Normal Sex Differentiation
Authors
INTRODUCTION
Sex differentiation is under strict genetic control. The basic difference between human males and females affecting every cell is the characteristic chromosomal combination: 46 chromosomes, including X and Y (46,XY), are essential for male cells and 46 chromosomes, with two Xs, for female cells. The difference originates from fertilization. In males, the oocyte is fertilized by a spermatozoon bearing the Y chromosome, in females, by a spermatozoon bearing the X chromosome. The oocytes always contain an X chromosome.
Y chromosomes are smaller than X chromosomes and contain fewer genes. The genetic inequality of X and Y chromosomes is compensated, if two X chromosomes are present within the cell nucleus, by heteropycnotic inactivation of much of one of the X chromosomes.
Within the interphase nuclei, the inactivated chromatin forms the X chromatin (Barr body). In normal 46,XX cells the inactivation is a random process affecting the paternally derived X chromosomes in 50% of cells and the maternally derived X chromosome in the rest. As the inactivation occurs at the stage of the blastocyst, it seems that sex differences are expressed early and that 46,XY blastomeres divide faster than 46,XX blastomeres.1 If more than two X chromosomes are present in the cell nucleus, the number of X chromatin bodies corresponds to the number of X chromosomes minus one.
A small distal portion of the heteropycnotic X chromosome escapes inactivation. This portion represents the pseudoautosomal segment of the X chromosome, which contains 2.5 megabases, homologous to the pseudosomal segment located on the distal portion of the short arm of the Y chromosome. During meiosis, the pseudoautosomal segment of the X and Y chromosomes pair and form chiasmata, permitting gene exchange.
Regarding sexual differentiation, the normal Y chromosome contains the sex reversal Y (SRY) gene, or testis-determining factor, which triggers testicular differentiation.2,3,4 The SRY gene is located on the short arm of chromosome Y in a subterminal region (Yp 21) and contains a domain of 80 amino acids (HMG box) that binds to DNA. The SRY gene activates a chain of other downstream genes related to the testicular morphogenic cascade. Although the SRY gene might be a mitotic regulator, it is not directly involved in testicular differentiation. Testicular development is possible in the absence of SRY in some 46,XX males and some true hermaphrodites.
The presence of the Y chromosome in interphase cell nuclei is manifested by the presence of Y chromatin. Y chromatin is a strongly fluorescent small chromatin cluster derived from the long arm of the Y chromosome. Its presence does not prove the presence of SRY.
CHROMOSOME X
Genes located on chromosome X are required for cellular survival. Many diseases, such as hemophilias A and B, adrenoleukodystrophy, colorblindness, Fabry's disease, G6PD anemia, and Duchenne muscular dystrophy, are X-linked. As regards sex development, the following genes are linked to the X chromosome:
- The androgen sensitivity gene (or the gene coding the androgen receptor)5,6 is located on the long arm of the X chromosome (Xq11–11.9) and has eight exons (A-H). Exon A regulates transcription, exon B and C are related to DNA binding, and exons E through H influence the binding of androgens. The gene determines the androgen sensitivity, and its presence in a normal form is required for male differentiation of the external genitalia. Different mutations of this gene are related to different phenotypic expressions, extending from incompletely virilized (but fertile) males to complete testicular feminization.
- The DAX 1 (DSS-AHG) gene,7,8,9 located at Xp21, codes the steroidogenic factor 1, an orphan nuclear receptor that binds to DNA without any ligand. Steroidogenic factor 1 activates P450-dependent steroidogenic enzymes in the gonads and adrenals and triggers secretion of antimüllerian hormone (AMH) within the testis. Dose-sensitive sex reversal is related to the DAX-1 gene. Its deletion is followed by hypoplasia of the adrenals and testes. Doubling of the dose-sensitive sex reversal locus in 46,XY persons is related to male gonadal dysgenesis.
- The FMR-1 gene9,10,11,12: The product of this gene is the protein related to fragility of the X chromosome. In males, fragility of the X chromosome is accompanied by increased proliferation of Sertoli cells, leading to macro-orchidism.
- Proliferation of granulosa cells is related to the presence of two X chromosomes. In X chromosome monosomy (45,X karyotype), the proliferation of granulosa cells is insufficient to provide a complete granulosa layer around oocytes. Consequently, oocytes degenerate at the stage of follicular differentiation.
- Some genes related to the development of ovarian follicles (granulosa cells?) are located on the long arm of the X chromosome (Xq2–6). Interstitial deletions of this region are observed in patients affected by early ovarian failure.
AUTOSOMAL GENES AFFECTING SEX DIFFERENTIATION
AMH gene13,14: The gene coding AMH is located on chromosome 19 (19p13.2–13.3). Prenatally, this gene is activated exclusively in differentiated embryonal Sertoli cells starting around day 45. Its presence may be used as marker of Sertoli cell differentiation. SRY is considered to be a promotor of the AMH gene.
AMH receptor coding gene15,16: Located on chromosome 12 (12q13, its product represents a receptor protein of AMH.)
WT-1 gene17 (Wilms' tumor suppressor gene): Located at 11p13, its product is a transcription regulator. Absence of one allele of this gene (as in deletions at 11p13) causes Wilms' tumor, aniridia, genital malformations, and mental retardation (WAGR syndrome). Mutation of one allele is present in the Denys-Drash syndrome (renal dysplasia, Wilms' tumor, and in some cases testicular dysgenesis and ambiguous external genitalia).18
SOX-9 gene19: Located at 17q24. The absence of this gene is observed in camptomelic syndrome (bending of long bones; 50% of affected children die in the perinatal period, the remaining 50% during the first year). In males, there is testicular dysgenesis and female or ambiguous external genitalia. Cleft palate is present in 80%. In homozygotes, the syndrome is lethal. The SOX-9 gene product is a DNA-binding protein with a homeobox exhibiting 60% homology with the HMG box of the SRY gene. The SOX-9 gene interferes with SRY. In females affected by camptomelic syndrome, the ovaries are normal.
CHROMOSOMAL DELETIONS
Terminal deletions of the short arm of chromosome 9 (9p-) and deletions of the long arm of chromosome 10 (10q-)20,21 are related to syndromes of testicular dysgenesis and ambiguous external genitalia. The 9q- may include the absence of the steroid P450c17 coding gene located at 9q22.
Genes Coding Factors Involved in Steroidogenesis
Steroid conversions occur in cells derived from steroidogenic mesenchyme. The following genes are involved in steroidogenesis influencing sexual differentiation:
Steroid acute regulatory (StAR) gene22,23,24: This gene codes the steroid acute regulatory protein, which binds cholesterol liberated by luteinizing hormone or human chorionic gonadotropin from cytoplasmic lipoid droplets and transports cholesterol from the outer to the inner mitochondrial membrane. Defects of this gene are related to cortical adrenal hyperplasia and male pseudohermaphroditism characterized by storage of cholesterol within the adrenal cortex and a marked deficit of steroid P450scc. Cholesterol is the basic substrate for steroidogenesis.
P450 cholesterol side chain cleavage (P450scc)25,26: In humans, the defects of this enzyme have not been satisfactorily proved. Low activity of P450scc in cases of cortical adrenal hyperplasia and male pseudohermaphroditism with cholesterol storage within the adrenals and Leydig cells is usually related to defects in the StAR gene.
3β-hydroxysteroid dehydrogenase (HSD) (and delta 5,4 isomerase) type III: This gene is located on chromosome 9 and is activated in the adrenals and gonads.
P450c1727,29: This gene, which codes for both steroid c17 hydroxylase and 17,20 lyase, is located at 10q24–25. The dissociation of the hydroxylating effect and lysis effect is related to the presence of histidin in position 347 and glutamine in position 358. 17β HSD (17β-dehydrogenase)30: Type I is active in ovaries and the placenta, type III in the testes. Located at 9q22.
SRD 5A (gene coding testosterone reductase)31: Type I, related to puberty, is located at 5p15; type II is active in the fetal external genitalia and the prostate and is located at 2p23.
CYP 19 gene32: Codes the steroid aromatase, which represents an enzyme complex bound to the endoplasmic reticulum; expressed in Sertoli cells, Leydig cells, granulosa cells, and trophoblast. The gene contains nine coding exons.
CELLS PARTICIPATING IN GONADAL DEVELOPMENT; THE INDIFFERENT GONADS
The indifferent primordia of gonads are longitudinal streaks of cellular tissue known as the genital ridges. They are present on the medioventral surface of the urogenital ridges in embryos 35 to 42 days old (7 to 12 mm crown rump length [CRL]). After the genital ridges become evident, the original urogenital ridges are called the mesonephric ridges. The blastema of each genital ridge consists of primordial germ cells (PGCs), mesoblastic (coelomic) cells, and mesenchymal fibroblasts.
Primordial Germ Cells
All gametes, testicular as well as ovarian, are derived from PGCs. Determination of germ cells occurs early in development, within the inner cell mass of the blastocyst. Stewart and Mintz33 injected cells of mouse embryonal carcinoma cultivated in vitro into mouse blastocysts and observed their differentiation into germ cells. This proves that early somatic cells are converted into germ cells. Hertig and associates34 claimed that one of the cells of the eight-cell inner cell mass of a human 4.5-day-old blastocyst might be a germ cell. Jirásek (unpublished data) found a group of several PGCs in the ectoblast adjacent caudally to the ectoblast of the germ disc of the bilaminar embryo with the primary yolk sac (Fig. 1).
|
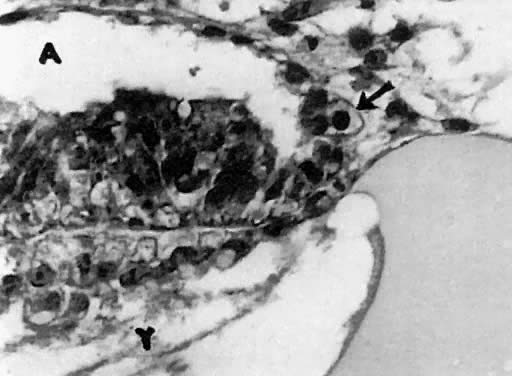
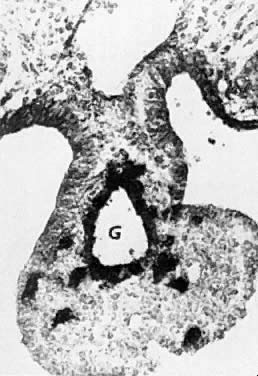
Subsequently, during rupture of the primary yolk sac, PGCs migrate into the connecting stalk and during closure of the secondary yolk sac colonize endoblasts adjacent to the connecting stalk. In early trilaminar embryos, PGCs are present in the endoderm of the posterior wall of the yolk sac and allantois.35 As the dorsal portion of the yolk sac transforms into the gut, PGCs begin to migrate from the endoderm to the urogenital ridges (Fig. 2 and Fig. 3). In somite embryos (25 to 40 days old), PGCs migrate using ameboid movement36 through the mesentery to the medioventral area of the urogenital ridges. PGCs proliferate mitotically beginning at their appearance and during the period of migration. The number of PGCs reaching the genital ridges is estimated to be 1,000 to 2,000. Within embryonal gonads, PGCs transform into spermatogonia or oogonia.
|
|
During migration, many PGCs do not find their way to the gonads.37 These lost extragenital cells are found in the adrenocortical blastema, in the mesonephros, and in the retroperitoneal mesenchyme. Germ cells located outside the gonads in the absence of special supportive cells degenerate.
Mesoblastic (Coelomic) Cells of the Genital Ridges
Coelomic cells proliferate near the attachment of the dorsal mesentery of the digestive tube. The cells on the surface contribute to the peritoneal lining or to the surface epithelium of gonads; the cells under the surface contribute the steroidogenic mesenchyme. The retroperitoneal steroidogenic mesenchyme located medially and cranially turns into the adrenocortical primordium; the steroidogenic mesenchyme adjacent to the medioventral surface of the urogenital ridges contributes the steroidogenic cell progenitors of the genital ridges (indifferent gonads). The progenitors of steroidogenic cells contribute most of the gonadal blastema, and in terms of gonadal differentiation contribute supportive cells (Sertoli cells, granulosa cells) as well as steroidogenic interstitial cells (Leydig cells, thecal cells).
Mesenchymal (Desmogenic) Fibroblasts of the Genital Ridge
Desmogenic fibroblasts contributing connective tissue of the gonads originate from the interstitium of the urogenital ridges. They mix with proliferating coelomic cells.
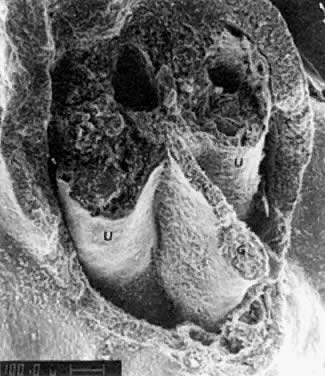
PGCs, coelomic cells (surface and steroidogenic), and mesenchymal fibroblasts contribute the genital ridges (indifferent gonads), which extend in human embryos 40 to 44 days old ( Jirásek stage 7–4) from T7 cranially to L4, S1 in males and S2 in females. The genital ridges are located on the surface medially to the mesonephric ridges. The genital and mesonephric ridges are anchored caudally to the inguinal areas by mesenchymal cords, the future gubernaculum of the testis or the round ligament of the uterus. The genital ridges represent the indifferent gonads (Fig. 4, Fig. 5, and Fig. 6).
|
|
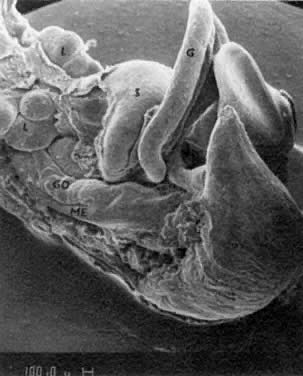
|
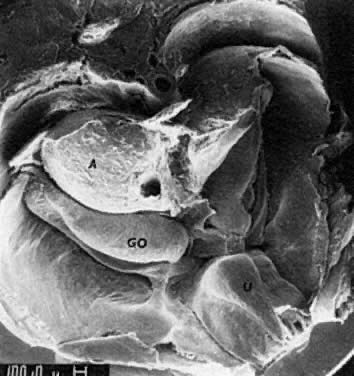
TESTICULAR DEVELOPMENT
Embryonal Testis (SRY-Related Morphogenic Cascade of Primary Testicular Development)
The first stage is mitotic regulation of PGCs. In 46,XY embryos 42 to 45 days old ( Jirásek stage 7–5), PGCs interact with adjacent coelomic cells (cell-to-cell interaction). PGCs temporarily lose glycogen, alkaline phosphatase, and their round shape and join the undifferentiated supportive cells.38,39 The consequence of this interaction is lifelong mitotic proliferation of germ cells (spermatogonia). Mitotic regulation of germ cells precedes differentiation of supportive cells into embryonal Sertoli cells (Fig. 7).
|
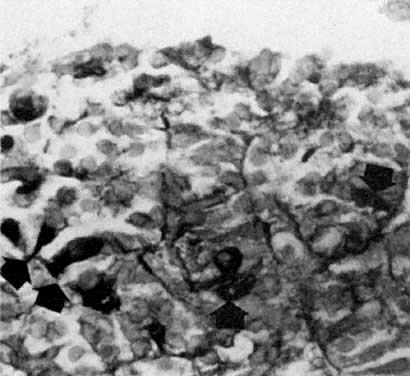
The second stage, differentiation of Sertoli cells, includes transformation of steroidogenic small round mesenchymal cells with spheric nuclei into cylindrical embryonal Sertoli cells with elongated nuclei exhibiting differentiation of an apical (supranuclear) domain and a basolateral domain. Differentiation of the supranuclear zone is characterized by the presence of acid phosphatase. Differentiation of the basolateral domains of Sertoli cells is related to the aggregation of Sertoli cells into testicular cords. PGCs (spermatogonia) are incorporated into cords regardless of Sertoli cell differentiation.
The character of the cords depends on the surrounding mesenchyme: the portions of the testicular cords located within the gonadal blastema give rise to the seminiferous cords, and the portions adjacent to the mesonephros, located within the mesonephric mesenchyme, give rise to the cords of the rete testis (Fig. 8). Embryonal Sertoli cells produce AMH.14,15
|
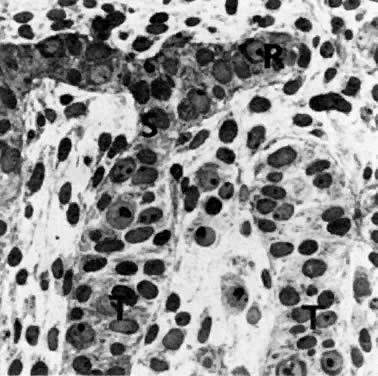
The barrier between the germinal testicular compartment and the extracellular compartment of the interstitium is formed by the specific membranes under the testicular cords. These membranes are contributed both by Sertoli cells and by the mesenchymal desmogenic cells of the interstitium.
The interstitium of the embryonal testis (days 45 to 56 after conception) contains a mixture of desmogenic fibroblasts and undifferentiated steroidogenic progenitor cells that do not produce any androgens. The interstitium is filled with glucosaminoglycans synthesized under the influence of AMH. The glucosaminoglycans are related to the regression of the müllerian ducts.40
At the end of embryonal period, desmogenic fibroblasts concentrate under the surface epithelium and contribute the primordium of the tunica albuginea.
Fetal Testis
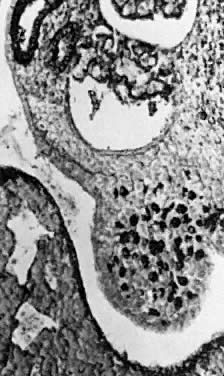
The stage of fetal testis is characterized by the presence of interstitial Leydig cells (Fig. 9), which produce androgenic steroids, predominantly testosterone. Steroidogenesis is controlled by the genes of steroid conversion.
|

Precursors of Leydig cells are progenitor steroidogenic cells scattered among desmogenic fibroblasts in the interstitium of embryonal testes. Transformation of steroid-producing precursor cells into steroidogenic cells is characterized by the presence of acid phosphatase in the Golgi zone. The process includes the human chorionic gonadotropin-luteinizing hormone (hCG-LH) receptors and binding of hCG-LH. Steroid-producing Leydig cells are epithelioid cells located in the testicular interstitium. They first appear around day 60 of development at the end of the embryonal period.38 Their numbers increase until the end of third month and decrease thereafter until birth. Characteristic enzyme activity of the fetal Leydig cells, which is easy to demonstrate histochemically, is the 3β-OH steroid dehydrogenase. The main steroid produced by fetal Leydig cells is testosterone.41 Its principal role is to support the growth of the epididymis and ductus deferens and to control (after conversion to dihydrotestosterone [DHT]) the growth and differentiation of the prostate and the male external genitalia. Prenatal sex-related differentiation of the central nervous system is also related to exposure to testicular androgens.42
During the prenatal period, within the fetal testes, Sertoli cells fail to mature and are insensitive to androgens. Fetal spermatogonia divide slowly mitotically and never enter meiosis. After birth, Leydig cells disappear due to insufficient LH support (inactive testis of a child) and reappear at the beginning of puberty. Mature Sertoli cells, which become androgen-sensitive, appear consequently. The prepubertal testis is converted (related to stimulation of LH and follicle-stimulating hormone [FSH]) into an active adult organ exhibiting steroidogenic Leydig cells, luminization of seminiferous tubules with mature Sertoli cells, mitotic proliferating spermatogonia, and terminally differentiated meiotic spermatocytes, prespermatids, and spermatids, as well as mature spermatozoa. In healthy males, germinal proliferation occurs from puberty until death.
Testicular Descent
Subsequent to the transformation of the genital ridges into embryonal and fetal testes and the degeneration of the mesonephros, the testes descend into the pelvis. They are anchored by the caudal ligaments of the urogenital ridges to the labioscrotal swellings of the indifferent external genitalia. Later, as the scrotum is formed, the caudal ligaments of the urogenital ridges transform on each side into the testicular gubernaculum. The gubernaculum contains connective tissue rich in glucosaminoglycans, whose differentiation is influenced by testosterone.
During the sixth fetal month, a diverticulum of the abdominal wall evaginates along the gubernaculum through the inguinal canal into the scrotum. The external envelope of the evagination is the tunica vaginalis testis et funiculi spermatici, which contains the internal spermatic fascia, detached from the fascia of the transverse abdominal muscle, and m. cremaster with the fascia cremasterica, which are detached together from the internal oblique abdominal muscle and its fascia. The external spermatic fascia is formed from the fascia of the external oblique abdominal muscle. Shortly before birth, the testes descend retroperitoneally through the inguinal canal into the scrotum as the gubernaculum shortens, and they become located retroperitoneally aside the scrotal diverticulum.
OVARIAN DEVELOPMENT
The morphogenic sequence of ovarian development is divided into two basic periods: prefollicular and follicular. The prefollicular period precedes differentiation of primary isolated ovarian follicles and includes mitotic proliferation of oocytes and their entrance into the meiotic prophase. The follicular period is related to the interruption of meiosis I, after termination of the prophase, and to the differentiation of isolated ovarian follicles with a distinct granulosa layer. The first phase does not require the presence of two X chromosomes (as observed also in 45,X fetuses).43
The prefollicular phase of ovarian development has two stages: the embryonal ovary and the early fetal ovary.
Embryonal Ovary
The embryonal ovary40 is present in embryos of 18- to 40-mm CRL and is characterized by mitotic proliferation of germ cells (oogonia) within the genital ridges. Simultaneously, the blastema of the indifferent gonad differentiates into epithelial cords and interstitium (Fig. 10). The interstitium contains desmogenic fibroblasts, the cords that form the rete adjacent to the mesonephros, and medullary cords, which are incompletely separated from the surface epithelium. Medullary cords contain undifferentiated supportive cells and oogonia. The PGCs located in contact with and under the surface epithelium undergo an intensive mitotic proliferation, resulting in the formation of cortical cords composed almost exclusively of oogonia. Embryonal ovaries are characterized by intensive mitotic proliferation of oogonia, which do not interact with other cells. The ovarian cords result from differentiation of the connective tissue. By definition, no meiotic activity occurs in the embryonal ovary. The mitotic wave of oogonia exhausts their entire mitotic capacity during fetal development.
|
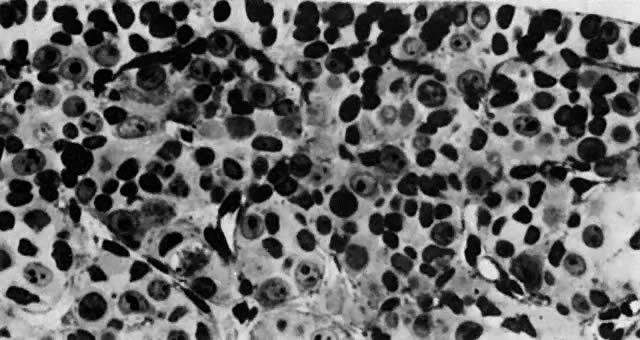
Early Fetal Ovary
At the beginning of meiosis I, meiotic oocytes appear in the medullary cords and the deepest portions of the cortical cords in fetuses of about 40 mm CRL (end of week 10). Oocytes entering the meiotic phase form clones and are interconnected by cytoplasmic bridges. All interconnected oocytes are at the same stage of meiotic prophase (Fig. 11). Leptotene, zygotene, and pachytene oocytes are present by week 11; diplotene oocytes appear a week later.
|

Simultaneously, as the “oldest” oocytes enter meiosis, the “young” oogonia underneath the surface epithelium continue mitotic proliferation. In early fetal ovaries, there are no primary isolated follicles with a complete granulosa layer and connective tissue envelope.
In fetuses affected by X-chromosome monosomy, normal ovarian development proceeds until this early fetal stage. The second phase—the follicular phase—of ovarian development is related to the interruption of meiosis I after the prophase has been completed, and to the differentiation of isolated primary follicles. During the phase of follicular differentiation, the stages of late fetal ovary and perinatal ovary are distinguished.
Late Fetal Ovary
After the oocytes reach the diplotene stage of the first meiotic prophase, connective tissue invades groups of meiotic oocytes, and at the same time granulosa cells—steroidogenic progenitors of coelomic origin—spread over the surface of the oocyte and contribute an epithelial granulosa layer around the oocyte (Fig. 12). At this stage, the granulosa cells interact with the oocyte and interrupt the meiosis. The nuclei of oocytes return to an interphase state. If the proliferation of granulosa cells is inadequate, oocytes come into direct contact with the connective tissue and undergo degeneration (atresia).
|
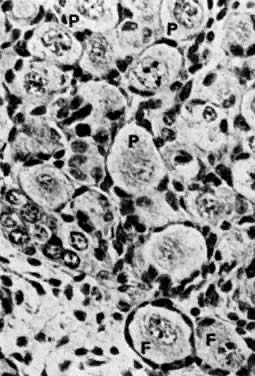
Although the primary isolated follicles differentiate in the deepest portions of the medullary and cortical oocytes (the “oldest” oocytes), the superficially located oogonia continue mitotic division, differentiate into oocytes, and enter the meiotic prophase. There are no steroid-producing cells around primary ovarian follicles.
The interval between the end of the first meiotic prophase and metaphase lasts until ovulation of the follicle, occurring throughout the reproductive years (i.e., 12 to 50 years of age). The formation of complete follicles requires the presence of at least two X chromosomes. In late fetal ovaries, the number of oocytes44 is estimated to be 6 million; at birth, about 2 million oocytes are present, incorporated into follicles. Both ovaries of a 20-year-old woman are thought to contain 400,000 oocytes. Of these, only about 400 are ovulated during the entire reproductive period of a woman.
The formation of follicles within late fetal ovaries in the presence of two X chromosomes represents a rescue operation, breaking meiosis I and saving some oocytes before termination of meiosis and consequent degeneration for a maximum of 50 to 60 years. In normal fetal ovaries, only 30% of the oocytes that are formed reach the stage of primary follicles.
Perinatal Ovary
The growth and cavitation of the follicles are characteristic aspects of the perinatal ovary. All stages of folliculogenesis are present in perinatal ovaries during the second postnatal month: primary resting follicles with flat granulosa cells, primary growing follicles with cylindrical granulosa cells, compact growing follicles with a multilayered granulosa, and vesicular (cavitated) follicles. Epithelioid thecal cells containing the 3β-hydroxysteroid dehydrogenase first appear in perinatal ovaries around follicles with a multilayered granulosa at the stage of beginning cavitation beneath the cumulus—oocyte complex. Follicular cavitation occurs in newborns after separation from the placenta.
Drafting of oocytes begins in the perinatal ovaries. As the production of hypophyseal gonadotropins is discontinued, shortly after birth, the perinatal ovary changes during the first postnatal year into the inactive or prepubertal ovary of a child. During childhood, follicular drafting continues, and the drafted follicles (in the absence of gonadotropins) develop to the stage of cavitated follicles. There are, however, no thecal cells around the growing follicle. The absence of thecal cells is a characteristic feature of the ovaries of a child. During puberty, the ovary gradually begins to produce steroids under the influence of FSH and LH. In addition to drafting, in the adult ovaries oocytes grow and are selected, and some are ovulated. Ovulated follicles change into the corpora lutea. As the follicles become exhausted, the adult ovary changes into a menopausal ovary completely deprived of follicles (at the age of 50 to 60 years).
Differences Between Male and Female Gonadal Development
The most important difference between males and females is that there is lifelong proliferation of spermatogonia, but the proliferation of oogonia is limited to a short prenatal period. The analysis of morphogenesis suggests that the early differentiation of PGCs, which are not incorporated into germ layers, inhibits almost all their genes related to differentiation. The basic programming of germ cells is related to their migration into gonads and to a programmed number of mitoses followed by meioses. If there is one germ cell in the eight-cell inner cell mass of the blastocyst, and if the population of germ cells reaching the genital ridges is about 1,000, there are about 11 mitotic generations of PGCs. Within the ovaries, if the population of oocytes is around 5 million, there are an additional 12 to 14 generations of oogonia. The entire mitotic capacity of the oogonia is exhausted by this mitotic division; thereafter, germ cells undergo terminal differentiation into oocytes, which enter meiosis. The prophase of meiosis I prenatally creates a unique combination of genes from the paternal and maternal chromosomal sets.
The formation of ovarian follicles, which is related to the presence of two X chromosomes, is based on interaction between granulosa cells and meiotic oocytes. Meiosis in oocytes is interrupted after the prophase has been completed, and some of the oocytes are saved for a period of no longer than 55 years. Therefore, the formation of ovarian follicles represents a rescue operation, saving mitotic oocytes.
In testicular development, the mitotic programming of male germ cells is modified by the cell-to-cell interaction of germ cells with the progenitors of gonadal supportive cells. This interaction between male germ cells and undifferentiated supportive cells changes the mitotic program of male germ cells, extending their mitotic proliferation for the man's entire life span and preventing them from undergoing terminal differentiation and meiosis before puberty.
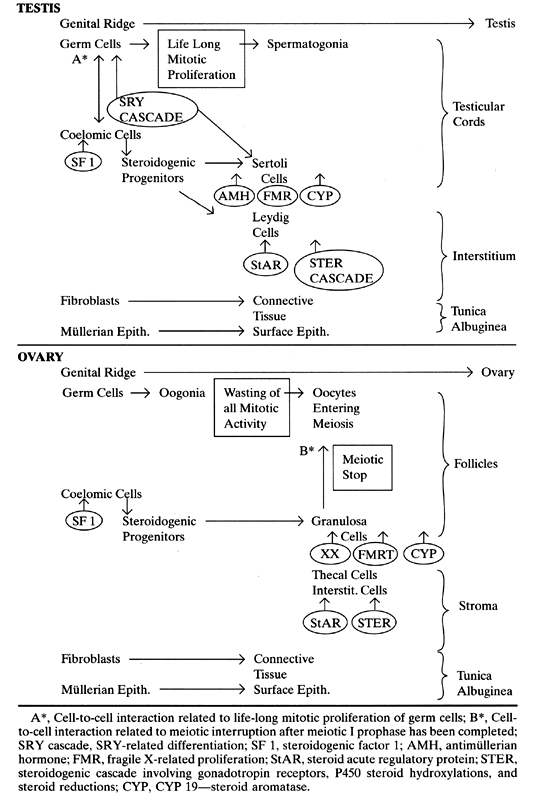
Proposed gene activations and a comparison of testicular and ovarian development are summarized in Table 1.
TABLE 1. Genes and Gonadal Development
DEVELOPMENT OF GENITAL DUCTS
The urogenital sinus, mesonephros, mesonephric ducts, and paramesonephric ducts all participate in the development of genital ducts.45
Urogenital Sinus
The hindgut is an endodermal pocket closed by the cloacal membrane. By the end of the fourth developmental week, the mesonephric ducts open laterally into the hindgut, thus turning the terminal portion of this organ into the cloaca. The cloaca becomes subdivided by a frontally oriented urorectal septum, contributing the primitive perineum, into a dorsal anorectal canal and a ventral primitive urogenital sinus (with openings of the mesonephric ducts). The cloacal membrane closing the hindgut splits into the anal membrane, closing the anorectal canal, and the urogenital membrane, closing the urogenital sinus. The urogenital membrane extends anteriorly to the tip of the genital tubercle, contributing the urethral plate. The urogenital membrane ruptures, leaving the opening of the urogenital sinus (beginning at 8 weeks in 20-mm embryos). The anal membrane disintegrates several days later, leaving the anal opening. The upper portion of the primitive urogenital sinus located above the openings of the mesonephric ducts is known as the vesicourethral primordium, and the portion located below the openings of the mesonephric ducts is known as the definitive urogenital sinus. The urogenital sinus, with its accompanying mesenchyme, is closely related to the formation of the external genitalia, the vaginal vestibule, and its glands in females and the prostate, bulbourethral glands, and the cavernous urethra in males.
Mesonephric (Wolffian) Ducts and Mesonephros
The uropoietic organs (pronephros, mesonephros, and metanephros) are derivatives of the intermediary mesoderm located between the somites and the unsegmented mesoderm of the somatopleura and the splanchnopleura. Rudimentary pronephric nephrons are located lateral to the cervical segments. Several epithelial buds (one per segment) evaginate from the cervical glycogen-rich surface ectoderm, detach, and assemble lateral to the groups of pronephric cells.40,46 Consequently, ectodermal cells of the neighboring segment join and contribute a longitudinally growing pronephric duct. During mesonephric development (C6 to L3), all mesonephric nephrons enter this pronephric duct, which becomes the mesonephric duct. Mesonephroi (the plural of mesonephros) are located lateral to the dorsal mesentery in the urogenital ridges. Both mesonephric ducts enter the primitive urogenital sinus, which is not detached from the hindgut (5- to 6-mm embryos, fifth week after conception). By the end of the fifth week after conception, ureteric buds evaginate from the terminal portion of the mesonephric duct and grow into the mesonephric blastema, located in L4 and L5, thereby inducing the differentiation of nephrons of the definitive kidneys.
As the urogenital ridges transform into mesonephric ridges and genital ridges, the mesonephric ridges located lateral to the gonads may be divided into epigenital and paragenital portions. The epigenital portions contain 5 to 12 mesonephric nephrons, connecting the rete of the gonads.
In males, the epigenital mesonephric tubules develop into the ductuli efferentes of the epididymis (Fig. 13 and Fig. 14). The paragenital tubules degenerate and are known as the paradidymis.
|
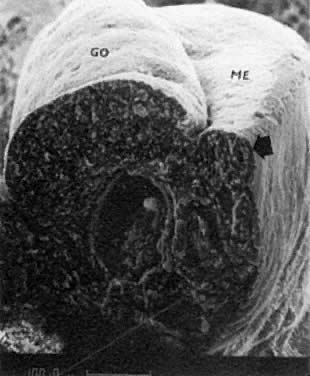
|
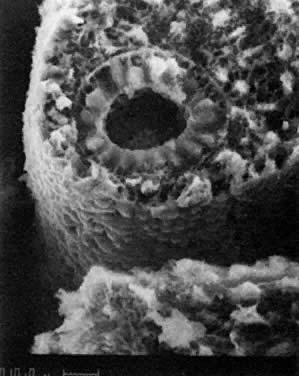
In females, both the epigenital and the paragenital nephrons transform into the irregular tubules of the epo-ophoron and paro-ophoron. Growth of the mesonephric ducts and of the epigenital mesonephric tubules is testosterone-dependent. In males, in the presence of testosterone and its receptors in target tissues, the mesonephric ducts give rise to the ductus epididymis, ductus deferens, and ductus ejaculatorius. The vesicular glands are formed as evaginations from the terminal portion of the mesonephric ducts. In the absence of testosterone, the mesonephric ducts disintegrate. The persisting remnants of the cranial portion of the mesonephric ducts in males are known as the appendix of the epididymis or, in females, the appendix of the ovary. Remnants of the mesonephric ducts present in females within the plica lata or along the vaginal wall (sometimes reaching the vaginal vestibule) are known as Gartner's canals (ducts).
Paramesonephric (Müllerian) Ducts
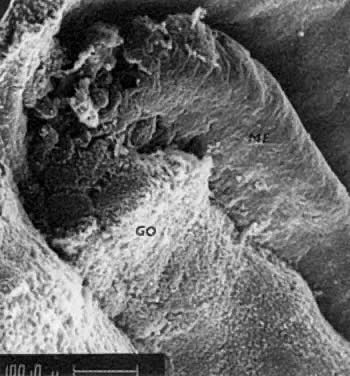
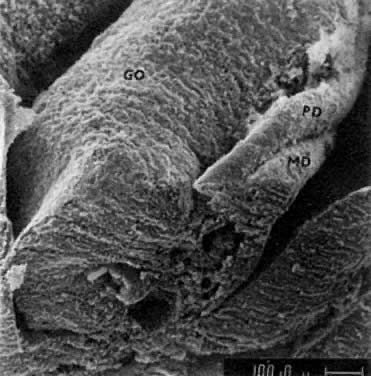
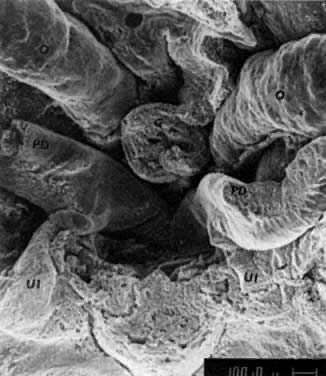
The paramesonephric ducts originate in 13- to 15-mm embryos (44 to 48 days after conception) as invaginations lined by coelomic cells of the gonadal surface epithelium, growing from the upper pole of the genital ridge to the mesonephric ridge (Fig. 15). This epithelium invaginates lateral to the superior blind end of the mesonephric duct, located in the regressing mesonephric ridge.47 The invaginating cellular cord of the paramesonephric duct grows caudally in contact with the mesonephric duct, using the latter duct as a guiding structure.46,48 The paramesonephric ducts cross the mesonephric ducts ventrally (Fig. 16), approach near the midline, and run parallel to the urogenital sinus. Luminization of the paramesonephric ducts proceeds craniocaudally. Cranially, the ducts open into the peritoneal cavity near the cranial poles of the gonads (Fig. 17 and Fig. 18). Caudally, the blind ends of both ducts contact the endoderm of the urogenital sinus (without breaking through) between the openings of the mesonephric ducts in the elevated area known as the müllerian tubercle. Parallel segments of both paramesonephric ducts are separated temporarily by a septum. As the septum disintegrates, the caudal portions of both paramesonephric ducts fuse into a single uterovaginal canal (during weeks 9 to 10 after conception) (Fig. 19).
|
|
|

|

|
Female Transformation of Paramesonephric Ducts
Each paramesonephric duct may be divided into an upper and lower portion. The upper portion develops into the ovarian tube, and the lower portions of both ducts fuse during the ninth week into a single uterovaginal canal (see Fig. 19). The uterovaginal canal is composed of a uterine and a vaginal segment. In the uterine segment, the mesenchyme accompanying the mesonephric ducts fuses with the mesenchyme of the paramesonephric ducts and gives rise to the uterine wall, mainly myometrium; at the same time, the epithelium of the paramesonephric ducts contributes the surface lining and epithelium of the glands.
The vaginal segment of the uterovaginal canal contacts the endodermal epithelium of the urogenital sinus in the area of the müllerian tubercle. Paired endodermal outgrowth (sinovaginal bulbs) enfold the tip of the uterovaginal canal, and multilayered epithelium develops around the cylindrical paramesonephric epithelium of the vaginal segment of the uterovaginal canal, resulting in the formation of the vaginal plate. As multilayered epithelium expands around the lower end of the uterine mesenchymal condensation, vaginal fornices are formed, and the simple paramesonephric mesodermal cylindrical epithelium located centrally in the vaginal plate degenerates. The vaginal lumen is formed by a dehiscence of the central layers of the multilayered epithelium of the vaginal plate. The proximal four fifths of the vagina is formed around the vaginal portion of the uterovaginal canal; the caudal fifth arises entirely from the endodermal sinovaginal bulbs.
In some areas where the cylindrical epithelium of the uterovaginal canal is not incorporated into the vaginal plate and remains at its periphery, persistent islands of cylindrical ( müllerian) epithelium may give rise to vaginal adenosis. Mesenchymal remnants of the müllerian tubercle located between the vagina and the urogenital sinus contribute to the hymen.
Regression of Paramesonephric Ducts in Males
In males, the central paramesonephric ducts degenerate, leaving only remnants of their cranial and caudal ends. The regression of each paramesonephric duct is closely related to the development of the ipsilateral testis. The decisive factor in this process ( müllerian inhibiting factor [MIH]) is produced by embryonal Sertoli cells and seems to be related to a substance rich in glucosaminoglycans that fills the interstitial spaces of the embryonal testes.42 The testicular glucosaminoglycans with MIH diffuse along the caudal gonadal ligament, which crosses the paramesonephric and mesonephric ducts. The epithelial cells of the paramesonephric ducts, exposed to MIH and testicular glucosaminoglycans, swell and rupture, and consequently liberated acid hydrolases destroy the ductal cells. Once initiated, the regression of the paramesonephric ducts extends cranially as well as caudally. The cystic cranial portion of the paramesonephric duct, corresponding to the infundibulum of the oviduct, changes into the testicular appendix (hydatid of Morgagni). The caudal portions of the uterovaginal canal remain preserved within the prostate as the prostatic utriculus.
MORPHOGENESIS OF THE EXTERNAL GENITALIA
Indifferent Stage
The external genitalia originate from mesenchyme located around the cloaca (the anal portion of the hindgut and the primitive urogenital sinus), enclosed by the cloacal membrane. The cloacal area is elevated and represents the genital tubercle of classic embryology. The cloacal membrane is sagittally oriented, is anchored anteriorly to a small glandar tubercle, and is delineated laterally by cloacal folds, ending posterolaterally as anal hillocks (Fig. 20). As the glandar primordium of the phallus develops, the anterior portion of the cloacal membrane provides a sagittally oriented epithelial plate (plug), and the corporal portion of the cloacal membrane is transformed into a depressed urethral groove between the cloacal folds.
|
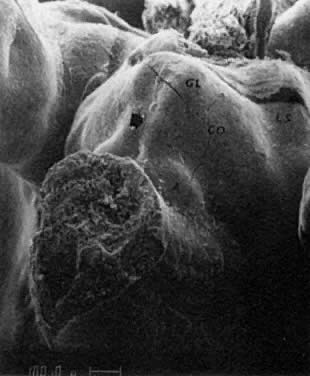
The rectum becomes separated from the urogenital sinus by a peritoneal fold known as the urorectal septum. Consequently, the cloacal membrane becomes divided by the primitive perineum into the genital membrane anteriorly and the anal membrane posteriorly. The genital membrane disintegrates in 18- to 20-mm embryos (8 weeks after conception). The anal membrane ruptures in 20- to 22-mm embryos. The derivative of the genital tubercle anterior to the opening of the urogenital sinus is properly known as the phallus. Mesenchyme located dorsolateral to the urethral groove contributes the cavernous bodies, and mesenchyme in the urethral folds becomes the spongy body of the urethra. Labioscrotal swellings are cutaneous lobes lateral to the genital tubercle (Fig. 21). The caudal ligaments of the urogenital ridges (testicular gubernaculum in males, round ligament of uterus in females) are anchored in their mesenchyme.
|
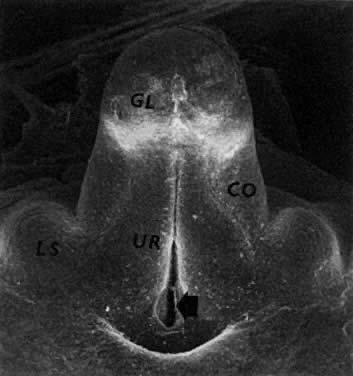
Masculinization
Masculinization of the external genitalia consists of the transformation of the phallus and labioscrotal swellings, respectively, into the penis and scrotum. During the masculinizing process, the urogenital distance increases and the labioscrotal swellings become interposed between the anus and the opening of the urogenital sinus. The labioscrotal swellings then fuse posteroanteriorly into the scrotum in a zipperlike fashion as the scrotal raphe is formed (Fig. 22). As the rims of the urethral groove fuse (posteroanteriorly), the cavernous urethra is contributed by the urethral folds. The tip of the cloacal membrane, preserved on the glandar primordium for a considerable time as the epithelial plug, becomes luminized by the dehiscence of the epithelium. This process is related to the formation of the preputial lamella, frenulum, and navicular fossa.
|
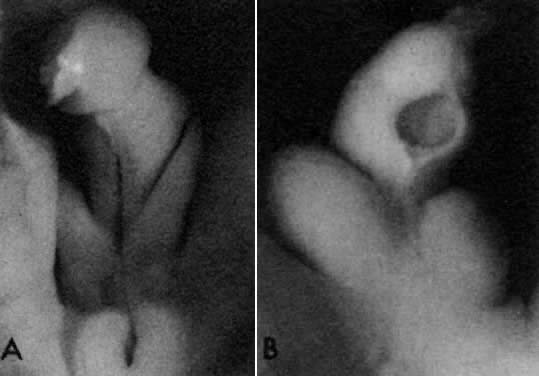
Masculinization is a dihydrotestosterone-dependent process taking place in 35- to 90-mm fetuses during weeks 9 to 12 after conception. Male accessory organs, such as the prostate and the bulbourethral glands, originate from endodermal buds of the urogenital sinus growing into a specific urogenital mesenchyme.
Feminization
Feminization of the external genitalia consists of the conversion of indifferent genitalia with a phallus and labioscrotal swellings into a female vulva with a clitoris, labia minora, labia majora, and vaginal vestibule (Fig. 23).
|
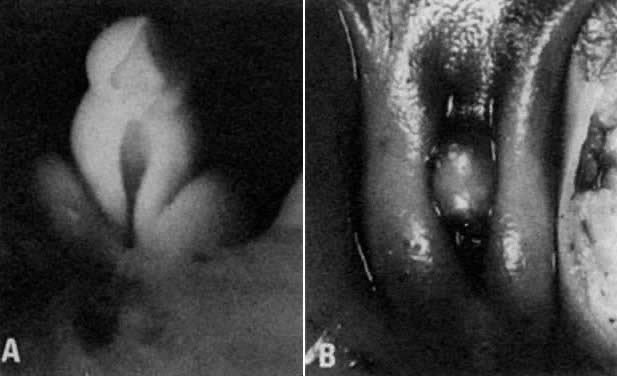
In contrast to masculinization, during the feminizing process there is no increase in the anogenital distance and no fusion of the labioscrotal swellings, nor of the rims of the urethral groove. The phallus bends ventrally, and its growth is slow. A sagittal lengthening transforms the urogenital sinus into the vaginal vestibule, and the vaginal and urethral openings become separated. The derivative of the genital tubercle located above the separated urethral and vaginal openings is the clitoris. The rims of the urethral groove become the labia minora, the labioscrotal swellings the labia majora.
Feminization of the external genitalia results from the absence of androgens or from androgen insensitivity related to various disorders of the androgen receptor. Female accessory sex glands, such as the glandulae vestibulares majores (Bartholin's glands), minores, and urethrales, originate from endodermal buds growing from the epithelium of the urogenital sinus into adjacent mesenchyme.
Similarities in the development of male and female genital organs are evident from Table 2.
TABLE 2. Homologues in Male and Female Genital Systems
Indifferent Primordia | Male | Female |
Genital ridge | Testis | Ovary |
Caudal ligament | Gubernaculum | Ligament of ovary and round |
Mesonephros |
| ligament of uterus |
Epigenital portion | Efferent ducts of epididymis | Epoophoron |
| (caput epididymis) |
|
Paragenital portion | Paradidymal ducts | Paroophoron |
Mesonephric duct | (Appendix of epididymis) | (Appendix of ovary) |
| Duct of epididymis | Gartner's duct |
| Ductus deferens |
|
| Ductus ejaculatorius |
|
| Seminal vesicle |
|
Paramesonephric ducts |
|
|
Paired cranial portions | Appendix of testis | Uterine tubes |
Uterovaginal canal | Utriculus prostaticus | Uterus, upper four fifths of vagina |
Müllerian tubercle | Colliculus seminalis | Hymen |
Urogenital sinus | Prostatic and membranous | Lower fifth of vagina |
| urethra | Vaginal vestibule |
| Prostate | Urethral and paraurethral |
Indifferent external genitalia |
| (Skene's) glands |
Genital tubercle |
|
|
Pallus | Penis | Clitoris |
Glans | Glans of penis | Glans of clitoris |
Corpus with urethral | Corpus of penis with cavernous | Crura of clitoria |
groove | urethra | Labia minora |
Labinoscrotal swellings | Scrotum | Labia majora |
REFERENCES
Pergament E et al: Sexual differentiation and preimplantation cell growth. Hum Reprod 9: 1730–1732, 1994 |
|
Sinclair AH et al: A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 346: 240–244, 1990 |
|
Berta P et al: Genetic evidence equating SRY and the testis determining factor. Nature 348: 448–450, 1990 |
|
Giese K, Page J, Grossckedl R: Distinct DNA-binding properties of HMG domain of murine and human SRY. Proc Natl Acad Sci USA 91: 3368–3372, 1994 |
|
Quigley C et al: Androgen receptor defects: Historical, clinical and molecular perspective. Endocrine Rev 6: 271–321, 1955 |
|
Pinsky L et al: Lessons from androgen receptor gene mutations that cause androgen resistance in human. In Hughes IA (ed): Sex Differentiation: Clinical and Biological Aspects. Frontiers in Endocrinology, Vol 20, Serono Symposia, 1996 |
|
Luo X, Ikeda Y, Parker KL: A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell 77: 481–490, 1994 |
|
Shen WH et al: The orphan nuclear receptor SF-1 regulates MIS expression. Links to the sex determining pathway. Cell 77: 651–661, 1994 |
|
Ingraham HA et al: The nuclear receptor steroidogenic factor 1 acts at multiple levels of the reproductive axis. Genes Dev 8: 2302–2312, 1994 |
|
Verlerk AJ et al: Identification of a gene (FMR-1) containing a CGG repeat coincident with breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65: 905–911, 1991 |
|
Nistal M et al: Macro-orchidism. Light and electron microscopic study of four cases. Hum Path 23: 1011–1018, 1992 |
|
Laxova R: Fragile X syndrome. Adv Pediatr 41: 305–315, 1994 |
|
Lee MM, Donahoe PK: Müllerian inhibiting substance: A gonadal hormone with multiple functions. Endocrin Rev 14: 152–164, 1993 |
|
Imbeand S et al: Molecular genetics of persistent müllerian duct syndrome: A study of 19 families. Hum Mol Genet 3: 125–131, 1994 |
|
Clemente di N et al: Cloning expression and alternate splicing of the receptor for anti-müllerian hormone. Mol Endocrinol 8: 1006–1020, 1994 |
|
Imbeand S et al: Insensitivity to anti-müllerian hormone due human anti-müllerian hormone receptors. Nature Genet 11: 382–388, 1995 |
|
Glaser T et al: A highly polymorphic locus cloned from the break point of a chromosome 11p13 deletion associated with the WAGR syndrome. Genomic 5: 880–893, 1989 |
|
Pelletier J et al: Germline mutations in the Wilms tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash Syndrome. Cell 67: 437–447, 1991 |
|
Wagner T et al: Autosomal sex-reversal and camptomelic dysplasia are caused by mutations in and around the SRY-related gene SOX-9. Cell 79: 1111–1120, 1994 |
|
Benett CP et al: Deletion 9p- and sex reversal. J Med Gen 30: 518–520, 1993 |
|
Wikie OAM et al: Complete and partial XY sex reversal associated with terminal deletion of 10q. Am J Med Genet 46: 597–600, 1993 |
|
Lin D et al: Normal genes for cholesterol side chain cleavage enzyme, P450scc, in congenital adrenal hyperplasia. J Clin Invest 88: 1955–1962, 1991 |
|
Lin D et al: Role of steroidogenic acute regulatory protein in adrenal and gonadal steroidogenesis. Science 267: 1828–1831, 1995 |
|
Jeffcoate CR et al: Regulation of cholesterol movement to mitochondrial cytochrome P450scc in steroid hormone synthesis. J Steroid Biochem Molec Biol 43: 751–767, 1992 |
|
Yang X et al: Inherited congenital adrenal hyperplasia in the rabbit is caused by a deletion in the gene encoding cytochrome P450 cholesterol side-chain cleavage enzyme. Endocrinology 132: 1977–1982, 1993 |
|
Lin D et al: Normal genes for cholesterol side chain cleavage enzyme, P450scc, in congenital lipoid adrenal hyperplasia. J Clin Invest 88: 1955–1962, 1991 |
|
Resume E et al: Structure and expression of a new complementary DNA encoding 3 beta-hydroxysteroid dehydrogenase delta 5–4 isomerase in human adrenals and gonads. Mol Endocrinol 5: 1147–1157, 1991 |
|
Kitamura M, Buczko E, Dufan M: Dissociation of hydroxylase and lyase activities by site-directed mutagenesis of rat P450c17. Mol Endocrinol 15: 1373–1380, 1991 |
|
Geller DH, Mendonca BB, Miller WL: The molecular basis of isolated 17,20 lyase deficiency. Pediatr Res 39 (part 2):89A, 1996 |
|
Anderson S et al: Molecular genetics and pathophysiology of 17β-hydroxysteroid dehydrogenase 3 deficiency. J Clin Endocrin Metab 81: 130–136, 1996 |
|
Russel DW, Wilson JD: Steroid 5α-reductase. Two genes, two enzymes. Ann Rev Biochem 63: 25–61, 1994 |
|
Nelson DR et al: The P450 superfamily: Update on new sequences, gene mapping, accession numbers, early trivial names of enzymes and nomenclature. DNA Cell Biol 12: 1–51, 1993 |
|
Stewart TA, Mintz B: Successive generation of mice produced from established culture of euploid teratocarcinoma cells. Proc Natl Acad Sci USA 78: 6314–6321, 1981 |
|
Hertig AT, Rock J, Adams EC: A description of 34 human ova within the first 17 days of development. Am J Anat 98: 435, 1956 |
|
Witschi E: Migration of germ cells of human embryo from the yolk sac to the primitive gonadal folds. Contrib Embryol Carnegie Inst Wash 32: 67, 1948 |
|
Blandau RJ, White BJ, Rumery RE: Observation on the movements of living primordial germ cells in the mouse. Fertil Steril 14: 482, 1963 |
|
Jirásek JE: Die Verteilung der Urgeschlechtzellen in den Keimdrüsen menschlicher Feten. Acta Histoch (Jena) 13: 220, 1962 |
|
Jirásek JE: The relationship between the structure of the testis and differentiation of the external genitalia and phenotype in man. Ciba Foundation Colloquia on Endocrinology, Vol 16, Endocrinology of the Testis, 1967 |
|
Jirásek JE. Development of the Genital System and Male Pseudo-hermaphroditism. Baltimore, Johns Hopkins, 1971 |
|
Jirásek JE: Reproductive embryology. In Simpson JL (ed): Disorders of Sexual Differentiation. New York, Academic Press, 1976:51–110 |
|
Siiteri PK, Wilson JD: Testosterone formation and metabolism during male sexual differentiation in the human embryo. J Clin Endocrin Metab 38: 113, 1974 |
|
Pilgrim C, Reisert I: Differences between male and female brains: Developmental mechanisms and implications. Horm Metab Res 24: 353–359, 1992 |
|
Jirásek JE: Human sex differentiation: Gonadal histogenesis. In Hammer JE, Jenning BR (eds): The 1987 Distinguished Visiting Professorship lectures. Memphis, University of Tennessee, 1987 |
|
Baker TD: Oogenesis and ovarian development. In Balin H, Glasser SR (eds): Reproductive Biology. Amsterdam, Excerpta Medica, 1973 |
|
Felix W: The development of the urogenital organs. In Keibel F, Mall FP (eds): Manual of Human Embryology. Philadelphia, Lippincott, 1912 |
|
Jirásek JE: Genital ducts and external genitalia. Development and anomalies. Birth Defects 716: 131, 1971 |
|
Jirásek JE: Atlas of Human Prenatal Morphogenesis. Boston, Martinus Nijhoff, 1983 |
|
Gruenwald P: The relations of the growing müllerian duct to the wolffian duct and its importance for the genesis of malformations. Anat Rec 81: 1, 1941 |