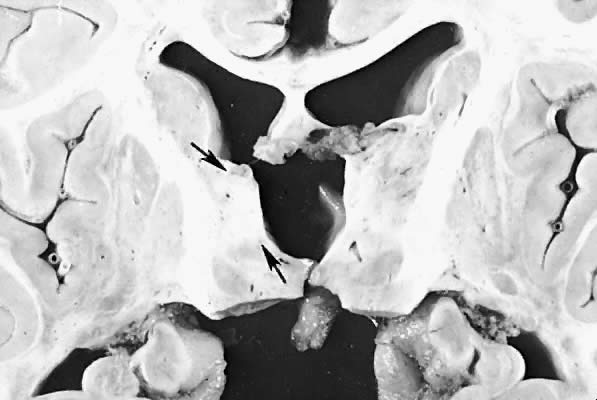
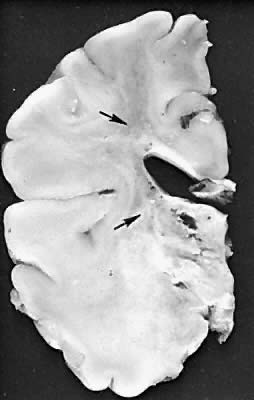
Five major neuropathologic patterns of injury are summarized in Table 2. Distribution of the lesions reflects a combination of regional circulation and metabolic factors that change with the gestational age of the infant. Both the degree of myelination and the presence of excitatory amino acid binding sites also may play a role in the pathophysiology of tissue damage.6 The primary lesions seen on autopsy in term newborns are in the cortex and basal ganglia (Fig. 1). In the premature infant, however, lesions generally occur in the germinal matrix and periventricular regions of the brain (Fig. 2). Efforts to resuscitate the infant should be directed toward adequate ventilation and perfusion, correction of accompanying metabolic acidosis, maintenance of normal blood pressure and glucose, and treatment of seizures. Antidiuretic hormone may be secreted inappropriately after a large cerebral insult, and fluid overload should be avoided. Serum osmolarity and sodium concentration should be monitored to avoid the consequence of cerebral edema and seizures. Although antiedema agents may reduce intracranial pressure, it is unclear whether they improve long-term neurologic sequelae.4 Seizures in the hypoxic-ischemic setting may be refractory to anticonvulsant therapy, but treatment is recommended to reduce the incidence of apnea and hypertension. Handling should be minimized to reduce the incidence of hypoxemic episodes, especially in the premature neonates.
TABLE 2. Neuropathologic Patterns of Hypoxic-Ischemic Injury
Pattern | Distribution |
Selective neuronal necrosis | Hippocampus, cerebellum, brainstem |
Status marmoratus of the basal ganglia | Basal ganglia, thalamus |
Parasagittal cerebral injury | Cerebral cortex, subcortical white matter |
Focal and multifocal brain injury | Cerebral cortex |
|
|
SPINAL CORD INJURY
Despite a decrease in the incidence of spinal cord injury sustained by delivery, perinatal damage may be responsible for 11% of the cord injuries seen in the pediatric population.7 Fetal malposition during delivery subjects the cord to longitudinal stretching and torsional forces. Spinal cord lesions include epidural hemorrhages; meningeal lacerations; and trauma to the arteries, ligaments, musculature, and nerve roots. In the majority of infants, spinal cord injury occurs after vaginal delivery in the breech position, yet cases have been reported after cephalic delivery.
The infant spine is more deformable than the adult spine because of the elasticity of the interspinous ligaments, posterior joint capsule, and cartilaginous end plates.7 This elasticity makes the infant spine more susceptible to hyperextension injury. Hyperextension of the fetal head in breech presentation, also referred to as star gazing fetus, has been associated with neurologic complications when the infant is delivered vaginally. Injuries include vertebral artery compression, spinal cord torsion and traction, and quadriparesis. The dura is the major supporting structure of the spinal cord. If a “pop” is heard during the delivery, it may signify the rupture of the dura mater and significant spinal cord injury.
In most breech deliveries, the mechanism of injury is traction, and the lesion usually is located in the lower cervical or upper thoracic cord (Fig. 3). Along with hypotonia, the infant with a lower cervical injury may have diaphragmatic breathing and Horner's syndrome. Lower thoracic lesions result in flaccid paralysis of the lower extremities and bowel and bladder dysfunction. Expulsion of urine after suprapubic pressure confirms bladder paralysis. Lack of perceptible response to pain can help localize the level of injury.
|
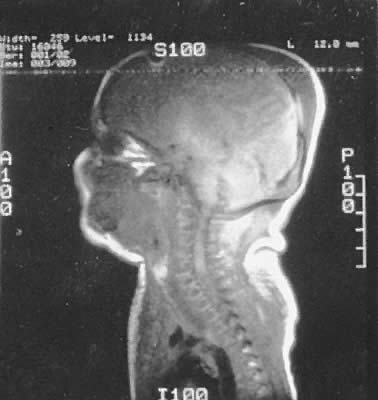
Spinal cord injury from cephalic presentation usually is located in the upper to mid-cervical region. The uncinate processes are flat in the neonatal spine and are insufficient in withstanding excessive torsional forces.7 High cervical lesions may result in paralysis of the diaphragm and quadriplegia. A lesion above the fourth cervical vertebrae can be fatal unless respirations are supported.
Although the most common mechanism of spinal cord injury is traction, vascular compromise results in cord ischemia. It is postulated that damage to the vertebral arteries by subluxation and dislocation can compromise the lumen of the arteries as they ascend through the foramina transversaria.8 The consequent thrombosis may result in brain-stem or upper cervical cord ischemia. Cord infarction is seen in newborns undergoing umbilical artery catheterization. The artery of Adamkiewicz is the major segmental artery to the thoracolumbar cord, arising between the level of T-10 and T-12. If the catheter tip is placed in this region, embolization of the artery may occur and result in acute paraplegia.
Signs of spinal shock are flaccidity, loss of spontaneous movements, and absence of stretch reflexes. Such features may falsely be attributed to birth asphyxia or brain injury. The differential diagnosis of spinal cord injury at birth includes cerebral anoxia, myelodysplasia, infantile spinal muscular atrophy, and cerebral palsy. Bowel and bladder dysfunction and a sensory level to pinprick examination suggest a spinal cord injury.
Radiologic studies may be normal in infants with spinal cord injury.7 Plain films may show subluxation, fracture, or anomalies, but the spinal cord can be ruptured by longitudinal distraction without disruption of the vertebral column. Infants must be examined frequently for signs of spinal cord damage, even if there is no radiographic evidence of injury.
The treatment of neonatal spinal cord injury has not been well-established. A study performed by Bracken and coworkers9 concluded that treatment with methylprednisolone within the first 8 hours of injury is beneficial, but the study excluded children younger than 13 years of age.
BRACHIAL PLEXUS INJURY
Brachial plexus palsy is a well-known complication of both breech and cephalic injury. Although obstetric trauma is the most common cause of birth-related brachial plexus injury, injuries also occur from intrauterine compression, exostosis of the first rib, neoplasm, and neck compression.10 The incidence of obstetric plexus palsy ranges from 0.19 to 2.6 per 1000 live births.11,12,13,14 Predisposing factors include large birth-weight infants with shoulder dystocia, forceps or vacuum assistance, and breech presentation.13,14,15 Obstetric injury can result from traction of the plexus, root avulsion, or transection of the nerves as they join to form the plexus. Stretch injuries have a good prognosis, whereas those due to avulsion may not recover function.
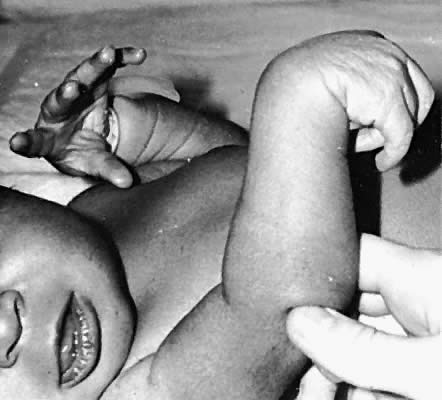
Injury to the upper segments of the brachial plexus, C-5 and C-6, is termed Erb's palsy. The diagnosis is suspected when the arm is held in the characteristic posture of adduction with internal rotation (Fig. 4). This is caused by paralysis of the deltoid, supraspinatus, and infraspinatus. The elbow is extended and the forearm is pronated because of weakness of the biceps and brachioradialis muscles. This results in the “waiter's tip” hand (Fig. 5). Despite the typical hand posture in Erb's palsy, wrist and hand movements are preserved. The bicep stretch reflex is absent, but the triceps reflex is spared.
|

|
Upper-middle trunk brachial plexus palsy involves cervical segments C5-C7. The affected arm is held in a position similar to that of Erb's palsy, but there is weakness of wrist flexion and extension.10 The biceps, triceps, and brachioradialis stretch reflex are absent.
Klumpke's palsy involves the lower segments of the brachial plexus. The injury results in a clawlike deformity of the hand (Fig. 6). Injury to the C-8 and T-1 spinal nerve fibers results in distal weakness, with flexion of the elbow, extension at the wrist, and hyperextension at the metacarpophalangeal joints. The triceps reflex is absent. Horner's syndrome occurs on the ipsilateral side when the injury involves the sympathetic fibers at the root level.
|

A complete brachial plexus palsy involves injury to nerve fibers C-5 through T-1. The diagnosis is suspected when the arm is limp and motionless. The fingers are flexed because of the tenodesis effect at the wrist.16 Horner's syndrome usually is present.
Associated lesions include clavicle fracture, humerus fracture, Horner's syndrome, and phrenic nerve paralysis. Because 80% to 90% of phrenic nerve paralysis is associated with brachial plexus injury, a chest radiograph is warranted when there is asymmetric diaphragmatic movement.17 Difficulty sucking, intermittent stridor, or xiphoid retractions should alert the physician to possible hypoglossal nerve paralysis or recurrent laryngeal injury.
Management of obstetric brachial plexus palsy is aimed at preventing contractures and promoting better skeletal development. After an initial period of immobilization against the abdomen to minimize swelling or hemorrhage or both, passive range-of-motion exercises are recommended, beginning 1 week after the injury. These exercises include shoulder rotation; elbow flexion and extension; wrist flexion and extension; finger flexion and extension; and thumb opposition, abduction, and adduction.14 Although electrophysiologic studies may distinguish the extent of the lesion, CT myelography is useful in evaluating nerve root injuries and detecting pseudomeningoceles.18 Favorable prognostic signs include early onset of recovery and involvement of only the proximal upper extremity. The indication and timing of operative intervention remain controversial.
FACIAL NERVE PALSY
Facial weakness in newborns can be divided into two broad categories: developmental and acquired. Etiologies of developmental facial paralysis include Mobius syndrome, hypoplasia of the depressor anguli oris muscle, and hemifacial macrosomia. Despite earlier observations, acquired facial nerve palsy often is the result of obstetric forceps.19 Other risk factors predictive of facial nerve injury include primiparity, birthweight exceeding 3500 g, and prolonged second stage of labor.20 The obstetric position of the fetus also is implicated; occipito left positions can have left facial palsies.21
A complete unilateral facial palsy in the newborn is not difficult to recognize (Fig. 7). At rest, the affected eye may fail to close and the nasolabial fold is flat. When the infant cries, only the normal side of the mouth droops. The lesion is believed to be the result of extracranial compression after the nerve exits the stylomastoid foramen. The clinical signs of a partial facial paralysis may be more subtle, with the infant showing only asymmetric wrinkling of the forehead or unequal elevation of the corner of the mouth (Fig. 8). The prognosis of an infant with facial nerve trauma generally is favorable, with nearly 90% resolving completely.19 A careful birth history and physical examination should help distinguish between developmental and acquired lesions. Electrodiagnostic studies are valuable in persistent cases to quantitate the injury and distinguish muscle agenesis or nuclear aplasia.22
|

|

SKULL FRACTURE
Linear skull fractures are relatively common in the newborn and can occur in association with cephalohematomas and other external evidence of birth trauma. If the infant is stable, no therapy is necessary. Intracranial complications are rare, but the physician should be aware of the possibility of epidural or subdural hemorrhage or the development of a leptomeningeal cyst over subsequent weeks. Follow-up skull radiographs at several months are recommended to ensure that healing has taken place and the fracture has not increased in width. The leptomeningeal cyst occurs as a protrusion of the arachnoid through the defect in the dura and skull. It may present as a pulsatile mass within the scalp and is detected easily by transillumination of the affected area. Surgical excision is recommended.
Depressed fractures occur secondary to obstetric trauma and extreme molding of the fetal skull on the maternal symphysis or ischial spines during labor. The surface of the skull is buckled inward without loss of bone continuity, similar to that of an indented Ping-Pong ball. Displacement of the bone greater than 5 mm is considered sufficient to impinge on the cerebral cortex and impair local blood supply. The management of neonatal depressed skull fractures is unclear, and it is believed that spontaneous elevation occurs over a period of months. Nonsurgical methods such as the use of vacuum breast pump, digital manipulation, or obstetric vacuum extractor have been successful in the treatment of these lesions.23 Surgical evaluation is recommended if there is radiologic evidence of bone fragments within the brain parenchyma or signs of raised intracranial pressure, neurologic deficit, or cerebral spinal fluid beneath the galea aponeurotica, implying dural laceration.
EXTRACRANIAL HEMORRHAGE
Caput Succedaneum
Caput succedaneum is an ecchymotic, edematous swelling superficial to the periosteum, extending across suture lines. Observed hours after vaginal delivery, it results from compression on the presenting portion of the infant against the cervix. Although commonly located on the vertex of the head, edema may extend into the supraorbital tissues and cause facial discoloration and distortion. The blood resolves over several weeks, and aspiration of the involved spaces is contraindicated because of the high risk of infection. Phototherapy may be required if hyperbilirubinemia develops, but often no specific intervention is necessary.
Cephalohematoma
Cephalohematoma refers to a subperiosteal hemorrhage confined within suture boundaries. The subperiosteal bleeding is slow, resulting in an increasing size after birth. Causal factors include obstetric trauma, primiparity, and fetal skull size. Occasionally, cephalohematomas are associated with an underlying skull fracture. The majority of these fractures are linear and clinically insignificant. Although uncommon, a massive cephalohematoma may result in enough blood loss to cause anemia and serve as a source of bilirubin production. The majority of the lesions reabsorb within the first few months of life, and calcification may appear after the second week.
INTRACRANIAL HEMORRHAGE
Subdural Hemorrhage
Improvement in obstetric technique has reduced the incidence of subdural hemorrhage. Tentorial subdural hemorrhage may occur in normal deliveries and remain undiagnosed in the infant with spontaneous recovery. Subdural bleeding results from rupture of the bridging veins overlying the cerebral hemispheres or dural sinus tears. Abnormal pressure on the skull from forceps, vacuum, or breech presentation may result in tearing of the tentorial attachments to the falx and petrous bones. Small tears or rupture of the bridging veins can result in subacute symptoms, presenting hours to days after delivery. Hemorrhage from a major tear of a dural sinus can extend into the posterior fossa, compress the cerebellum and obstruct the fourth ventricle, and result in brain-stem compression.
An infant with a molded head in whom signs of intracranial hypertension develop after a symptom-free interval should raise the suspicion of tentorial tear and posterior fossa hemorrhage. Clinical symptoms include irritability, weak or high-pitched cry, vomiting, and pallor. On examination, the infant often is hypotonic with a tense fontanelle and may show an asymmetry of motor function. Seizures are frequent. Neonates should be monitored for early signs of brain-stem compression: unequal pupils, bradycardia, irregular respiration, and oculomotor paralysis.
Computed tomography is the technique of choice to verify subdural hemorrhage and should be obtained as quickly as possible in symptomatic infants. Surgical evacuation of a posterior fossa subdural bleed is necessary when brain stem signs occur. Midline shift and transtentorial herniation from a subdural hemorrhage over the cerebral convexity is alleviated by tapping the subdural space. Repeated taps should not be performed if the infant is asymptomatic. Conservative treatment is advocated in the stable neonate.24
The prognosis of an infant with subdural hemorrhage depends on the degree of hemorrhage. Neonates with laceration of the falx or dural sinus often have hydrocephalus and other sequelae develop. A more favorable outcome can be expected in those infants with a convexity subdural hemorrhage.
Subarachnoid Hemorrhage
Subarachnoid hemorrhage in the newborn is unlike the dramatic arterial hemorrhage seen in the adult population. It originates from the vascular channels that are remnants of anastomoses of the leptomeningeal arteries or the bridging veins within the subarachnoid space.23 It is thought to result from physiologic trauma or hypoxic event during delivery.
Because the bleed usually is venous in origin, clinical signs may be minimal or absent, and the hemorrhage often is self-limited. Seizures can occur in an otherwise healthy infant interictally.23 The convulsions begin on the second day of life. In rare instances, neonates with large subarachnoid hemorrhages have respiratory disturbances, brain stem dysfunction, and coma develop.
The diagnosis of symptomatic subarachnoid hemorrhage is based primarily on the CT findings. Other causes of blood in the subarachnoid space (hemorrhage associated with intraventricular or intracerebral bleeding) are excluded by CT scan. A lumbar puncture should support the clinical suspicion with elevated red blood cells and protein, xanthochromia, and hypoglycorrhachia. Management is supportive, and hydrocephalus secondary to impaired cerebrospinal absorption occurs in some cases.
Intraventricular Hemorrhage in Term Infants
Although this lesion is more frequent in preterm infants, intraventricular hemorrhage is detected by CT scan in full-term neonates. It may originate from the germinal matrix, the veins of the choroid plexus, or both. Perinatal asphyxia and trauma often are clinical correlates. Presentations are variable and consist of an altered level of consciousness, respiratory disturbances, seizures, and hemiparesis or quadriparesis. Posthemorrhagic hydrocephalus is common, and a ventriculoperitoneal shunt may be required. Diagnosis is made by ultrasound or CT scan. Treatment is similar to that of a preterm infant with intraventricular hemorrhage.
MYASTHENIA GRAVIS
Myasthenia gravis is a disorder of postsynaptic neuromuscular transmission and is included in the differential diagnosis of a hypotonic infant. Transient neonatal myasthenia occurs in approximately 21% of infants born to mothers with acquired myasthenia gravis.25 Although the syndrome is believed to be caused by the passive transfer of acetylcholine receptor antibodies, the pathophysiology remains unclear, and the syndrome occurs in infants who are antibody-negative. Infants of mothers with acquired myasthenia gravis are at risk for development of transient neonatal myasthenia gravis.25
The majority of infants with transient myasthenia have symptoms develop within the first 3 days after birth. Common clinical findings include shallow respiration, poor suck, hypotonia, and weak cry. Ptosis and ophthalmoplegia are less common. Mechanical respiration and gavage feedings are necessary in up to one third of the patients.25 Although some studies have correlated the severity of symptoms with the infant's antibody titers, others have not confirmed these findings.25
Genetic defects causing myasthenia gravis in the neonate are rare. These infants are born to mothers without myasthenia gravis, and antibodies against the acetylcholine receptor protein are not detected. Two clinical syndromes are recognized: familial infantile myasthenia and congenital myasthenia.26 The clinical features are similar to those of transient neonatal myasthenia, but long-term therapy may be necessary.
When the mother has myasthenia gravis, the diagnosis of transient neonatal myasthenia is not difficult. In addition to detecting high-serum titers of AChRP antibody in the newborn, an anticholinesterase agent should be administered. A subcutaneous or intravenous injection of edrophonium chloride (Tensilon), 0.15 mg/kg of body weight, should show a temporary reversal of an unequivocal neurologic deficit, such as sucking or swallowing difficulties. Repetitive nerve stimulation is recommended in combination with the Tensilon injection, especially in infants born to mothers with acquired AChRP antibody-negative myasthenia gravis.
Management of transient neonatal myasthenia gravis includes careful attention to airway maintenance, postural drainage, and nutritional status. Gavage feedings and mechanical ventilation are recommended in newborns with severe generalized weakness and respiratory distress. Most require anticholinesterase medication that can be discontinued slowly when the infant no longer has symptoms.
REFERENCES
Robertson NJ, Edwards AD: Recent advances in developing neuroprotective strategies for perinatal asphyxia. Curr Opin Pediatr 10: 575, 1998 |
|
Depp R: Perinatal asphyxia: Assessing its causal role and timing. Semin Pediatr Neurol 2: 3, 1995 |
|
Carter BS, Haverkamp AD, Merenstein GB: The definition of acute perinatal asphyxia. Clin Perinatol 20: 287, 1993 |
|
Roland EH, Hill A: Clinical aspects of perinatal hypoxic-ischemic brain injury. Semin Pediatr Neurol 2: 57, 1995 |
|
Hill A, Volpe JJ: Perinatal asphyxia: Clinical aspects. Clin Perinatol 16: 435, 1989 |
|
Latchaw RE, Truwit CE: Imaging of perinatal hypoxic-ischemic brain injury. Semin Pediatr Neurol 2: 72, 1995 |
|
Rossitch E Jr, Oakes WJ: Perinatal spinal cord injury: Clinical, radiographic and pathologic features. Pediatr Neurosurg 18: 149, 1992 |
|
Painter MJ: Neurologic Sequelae of Birth. In Depp R, Eschenbach DA, Sciarra JJ (eds): Gynecology and Obstetrics. Philadelphia, Harper & Row, 1987 |
|
Bracken MB, Shepard MJ, Collins WF etal: Methylprednisolone or naloxone treatment after acute spinal cord injury: 1-year follow-up of data. J Neurosurg 76: 23, 1992 |
|
Alfonso I, Alfonso DT, Papazian O: Focal upper extremity neuropathy in neonates. Semin Pediatr Neurol 7: 4, 2000 |
|
Sherburn EW, Kaplan SS, Kaufman BA et al: Outcome of surgically treated birth-related brachial plexus injuries in twenty cases. Pediatr Neurosurg 27: 19, 1997 |
|
Laurent JP, Lee RT: Birth-related upper brachial plexus injuries in infants: Operative and nonoperative approaches. J Child Neurol 9: 111, 1994 |
|
Slooff ACJ: Obstetric brachial plexus lesions and their neurosurgical treatment. Microsurgery 16: 30, 1995 |
|
Shenaq SM, Berzin E, Lee R et al: Brachial plexus birth injuries and current management. Clin Plast Surg 25: 527, 1998 |
|
Gilbert A: Long term evaluation of brachial plexus surgery in obstetrical palsy. Hand Clin 11: 583, 1995 |
|
Clarke HM, Curtis CG: An approach to obstetrical brachial plexus injuries. Hand Clin 11: 563, 1995 |
|
Harris MC, Roth P: Neonatology. In Polin RA, Ditmar MF (eds): Pediatric Secrets. Philadelphia, WB Saunders, 1989 |
|
Birchansky S, Altman N: Imaging the brachial plexus and peripheral nerves in infants and children. Semin Pediatr Neurol 7: 15, 2000 |
|
Falco NA, Eriksson E: Facial nerve palsy in the newborn: Incidence and outcome. Plast Reconstr Surg 85: 1, 1990 |
|
Levine MG, Holroyde J, Woods JR et al: Birth trauma: Incidence and predisposing factors. Obstet Gynecol 63: 792, 1984 |
|
Hepner WR Jr: Some observations on facial paresis in the newborn infant: Etiology and incidence. Pediatrics 8: 494, 1951 |
|
McHugh HE, Sawden KA, Levitt MN: Facial paralysis and muscle agenesis in the newborn infant: Etiology and incidence. Arch Otolaryngol 89: 131, 1969 |
|
Volpe JJ: Neurology of the Newborn, p 293. 2nd ed. Philadelphia, WB Saunders, 1987 |
|
von Gontard A, Arnold D, Adis B: Posterior fossa hemorrhage in the newborn-diagnosis and management. Pediatr Radiol 18: 347, 1988 |
|
Papazian O: Transient neonatal myasthenia gravis. J Child Neurol 7: 135, 1992 |
|
Fenichel GM: Clinical Pediatric Neurology, p 159. 2nd ed. Philadelphia, WB Saunders, 1993 |