Physiology and Tests of Adrenal Cortical Function
Authors
INTRODUCTION
The adrenal cortex is the outer portion of the paired adrenal glands located adjacent to the kidneys. It can be divided into three zones which produce different steroidal hormones. The outermost layer is the zona glomerulosa which produces mainly alodsterone under the control of the renin-angiotensin system. Aldosterone is a mineralocorticoid which is responsible for sodium and potassium hemostasis. The zona fasciculata is the next layer and the zona reticularis is the inner most portion of the adrenal cortex. Both of these layers regulate the stress response by producing glucocorticoids in response to stimulation by adrenocorticotropin hormone (ACTH) produced in the pituitary gland. ACTH is stimulated by corticotropin-releasing hormone (CRH) produced in the hypothalmus. Androgens are also produced in zona fasciculata and reticularis.
ANATOMY
Normal adrenal glands are paired, pyramidally shaped, yellow-brown convoluted organs surrounded by fat. They are surrounded by a capsule and are located at the upper pole of the kidneys. After sudden death, the average weight of the adrenal in healthy persons is 4 g. In autopsy series, the average weight is 6 g. The increase in size at autopsy has been attributed to the stress of antecedent illness. The cortex accounts for 80% and the medulla 20% of the weight of the adult gland.
The adult cortex is divided into three histologic zones: zona glomerulosa, zone fasciculata, and zona reticularis. The subcapsular zona glomerulosa consists of discontinuous clusters of relatively small, lipid-poor, compact cells. Most of the adrenal cortex is composed of the zona fasciculata, which is characterized by clear, large, lipid-rich cells arranged in parallel cords, with long columnar vascular sinuses interposed. The clear cells of the zona fasciculata gradually blend with the more lipid-poor cells of the zona reticularis. In the zona reticularis, the cells are arranged in alveolar patterns, with vascular sinuses.
The blood supply to the adrenal consists of numerous arteries that usually arise from the aorta, the inferior phrenic arteries, and the renal arteries. Blood flow proceeds from the subcapsular region toward the central (medullary) portal vein. The short right adrenal vein drains directly into the inferior vena cava. In most persons, the left adrenal vein drains into the inferior phrenic vein before emptying into the left renal vein; in some, it enters the left renal vein directly.
EMBRYOLOGY
The adrenal cortex is derived from the mesoderm lining the posterior abdominal wall. The fetal cortex begins its development in the 5-week-old fetus as a collection of large, acidophilic cells located between the root of the dorsal mesentery and the developing gonad. The adrenal medulla is formed from the neural crest cells that migrate to the medial aspect of the cortex during the seventh week of fetal development. Concurrently, cells destined to form the permanent or adult adrenal cortex proliferate and eventually envelop the fetal cortex.
At birth, the adrenal glands are 10–20 times larger than the adult gland, relative to kilograms of body weight. The cortex consists of a small, outer permanent zone and a large, inner fetal zone. Only the zona glomerulosa and a thin layer of fasciculata can be delineated in the permanent zone. The fetal zone undergoes rapid involution after birth and is no longer present after 1 year of age. As the fetal zone atrophies, the zona fasciculata of the permanent cortex enlarges. However, the gland does not return to its weight at birth for 2 years. The zona reticularis differentiates by the end of 3 years.1, 2
The fetal adrenal is responsive to both ACTH and, during the first half of gestation, human chorionic gonadotropin (hCG). In vitro studies have shown that hCG augments the production of dehydroepiandrosterone sulfate (DHEAS) in adrenal tissue obtained from 12- to 17-week fetuses.3 At 20 weeks, the pituitary content of ACTH begins to rise rapidly, concomitant with an increase in the responsiveness of the pituitary to corticotropin-releasing hormone (CRH).4 In anencephalic fetuses with deficient ACTH secretion, the adrenal glands are of normal size until 20 weeks, and undergo atrophy thereafter. Thus, ACTH appears to be the major regulator of the fetal adrenal during the second half of pregnancy. ACTH appears to induce the expression of 3β-hydroxysteroid dehydrogenase near term in primates.1 ACTH receptors are present on cells of both inner fetal and outer permanent zones.2
The effect that ACTH has on stimulating fetal adrenal growth may be modulated by the extracellular matrix proteins, insulin-like growth factors (IGFs), and other proopiomenlanocortin (POMC)-derived peptides. Steroidogenic factor 1 (SF-1) is a transcription factor in the nuclear hormone receptor superfamily that is required for fetal growth. SF-1 is an orphan nuclear receptor that plays a role in adrenal and gonadal development. Mutation of this gene in humans and mice has produced genotypic females and males who are phenotypically female and lack adrenal glands.2, 3, 4 SF-1, which is expressed in all three layers of the adrenal cortex, has been shown to upregulate expression of cholesterol side chain cleavage, steroidogenic acute regulatory protein (StAR), and DAX-1.2 DAX-1 is also an orphan nuclear receptor which is expressed mainly in the zona glomerulosa. DAX-1 binds to SF-1. DAX-1 suppresses SF-1 induced gene expression.5, 6 Many other transcriptional factors play a role in the development of the adrenal gland.7
The fetal adrenal lacks significant 3β-hydroxysteroid dehydrogenase activity,5 and the primary product of the inner fetal zone is DHEAS. DHEAS is a precursor for placental production of estrogens. Fetal glucocorticoids and mineralocorticoids are synthesized in the outer permanent zone. Placental progesterone serves as a substrate.
DEVELOPMENTAL ANOMALIES
Accessory adrenal rests are a common developmental anomaly. As discussed by Phipps and Tagatz in a previous version of this chapter, they usually are found under the capsule of the kidney or liver, or in the broad ligament. They usually contain only adrenocortical tissue, but on rare occasions, adrenal medullary tissue is present. Steroid-producing tumors have developed in accessory adrenocortical rests. Hyperplasia of accessory adrenocortical tissue may explain the rare instances in which bilateral adrenalectomy does not cure Cushing's disease.
Rare congenital anomalies include adrenal hypoplasia and aplasia, often unilateral, usually affecting the right side and often associated with renal agenesis on the affected side. Congenital adrenal hypoplasia occurs in approximately 1:12,500 births.8 Genetic mutations in the SF-1 gene and the DAX-1 gene are known causes of adrenal agenesis. Adrenal insufficiency has been documented in three individuals with mutations in the SF-1 gene. Two patients exhibited XY sex reversal due to the lack of gonads.3 Over 50 different mutations in the DAX-1 gene are associated with variable onset X-linked adrenal failure. Patients with DAX-1 mutations develop hypogonadotropic hypogonadism at puberty.9
Midline fusion of the adrenals may be associated with other malformations of the urinary and genital systems, including renal fusion and müllerian anomalies.
ADRENAL STEROID SYNTHESIS
Cholesterol is the precursor for the three classes of steroids produced by the adrenal: the glucocorticoids (C21), mineralocorticoids (C21), and androgens (C19). Figure 1 shows the major enzyme pathways.
|
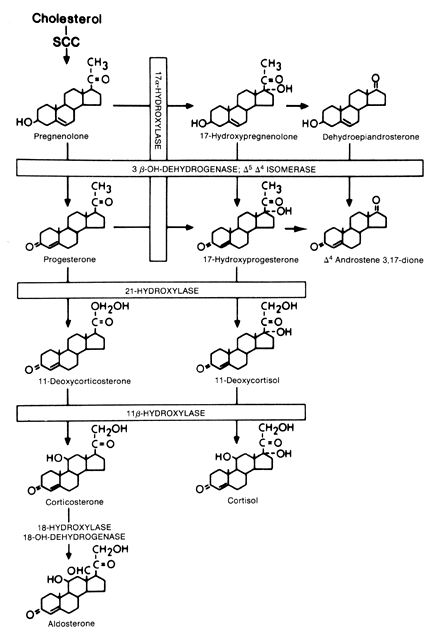
The rate-limiting step in cholesterol biosynthesis is side chain cleavage of a mitochondrial P450 enzyme (P450scc). This step is regulated by StAR which is stimulated by ACTH. Side chain cleavage by the P450 enzymye is a three-step enzymatic reaction that requires a flavoprotein, a nicotinamide adenine dinucleotide phosphate (NADPH)-dependent adrenodoxin reductase, and an iron-sulfur protein called adrenodoxin. All enzymes involved in cholesterol synthesis, except for 3β-hydroxysteroid dehydrogenase, are cytochrome P450 enzymes that require heme-containing membrane-bound mono-oxygenases. The 11β-hydroxylase enzyme (CYP11B1) and aldosterone sythetase (CYP11B2) are also mitochondrial P450 enzymes requiring adrenodoxin and adrenodoxin reductase, whereas the 21-hydroxylase and 17α-hydroxlase/17,20-desmolase enzymes are microsomal enzymes. The zona glomerulosa is the only region that expresses aldosterone synthetase (CYP11B2) which converts corticosterone to aldosterone and 17α-hydroxylase/17,20-desmolase is not active in the zona glomerulosa; therefore, only mineralocorticoids can be produced in this region.10
The zona glomerulosa is under control of the renin–angiotensin system, whereas the zona fasciculata and zona reticularis are under ACTH control. Therefore, the zona glomerulosa may be considered a separate gland. Cortisol, adrenal androgens, and mineralocorticoids other than aldosterone are secreted from the zona fasciculata and the zona reticularis, which function as a single unit. Unlike the zona glomerulosa, these zones lack the lacks aldosterone synthetase; thus, formation of the aldosterone (the most potent of the naturally occurring mineralocorticoids) occurs exclusively in the zona glomerulosa.10 This transformation of corticosterone to aldosterone does not appear to involve 18-hydroxycorticosterone as an intermediate product.11 Because the cells in the zona glomerulosa lack the enzyme 17α-hydroxylase/17,20-desmolase they are unable to synthesize glucocorticoids and androgens. Only the zona reticularis can sulfate DHEA to form DHEA-S which is the major circulatory androgen secreted by the adrenal gland.
The adrenal gland primarily uses cholesterol derived from plasma low-density lipoproteins (LDL) for steroidogenesis. LDL binds to specific cell surface receptors, internalized and moved to the lysosomes where it is converted to cholesterol. Cholesterol can also be made from the uptake of high density lipoproteins (HDL). There are also cell surface receptors for HDL which can transfer cholesterol into the adrenal cell without the uptake of HDL.12 Cholesterol can be synthesized from acetate in the adrenal gland. Additionally, both free and esterified cholesterol are stored in adrenal cells and may be used for de novo steroidogenesis.13
ADRENAL REGULATION
Corticotropin-releasing hormone
The central nervous system (CNS) controls the pituitary secretion of ACTH through the release of the peptide CRH. The 41-amino-acid sequence of CRH was derived from ovine hypothalamic extract by Vale and colleagues14 in 1981. Shortly thereafter, the structure of human CRH was determined,15 a structure that differed by only seven amino acids.
The major source of CRH is in the parvocellular neurons of the paraventricular nucleus of the hypothalamus.16, 17, 18 CRH is found elsewhere in the CNS, such as in the limbic system, the hindbrain, and the cerebrospinal fluid. It also can be detected in the pancreas, stomach, duodenum, adrenal medulla, and placenta, suggesting that it has diverse functions. After its synthesis in the parvocellular neurons as part of a larger peptide molecule, CRH is transported by means of axons through the median eminence, where it is secreted into the hypophyseal portal system.19
Hypophyseal portal blood concentrations of CRH are 150 times the peripheral circulating level. CRH induces the production and release of ACTH by binding to corticotrophs in the anterior pituitary via the type 1 CRH receptor which activates adenylate cyclase.20 CRH receptors are found in the neocortex, the limbic system, the adrenal medulla, and the sympathetic ganglia. The paraventricular nucleus also is the location of many of the vasopressin-containing cells that project to the posterior pituitary. Arginine vasopressin (AVP) acts via the V1B receptor to activate protein kinase C and is highly synergistic with CRH including in ACTH release.21, 22
Adrenocorticotropic hormone
Glucocorticoid synthesis and secretion is regulated by ACTH a single-chain polypeptide composed of 39 amino acids. ACTH appears to be produced by basophilic cells in the anteromedial zone of the anterior pituitary and the colloid cyst region.23 ACTH is synthesized as part of the large 241 amino acid precursor molecular POMC.24 This precursor molecule is present in the anterior pituitary, the hypothalamus, other parts of the brain, and several other sites. The manner in which POMC is processed into smaller peptides depends on the anatomic location.25 Other POMC products include α-, β-, and γ-melanocyte-stimulating hormone (MSH), β-lipotropin (β-LPH), and β-endorphin. α-MSH is composed of the 13 N-terminal amino acids of ACTH. β-LPH contains within its sequence the 18 amino acids of β-MSH. β-Endorphins are endogenous opioids that are a minor product of POMC in the pituitary, but a more substantial product in the hypothalamus (Fig. 2).
|
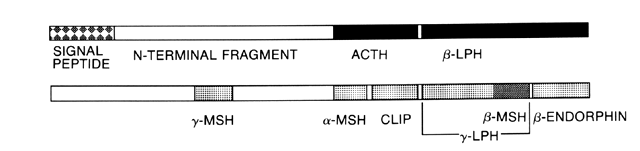
Of the 39 amino acids in ACTH, only the 24 N-terminal amino acids are necessary for full biologic activity. The half-life of ACTH is approximately 20 minutes.26 Cortisol secretion is maximal about 10 minutes after a pulse stimulus of ACTH. The magnitude of adrenal response varies with the magnitude and duration of the ACTH stimulus. A pulse achieving an ACTH concentration of 300 pg/ml results in maximal acute adrenal stimulation.
The hormone ACTH binds to its adrenal receptor in the zona fasciculata, which is coupled to adenylate cyclase, thereby resulting in phosphorylation of a cyclic adenosine monophosphate (cAMP)-dependent protein kinase. This protein kinase activates cholesterol ester hydroxylase, which breaks down cholesterol esters into free fatty acids and cholesterol. The cholesterol is transported to the mitochondria for steroid biosynthesis. MSH also activates cholesterol ester hydroxylase, but through a non-cAMP-dependent pathway. Protein kinase activation also results in the synthesis of a labile protein factor that leads to increased binding of cholesterol to P450scc in the mitochondria. ACTH can increase uptake of the precursor, LDL cholesterol (Fig. 3). In hypophysectomized rats, the transcription of P45021 and P45017α messenger ribonucleic acid (mRNA) increases 24 hours after exposure to ACTH treatment, whereas increases in mRNA transcription of P450scc, P45011β, adrenodoxin, and adrenodoxin reductase increase 36 hours after ACTH treatment. This temporal difference is created by synthesis of P45021 and P45017α as mature enzymes, whereas the others require transcriptional processing. This action of ACTH also appears to be cAMP dependent because the treatment of cell cultures with cAMP analogs produces a similar increase in mRNA.
|
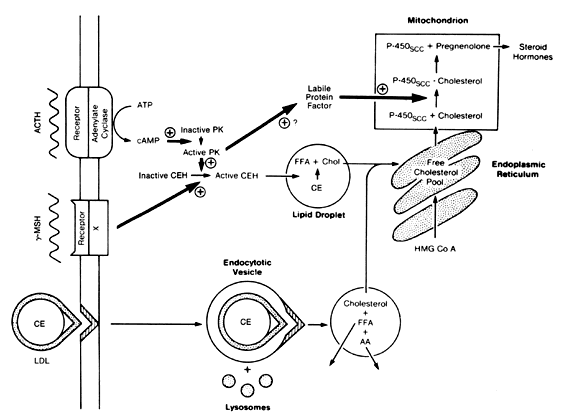
Cortisol and ACTH secretion exhibit significant fluctuation throughout the day.27, 28 As discussed by Phipps and Tagatz in a previous version of this chapter, ACTH is secreted in a pulsatile fashion, with diurnal variation. The pulses are more frequent and of greater magnitude in the early morning hours, shortly before the subject awakens. Then, secretion declines to a nadir that is reached in the evening before the onset of sleep. Blood levels of ACTH and cortisol are highest in the early morning and lowest in the evening in persons with the usual sleep–wake pattern. When the sleep–wake cycle is altered, the diurnal rhythm shifts after approximately 8 days to resume the relation to the sleep–wake pattern.
SF-1 and other transcriptional factors have been shown to regulate gene expression of the ACTH receptor, StAR protein, the receptor which transports of cholesterol and HDL (high density lipoprotein) into adrenal cortical cells and cytochrome P450 steroidogenic enzymes.2, 7
Specific stressful stimuli, including psychological stress, exercise, physical trauma, surgical procedures, hyperthermia, and hypoglycemia, result in augmented release of ACTH, presumably mediated through the CNS regulatory mechanisms discussed above.
Serotoninergic neurons are believed to play an important role in the control of ACTH release, especially since the suprachiasmatic nucleus contains the densest concentration of serotoninergic terminals in the CNS.29 In general, drugs that increase CNS serotoninergic activity induce an increase in the circulating levels of ACTH and related POMC products, such as β-lipotropin,30 with a subsequent increase in cortisol levels. Conversely, the potent serotonin antagonist, cyproheptadine, blocks ACTH and cortisol release and has been used to treat Cushing's disease.29 Endogenous opioids appear to play a role in CRH–ACTH–cortisol regulation; in humans, opioid agonists and antagonists appear to have inhibitory and stimulatory effects, respectively, on this axis.31 Some studies suggest a direct pituitary effect of opioids designed to reduce ACTH release.32 Catecholaminergic and other neuronal systems are involved as well. The ability of protein meals to increase cortisol levels is believed to reflect modification of CNS serotonin and catecholamine biosynthesis as a result of increased levels of tryptophan and tyrosine, respectively.33
Inhibitory glucocorticoid feedback plays an important role in regulating the CRH–ACTH–cortisol axis and appears to affect the pituitary, hypothalamus, and other regions of the CNS. Almost immediate feedback appears to involve cell membrane alterations that inhibit the action of CRH on ACTH release.34 This effect occurs as glucocorticoid levels increase and is virtually independent of the absolute magnitude of the levels.35 Delayed, longer-lasting effects of prolonged elevations of glucocorticoid levels appear to decrease the synthesis and release of CRH and ACTH by the hypothalamus and pituitary, respectively.34
Renin–angiotensin system
Angiotensin II, the end product of the renin–angiotensin system, is the pituitary stimulus for the synthesis and release of aldosterone by the zona glomerulosa.36 This process requires a complex relationship of hormone products produced by the liver, kidney, lung, and adrenal glands. Angiotensinogen, an α2-globulin synthesized in the liver, is cleaved by renin to release the decapeptide angiotensin I. The major site of the production of renin is in the juxtaglomerular kidney cells that surround the afferent arterioles that lead to the renal glomeruli. Two terminal amino acids are removed from angiotensin I by a converting enzyme primarily found in the lung, resulting in the formation of angiotensin II. Angiotensin II augments steroidogenesis in the zona glomerulosa by stimulating 20,22-desmolase and 18-hydroxylase activity, resulting in increased aldosterone secretion.
Under steady-state conditions, angiotensinogen is present in adequate concentrations, so alterations in the concentrations of angiotensin I, angiotensin II and, consequently, aldosterone are controlled by the release of renin. This system is largely regulated by renal perfusion pressure, a decrease of which appears to affect baroreceptors in the juxtaglomerular cells and thereby increase the amounts of renin to be released. Renin release is modulated by the sympathetic nervous system and chemoreceptors. The sympathetic nervous system stimulates renin secretion by reducing renal blood flow and by a direct effect on β-adrenergic receptors located on juxtaglomerular cells.37 Renin release additionally appears to be affected by the macula densa, a group of chemoreceptor cells responsive to changes in the concentration of sodium chloride in the distal convoluted tubule; the nature of this interaction is controversial.
Angiotensin II has other actions as well, including a powerful vasoconstrictor effect that results from its direct action on arteriolar smooth muscle. It is a potent dipsogenic agent and a stimulus for vasopressin release.37 Angiotensin II is degraded rapidly by angiotensinase in plasma and tissues, and has a half-life of approximately 60 seconds. Angiotensin III, a heptapeptide derivative of angiotension II, has actions similar to those of angiotensin II, but with less pressor activity. Angiotensin III is produced in low levels.38
The renin–angiotension–aldosterone system, acting in concert with vasopressin and atrial natriuretic hormone,39 regulates fluid and electrolyte balance within narrow limits. A fall in effective blood volume decreases renal perfusion, which affects angiotensin I, the majority of which is converted in one passage through the lung to angiotensin II. Angiotensin II constricts vascular smooth muscle and promotes aldosterone secretion from the adrenal gland by stimulating 20,22-desmolase and the conversion of corticosterone to aldosterone.40 The secreted aldosterone promotes sodium retention to restore normal blood volume. The direct effect of angiotensin II in elevating blood pressure and the indirect effect of aldosterone in restoring blood volume result in adequate renal perfusion and consequent suppression of renin secretion.
Potassium ions also play an important role in aldosterone regulation;41 high potassium levels lead to an increase in aldosterone production, and low levels result in a decrease. Physiologic amounts of ACTH can stimulate aldosterone production,42 but normally, this effect is of secondary importance. However, aldosterone has a diurnal pattern and an episodic secretion pattern that are similar to those of cortisol and that may be generated by other POMC pituitary derivatives.43, 44, 45 Indomethacin, a prostaglandin inhibitor, decreases plasma renin activity.46
The renin–angiotensin system is affected by pharmacologic amounts of estrogen. Women who use oral contraceptives have an increase in angiotensinogen levels that is attributable to an estrogen effect on the liver.47 Plasma renin levels decline, which is an apparent feedback effect of enhanced angiotensin II production secondary to the increased angiotensinogen levels. Thus, plasma renin activity (PRA), which depends on the concentrations of both angiotensinogen and renin, remains essentially normal.48 In the small percentage of women who have hypertension while taking oral contraceptives, this negative feedback effect and other normal compensatory mechanisms may be defective.
In pregnancy, there is an increase in angiotensinogen levels similar to that seen with oral contraceptive use. However, renin levels are greatly increased as well, resulting in a substantial increase in plasma renin activity.48 This increase in renin levels and the associated increase in angiotensin II levels appear to be necessary to maintain arterial pressure in the setting of the marked decrease in vascular sensitivity to angiotensin that characterizes the normal pregnant state. A substantial rise in plasma aldosterone levels occurs in association with these changes, but the effects of aldosterone on the distal renal tubule are antagonized by the high circulating levels of progesterone.
ACTIONS OF STEROID HORMONES
Steroid hormones bind to specific receptors in target tissues. Steroid hormone binding activates the receptor, causing a conformational change in the receptor and allowing the receptor to bind to DNA-binding sites called steroid-responsive elements. This change can activate or suppress a specific gene. If a gene is activated, mRNA will be transcribed and will then encode a specific protein product. The specificity of a hormone is due to its ability to bind to specific receptors. These receptors regulate gene production only in genes that can bind the activated receptor. The regulated gene product produces hormonal effects (i.e., regulation of metabolism by glucocorticoids, regulation of sodium and potassium by mineralocorticoids, anabolic effects by androgens).
Glucocorticoids
Cortisol secreted by the adrenal gland is the principle endogenous glucocorticoid in humans. Glucocorticoids originally were defined by their ability to raise blood glucose or increase liver glycogen levels in experimental animals. However, their actions include significant effects on other aspects of metabolism, renal function, and the immune, cardiovascular, and nervous systems.49 Further, the activity of the enzymes that are necessary for catecholamine synthesis by the adrenal medulla is regulated by adrenocortical glucocorticoids.50
The actions of cortisol involve increasing or decreasing protein synthesis in an effector cell. Free cortisol, in dynamic equilibrium with protein-bound forms, enters cells and binds to glucocorticoid receptors. The subcellular localization of the unoccupied glucocorticoid receptor is controversial. At one time, it was thought that unbound steroid receptors were localized to the cytoplasmic compartment, but more recent data favor a nuclear compartment localization. Whether glucocorticoid receptors differ from other steroid receptors on this point is not clear.51 The hormone receptor complex interacts with specific chromatin-binding sites on DNA, resulting in the induction or suppression of mRNA synthesis and, consequently, increased or decreased protein synthesis.
The effects of glucocorticoids on carbohydrate, lipid, and protein metabolism are closely related. In the liver, glucocorticoids increase the incorporation of pyruvate, lactate, and glycerol into glucose and glycogen production by altering enzymatic activity. The substrates for these reactions are lactate, which is derived from glycolysis in peripheral tissues; alanine, which is the primary gluconeogenetic amino acid released by increased protein catabolism; and glycerol, which is derived from the lipolysis of adipose tissue triglyceride. To compensate for the rise in blood glucose level, insulin production increases. Diabetes may occur in persons with a decreased capacity to produce insulin.
Further anti-insulin effects of glucocorticoids involve cellular resistance to the action of insulin. This effect decreases the entry of glucose into adipocytes, augmenting lipolysis and increasing levels of circulating free fatty acids. Glucocorticoids enhance the lipolytic effects of other hormones such as epinephrine. Not all adipocytes respond similarly; in humans with chronically elevated glucocorticoid levels, fat tends to be depleted in the extremities and augmented in the face, neck, and trunk (centripetal obesity).
Glucocorticoids have an antianabolic and a catabolic effect on muscle and protein. The rate of entry of amino acids into muscle is decreased, and the rate of egress is enhanced. All tissues with a protein matrix, such as connective tissue, skin, bone, and blood vessels, are affected. Glucocorticoid excess causes depletion of muscle mass; thin, fragile skin; decreased formation of bone matrix and osteoporosis; and poor wound healing. In children with endogenous or exogenous glucocorticoid excess, somatic growth is inhibited. In addition to its direct effects on protein metabolism, excess glucocorticoid inhibits the secretion of growth hormone.
In addition to suppressing growth hormone and ACTH secretion, elevations of endogenous or exogenous glucocorticoids suppress the release of the other major pituitary hormones by both hypothalamic and direct pituitary effects. Glucocorticoids blunt the thyroid-stimulating hormone response to thyrotropin-releasing hormone.52 In addition, the response to hypoglycemia of both growth hormone and prolactin is blunted.53 Further, one of the mechanisms by which cortisol excess causes gonadal dysfunction involves a suppressive effect on hypothalamic gonadotropin-releasing hormone pulse generator.54
Glucocorticoids also enhance neuronal excitation in the CNS, which is believed to be responsible for the psychosis that is seen in patients with glucocorticoid excess. These patients frequently exhibit alterations in electroencephalogram patterns. Conversely, cortisol deficiency often results in an inability to perform tasks that require mental concentration.
Alterations of epinephrine and norepinephrine secretion that are caused by glucocorticoids can affect blood pressure and myocardial function. Cortisol secretion is increased in response to most types of trauma and stress. Because patients with adrenal insufficiency cannot augment cortisol secretion in response to stress, they are susceptible to cardiovascular collapse and shock (addisonian crisis).
Glucocorticoids also affect renal function by increasing renal blood flow and the glomerular filtration rate and by increasing free water clearance. The latter effect is believed to result from a decrease in the permeability of the renal tubule, which is independent of the effect on glomerular filtration.
Pharmacologic doses of glucocorticoids have anti-inflammatory effects. Glucocorticoids stabilize lysosomes, decrease leukocyte diapedesis, and impair granuloma formation. Pharmacologic levels of glucocorticoids decrease the size of lymph nodes, thymus, and spleen, decrease antibody production, and lyse circulating lymphocytes. This immunologic effect of cortisol is the basis of many therapeutic regimens. However, this effect also reduces host resistance to infection.
Mineralocorticoids
Mineralocorticoid function (regulation of sodium and potassium levels) involves the action of steroids that are mediated by mineralocorticoid receptors. Aldosterone, which is secreted by the zona glomerulosa, is the primary mineralocorticoid in humans. It is regulated by the renin–angiotensin system. However, other steroids also may bind to the mineralocorticoid receptors, including 11-dedoxycortisol, corticosterone, 18-hydroxycorticosterone, and cortisol. Aldosterone and 9α-fluorocortisol (fludrocortisone), a synthetic mineralocorticoid most commonly used for replacement purposes, have potencies approximately 3000 and 125 times greater than that of cortisol, respectively.55 The mineralocorticoid hormone–receptor complex binds to a chromatin-specific DNA site to produce mRNA synthesis.
The primary target for aldosterone activity is the kidney. Autoradiographic studies of the renal nephron show specific aldosterone binding in the distal tubule and the cortical collecting tubule, intermediate binding in the medullar-collecting tubule, and no specific binding in the proximal tubule or the thick ascending limb.56 Studies show that protein secretion is stimulated by the hormone binding to the medullary collecting duct,57 whereas sodium transport is activated by the hormone binding to the cortical collecting duct.58 Sodium transport into the cell appears to occur through a sodium-potassium-adenosine triphosphatase (ATPase) action of mineralcorticoids.59
Several actions of mineralocorticoids have been identified in the genitourinary system. Aldosterone promotes the synthesis and secretion of kallikrein by the rat kidney. Kallikrein generates kinins, which are potent vasodilators, from the circulating precursor, kinogen.60, 61 Aldosterone also increases citrate synthesis in the rat and toad bladder, leading to increased ATP and possibly driving the sodium-potassium-ATPase pump.62 In studies of the toad bladder, aldosterone also increased the rate of free fatty acid synthesis, phosphorylase activity, and membrane phospholipid polyunsaturated fat content.63 In addition to these functions, aldosterone affects the ion content of feces,64 sweat,65 and saliva.66
Mineralocorticoid excess results in excessive sodium retention, expansion of extracellular fluid volume (weight gain), hypertension, and hypokalemia. Mineralocorticoid deficiency results in sodium depletion, loss of extracellular fluid, hypotension, and hyperkalemia.
Androgens
The adrenal gland secretes the androgens DHEAS, dehydroepiandrosterone (DHEA), androstenedione, and testosterone. Secretion from the adrenal gland directly or indirectly accounts for essentially all of the DHEAS, 80–90% of the DHEA, and approximately 50% of both the androstenedione and the testosterone present in the peripheral blood of women of reproductive age.67 Adrenal androgen secretion primarily is controlled by ACTH. The 24-hour secretory pattern of adrenal androgens parallels that of cortisol.68 Additionally, a separate anterior pituitary hormone may specifically stimulate adrenal androgen secretion.69 Further, prolactin appears to stimulate androgen production.70 Direct secretion of estrogen by the adrenal gland is negligible; isolated reports have suggested that the adrenal secretes small amounts of estrone and estradiol.71 The precise function of these androgens in women is not known, although their anabolic effects may be important. During childhood and the reproductive years, the circulating level of DHEAS increases. After menopause, it decreases with age.72 In postmenopausal women, the adrenal secretion of androstenedione and its subsequent peripheral aromatization is the principal source of the predominant estrogen, estrone.73 Adipose tissue is an important site for this conversion.74
CORTICOSTEROID-BINDING GLOBULIN
Ninety per cent of the cortisol in serum is bound to a circulating corticosteroid-binding (α2) globulin (CBG, or transcortin).75 CBG has a strong affinity for cortisol, and thus has been used as the basis for competitive protein-binding radioassays for cortisol. In addition, CBG has a high affinity for cortisone, corticosterone, 11-deoxycorticosterone, 11-deoxycortisol, progesterone, and 17-hydroxyprogesterone. Cortisol is not significantly displaced by other steroids because of their substantially lower concentrations or CBG-binding affinities. Most synthetic steroids are only weakly bound to CBG, except for prednisone and prednisolone. The half-life of CBG is approximately 5 days.
Cortisol is bound with high affinity to CBG, with low affinity to albumin, or unbound (free). The CBG-binding capacity for plasma cortisol is approximately 25 μg/dl. As cortisol levels increase beyond this point, binding to albumin, which is a high-capacity, low-affinity binding system for cortisol, becomes more important. Because only free cortisol is thought to affect cell function, it is proposed that CBG acts as a buffer to prevent rapid changes in the free levels of the hormones under non-steady-state conditions.76
However, new knowledge about CBG shows that its function may be more complex. It has sequence homology with the serine protease inhibitors (SERPIN). Proteins from this superfamily contain cleavage sites for serine proteases. Neutrophil elastase, a serine protease that is released from activated neutrophils, can cleave CBG in such a way that CBG is unable to bind cortisol. The hypothesis is that during inflammation, this effect could cause rapid release of free cortisol to the local tissue.77 In addition, membrane binding and internalization of CBG bound to cortisol has been shown in several tissues.77, 78
Synthesized in the liver, CBG has a molecular weight of 50–60 kD, depending on its sugar content. The protein originates from a single copy of the gene, which is located on chromosome 14. The CBG gene is located in a cluster of SERPIN genes with which it shows homology.79 The promoter region of the CBG gene contains sequences that are homologous to steroid-responsive elements (estrogen promoting, glucocorticoid inhibiting). The gene is translated into a single polypeptide chain to which five carbohydrate moieties are added. The carbohydrate composition does not influence steroid binding to CBG, but affects CBG binding to membrane receptors. During pregnancy, a variant of CBG is found. It is created by posttranslational modification of the sugar moieties.80
Concentrations of CBG are influenced by a number of factors. Administration of pharmacologic amounts of estrogen results in an increase in CBG and total cortisol levels, although the level of free cortisol may not rise. This effect is consistent with the general principle that, under steady-state conditions, the concentration of an unbound hormone is independent of plasma levels of binding proteins. Marked increases in CBG also occur during pregnancy, presumably due to the high circulating estrogen levels. Progestins and androgens appear to have little effect on CBG levels. Glucocorticoids suppress CBG levels, either because of high endogenous levels (e.g., Cushing's syndrome) or with exogenous dosing of glucocorticoids. Abnormally low levels of CBG also may be seen in patients with cirrhosis, hypertension, high-carbohydrate diets, pernicious anemia, and familial inheritance. Increased levels can be associated with malignant lymphoproliferative disorders.80
METABOLISM OF ADRENAL STEROIDS
Corticosteroids are cleared from the circulation by several mechanisms.81 The half-life of cortisol under physiologic conditions is approximately 66 minutes. The enzyme systems involved are located primarily in the liver, and most corticosteroid metabolites are excreted into the urine. A small fraction of the free or unbound cortisol present in plasma is not reabsorbed in the distal tubule of the kidney. This small, but diagnostically useful portion of the cortisol is excreted unchanged in the urine.
The major pathway for cortisol degradation starts with hepatic reduction of the double bond between C-4 and C-5 in the A ring, resulting in a hydrogen in the β-position of C-5, followed by reduction of the C-3 ketone moiety. This process yields tetrahydrocortisol, which is conjugated rapidly with glucuronic acid at the C-3 hydroxyl group to form a water-soluble metabolite that is excreted by the kidney. Thus, tetrahydrocortisol is the principal urinary metabolite of cortisol, but other metabolites also are produced. Aldosterone is metabolized in a fashion similar to that of cortisol; thus, its major urinary metabolite is tetrahydroaldosterone. Adrenal androgens are primarily excreted into the urine as 17-ketosteroids (17-KS).
LABORATORY EVALUATION OF ADRENAL FUNCTION
Evaluation of cortisol secretion
The first reliable method for measuring glucocorticoid levels is known as the Porter-Silber chromogen test. This fluorometric test measures cortisol and cortisone levels in the plasma, or their urinary metabolites, 17-OH corticosteroids (17-OHCS), by the intensity of a color change that occurs when the urine or plasma reacts with phenylhydrazine. Since the development of radioimmunoassays and competitive binding assays in the 1950s and 1960s, more accurate tests have been developed. The amount of hormone is quantified according to the degree that it displaces the tracer steroid from the antibody or binding protein. Radioimmunoassays have been developed for most of the major steroids derived from cholesterol. Basal abnormalities of hormone levels may indicate adrenal abnormalities. However, single determinations are affected by pulsatile secretion and circadian rhythm.
Circulating glucocorticoid levels have a diurnal rhythm. Therefore, a 24-hour urine collection is required for a reliable measurement of 17-OHCS level. Normal values range from 2 to 6 μg/24 hours in women and 3–10 μg/24 hours in men. The amount of 17-OHCS excreted is directly related to body mass. Obese subjects may exceed the normal values despite having normal adrenal function. Expressing urinary 17-OHCS as milliliters per gram of urinary creatinine level will correct for obesity. The normal value is 2–6.5 mg/g creatinine.
The 17-ketogenic steroids (17-KGS) rarely are used as a measure of cortisol secretion. 17-KGS constitute a greater percentage of cortisol metabolites than do 17-OHCS, but technical problems and the lack of specificity of 17-KGS make this measure less useful.
At 8 a.m., the normal cortisol level ranges from 7 to 22 ng/dl when measured by radioimmunoassay. This value is twice the level that is expected when the test is performed on plasma obtained at 8 p.m. In addition, cortisol is secreted episodically. Therefore, single estimations of plasma cortisol level without prior suppression are considered unreliable. Salivary measurements rather than plasma measurements have been suggested.82
The 24-hour determination of urinary free cortisol (UFC) level is a reliable test of the amount of cortisol excreted unchanged during a 24-hour period. It reflects not only daily production rates but also the portion that is free in the circulation. UFC can be measured by radioimmunoassay or competitive protein-binding radioimmunoassay. However, high performance liquid chromatography (HPLC) has become the method of choice. Normal values are less than 50 μg/24 hours with HPLC. It is particularly useful in distinguishing patients with obesity from those with Cushing's syndrome.83, 84 The determination of UFC is not useful in the diagnosis of adrenal hypofunction. Carbamazepine treatment and high fluid intake may falsely increase the UFC.82
Many extra-adrenal factors can affect cortisol measurement and lead to a false diagnosis of hyperfunction or hypofunction. Malnutrition and advanced age are associated with decreased cortisol production, whereas stress and obesity are associated with increased cortisol production.
In pregnancy, estrogen induces an increase in CBG concentration. This increase then causes an increase in plasma cortisol levels to maintain the same level of functional (free) cortisol. Therefore, levels of plasma cortisol are increased, but levels of urinary metabolites are normal. Administration of estrogens, especially high-dose oral contraceptives, will result in similar alterations in cortisol values. Other drugs can affect cortisol measurements as well. Any drug that induces hepatic enzymes, such as phenobarbital, may modify the metabolism of cortisol and lead to low urinary 17-OHCS levels. Also, liver and renal disease cause abnormalities in the excretion of cortisol.
Evaluation of adrenal androgens
The metabolites of adrenal androgens traditionally have been measured as urinary 17-KS. In normal women, important precursors for 17-KS are DHEAS, DHEA, and androstenedione; compounds with C-11 hydroxyl or ketone groups, such as cortisol or cortisone, also contribute. Drugs that cause fluorescent metabolites will yield falsely elevated values in a colorimetric assay. In disorders of excess adrenal androgen production, 17-KS excretion exceeds the normal range.
However, radioimmunoassay of specific androgens generally is available, often making 17-KS determinations unnecessary. DHEAS is exclusively an adrenal product, and its plasma level is a more specific measure of adrenal androgen production than is urinary 17-KS excretion. The total production rate of DHEAS has been estimated to be greater than 10 mg/day. In this laboratory, serum concentrations of DHEAS throughout the menstrual cycle were 82–338 μg/dl. The metabolic clearance rate of DHEAS is low, accounting in part for the long half-life of this steroid. The combination of significant production rate, low metabolic clearance, and long half-life results in substantial serum concentrations of DHEAS that are subject to limited fluctuation. These features of DHEAS and its exclusive derivation from the adrenal gland make serum DHEAS level a valuable marker of long-term adrenal androgen activity and raise the validity of a single serum determination of DHEAS concentration.85, 86, 87, 88, 89, 90, 91, 92, 93 Unlike testosterone and androstenedione, DHEAS cannot bind to serum sex-hormone-binding globulin, and most of the binding is to the high-capacity, low-affinity albumin bed.94, 95
In some cases of Cushing's syndrome, chronic hyperprolactinemia, and congenital adrenal hyperplasia (CAH), DHEAS level is elevated. Elevation of DHEAS level to greater than 700 ng/dl, with or without elevation of testosterone level, is strongly suggestive of an androgen-producing adrenal tumor. Although testosterone-producing adrenal adenomas associated with normal serum DHEAS level have been described, they are rare and may respond to gonadotropic stimulation. A normal serum DHEAS level essentially excludes the adrenal gland as the source of excessive androgen production.
Despite the apparent specificity of DHEAS as an adrenal marker, serum DHEAS levels may be moderately elevated in patients who experience chronic anovulation due to reproductive axis dysfunction.96, 97, 98, 99 Serum DHEAS levels usually do not exceed 700 μg/dl in these patients. The mechanisms underlying the anovulation-associated increase in serum DHEAS levels remain uncertain.
Elevation of serum DHEAS levels requires additional workup. A prolactin level should be obtained because elevations in prolactin levels stimulate adrenal androgen production. If the prolactin level is normal, a short-term ACTH stimulation test should be performed to exclude an attenuated form of CAH. Once attenuated CAH and hyperprolactinemia are excluded, and the serum DHEAS level is less than 700 μg/μl, the options are screening for the rare possibility of Cushing's syndrome or empirically suppressing the reproductive axis (with gonadotropin-releasing hormone agonists), given the likelihood that the elevated circulating levels of DHEAS represent a functional chronic anovulatory disorder. If the elevated DHEAS level is secondary to reproductive axis dysfunction, suppression of the reproductive axis over a period of 3–6 months should result in a reduction of serum DHEAS levels.100, 101, 102 However, if this decrease does not occur the patient may have an early, slowly progressing adrenal androgen-producing tumor (e.g., adenoma).
Evaluation of hypothalamic–pituitary–adrenal function
In general, evaluation of adrenal function requires more provocative testing. Diagnosis of hypercortisolism (Cushing's syndrome) is made by the finding of failure to suppress cortisol production by dexamethasone suppression test. The etiology of Cushing's syndrome is determined by evaluation of baseline circulating ACTH levels, the metyrapone test, and the CRH stimulation test. Diagnosis of an adrenal cause of hyperandrogenism can be made by an ACTH stimulation test. An ACTH stimulation test is required for the diagnosis of adrenal insufficiency.
PLASMA ADRENOCORTICOTROPIC HORMONE RADIOIMMUNOASSAYS
Plasma levels of endogenous ACTH can be measured by radioimmunoassay. ACTH is secreted in a pulsatile fashion, and its circadian rhythm is responsible for the circadian rhythm of cortisol secretion. Clinically, ACTH radioimmunoassays may measure not only the native 39-amino-acid ACTH molecule, but also structurally related POMC products.103 In normal, unstressed subjects, the level of ACTH late in the evening generally is less than 20 pg/ml, and morning values are in the range of 10–80 pg/ml.104 The plasma ACTH level can be helpful in determining the etiology of adrenocortical insufficiency105 and of Cushing's syndrome.
ADRENOCORTICOTROPIC HORMONE STIMULATION TESTS
The response of the adrenal gland to exogenous ACTH may be helpful from a diagnostic standpoint. In general, the synthetic ACTH analog cosyntropin, which consists of the 24 N-terminal amino acids of the native peptide, is used for testing purposes. The degree of response depends not only on the physiologic integrity of the gland but also on the degree of prior stimulation. This test is used to diagnose adrenocortical insufficiency by measuring cortisol levels or to diagnose CAH by measuring androgen precursor levels.
For the rapid ACTH test, one or two blood samples are collected to determine basal levels of plasma cortisol, followed by intravenous injection of 250 μg cosyntropin. Plasma may be sampled 30, 45, or 60 minutes after ACTH administration.106 With a normal response to stimulation, the plasma cortisol level will exceed 15 μg/dl and will exhibit an incremental rise of 5–7 μg/dl or more. A normal response excludes primary adrenocortical insufficiency. However, it does not necessarily exclude partial secondary adrenocortical insufficiency; some patients with mild or early ACTH deficiency may have sufficient basal ACTH production to prevent adrenocortical atrophy, yet lack the ability to activate this axis appropriately when stressed.
Prolonged ACTH stimulation also may distinguish primary from secondary adrenocortical insufficiency. One protocol for prolonged ACTH stimulation is collection of 24-hour urine specimens for 1 day before ACTH stimulation and the 2 days during ACTH infusion to determine the values of 17-OHCS and creatinine.107 A continuous intravenous infusion of 1600 μg cosyntropin, which is equivalent to the 160 units of ACTH originally used, is administered for 48 hours. It is not necessary to restrict diet or activity during the infusion. Normal subjects excrete more than 27 mg/24 hours 17-OHCS on the first day of the infusion, and more than 47 mg/24 hours on the second day. Patients with secondary adrenocortical insufficiency generally excrete more than 4 mg/24 hours on the first day of infusion, and more than 10 mg/24 hours on the second day. Patients with primary adrenocortical insufficiency usually excrete less than 3 mg/24 hours on the first day and less than 4 mg/24 hours on the second.
In patients with a high risk of primary adrenal insufficiency, glucocorticoid therapy may be initiated at the time of a rapid ACTH stimulation test or during a prolonged ACTH stimulation protocol. Dexamethasone is the glucocorticoid of choice; it is 25 times more potent than cortisol, and the amount required for treatment does not interfere with cortisol or 17-OHCS determination. Although dexamethasone suppresses the pituitary secretion of endogenous ACTH, it does not interfere with the response of the adrenal to exogenous ACTH, and therefore will not alter the results of the test.
The ACTH stimulation tests for the diagnosis of CAH involve the measurement of steroid precursors proximal to the enzymatic block. Various protocols have been used. Some require overnight dexamethasone suppression before testing, although most clinicians do not consider this step necessary. Levels of progesterone and 17-OH progesterone are elevated with CAH21, the level of circulating 18-hydroxycorticosterone is elevated with CAH11, and the 17-hydroxypregnenolone:17-hydroxyprogesterone ratio is elevated with CAH3β-HSD.
DEXAMETHASONE SUPPRESSION TEST
Dexamethasone, a potent synthetic glucocorticoid, is used in suppression tests because the small amount required to suppress ACTH does not interfere with the assays of steroids or steroid metabolites. A small single dose, 1 mg, is used in the overnight suppression tests to screen for Cushing's syndrome. However, false negatives may occur when cutoff values of 70–200 nmol/L are used.82 Liddle108 developed a higher dose dexamethasone test to confirm Cushing's syndrome and determine its etiology. These tests and their use in the diagnosis of Cushing's syndrome are discussed in detail in another chapter.
METYRAPONE TEST
Metyrapone decreases the adrenal secretion of cortisol, primarily by inhibiting the enzymatic activity of 11β-hydroxylase. In the normal person, the decline in circulating cortisol level activates the hypothalamic–pituitary unit to increase ACTH release. ACTH stimulates adrenal steroidogenesis, but because of the inhibition of 11β-hydroxylase, an excess of 11-deoxycorticosteroids, primarily 11-deoxycortisol (11-DOC), is secreted. The degree of metyrapone inhibition of cortisol synthesis is assessed by measuring serum cortisol level. The degree of resultant ACTH stimulation may be assessed by measuring the plasma level of ACTH directly, the level of 11-DOC in plasma or urine, or the urinary excretion of metabolites of 11-DOC, which are measured as 17-OHCS.
The metyrapone test is appropriate for assessment of hypothalamic–pituitary function in patients in whom there is no reason to suspect primary adrenocortical insufficiency. Otherwise, ACTH stimulation test should precede metyrapone testing because the administration of metyrapone may precipitate adrenal crisis with cardiovascular collapse. If the cortisol response to a rapid ACTH test is diminished, no additional information will be gained from the metyrapone test. In contrast to the ACTH stimulation test, the administration of dexamethasone or other glucocorticoids will invalidate the metyrapone test.
The patient must be hospitalized for the metyrapone test because of the possibility of precipitating adrenal crisis. During the administration of metyrapone, some patients with normal adrenal function will experience side effects, most commonly vertigo, nausea, and vomiting. Gastrointestinal symptoms are minimized by administering the metyrapone with food.
Many methods of metyrapone administration have been employed, including single-dose tests.84, 109 According to one protocol for the metyrapone test,110 beginning at 8 a.m., 750 mg metyrapone is given orally every 4 hours for a total of six doses. Plasma is taken at 8 a.m., immediately before the first dose; and again the next morning at 8 a.m., 4 hours after the last dose. The cortisol level after metyrapone administration should be less than 6–8 μg/dl if the test results are to be considered valid. In normal subjects, the 11-DOC level before the administration of metyrapone is approximately 1 ng/dl and the level after metyrapone administration is greater than 10.5 μg/dl. Patients with inadequate pituitary-adrenal reserve as a result of pituitary disease or long-term exogenous glucocorticoid therapy have 11-DOC levels after metyrapone of less than 8 μg/dl, with mean values of 2–3 μg/dl.
INSULIN-INDUCED HYPOGLYCEMIA TEST
Insulin-induced hypoglycemia stimulates the secretion of ACTH and cortisol. Testing with hypoglycemia provides results more rapidly than the metyrapone test, and it also provides an assessment of growth hormone secretion. Additionally, it is preferred by some authors over the metyrapone test as an evaluation of ACTH reserve.106, 111 Hypoglycemia is contraindicated in patients with cardiovascular disease, who are at risk for the development of arrhythmia, and in patients with epilepsy, who are at risk for the development of seizure. As is true of the metyrapone test, no useful information about the hypothalamic–pituitary–adrenal axis will be gained with insulin testing if the cortisol response to ACTH is subnormal.
For the test, the patient fasts overnight. A physician must be in attendance throughout the test to assess the mental status of the patient and administer glucose as necessary. A plasma sample is drawn at 8 a.m. for cortisol and glucose determination. A bolus of regular insulin 0.1 U/kg is given intravenously, and blood is sampled 30, 60, 90, and perhaps 120 minutes after the insulin dose. If pituitary disease is strongly suspected, the insulin dosage is decreased to 0.05 U/kg because severe hypoglycemia may result from the larger dose of insulin. Virtually all patients who achieve suppression of plasma glucose to the degree required for an adequate test will have tachycardia and diaphoresis. If the patient becomes confused, the test should be terminated immediately by infusing glucose.
Hypoglycemic stimulus is adequate if the glucose level falls to a level that is less than 50% of the baseline value. If the stimulus has been adequate, normal subjects exhibit a 5-μg/dl increment in plasma cortisol level and a concentration of greater than 15 μg/dl in the 60- or 90-minute sample.106 The best objective measure of the response of the hypothalamic–pituitary–adrenal axis to hypoglycemic stress may be the cortisol/glucose slope value, which is obtained by plotting serial cortisol values against the corresponding glucose values.109
In addition to its value in assessing ACTH reserve, the cortisol response to insulin-induced hypoglycemia may be useful for distinguishing patients with Cushing's syndrome from those with false-positive dexamethasone suppression test findings. Patients with Cushing's syndrome generally have blunted responses.84
CORTICOTROPIN-RELEASING HORMONE TESTING
Corticotropin-releasing hormone testing is replacing metyrapone and insulin-induced hypoglycemia testing to assess the hypothalamic–pituitary–adrenal access. The test is performed by intravenous injection of 1 μg/kg ovine or human CRH followed by measurement of baseline and stimulated serum ACTH and plasma cortisol levels. This test induces maximal ACTH and cortisol levels. Human CRH has a more rapid clearance than ovine CRH. The increment of cortisol is greater if the test is performed at 8:00 p.m., when the circulating cortisol levels are lowest, because the peak cortisol levels induced by the stimulation test are unchanged regardless of the time that the test is performed. Side effects of flushing, metallic taste, and increased respiratory rate have been reported. However, unlike the metyrapone test, it is considered safe to perform on an outpatient basis.82
The test has been useful in distinguishing patients with Cushing's disease, who have normal or exaggerated hormonal increases, from patients with ectopic ACTH secretion or non-ACTH-dependent Cushing's syndrome, who do not respond to stimulation.112, 113, 114, 115 The test also has been used in conjunction with bilateral inferior petrosal vein sampling to lateralize ACTH-secreting pituitary adenomas and rule out ectopic ACTH syndrome.116 ACTH response also may be blunted in patients with anorexia nervosa and primary affective disorders.117, 118 Its usefulness in adrenal insufficiency is limited. Patients with adrenal insufficiency due to hypothalamic abnormalities usually have greater increases in the ACTH level after stimulation compared with patients with pituitary abnormalities.119
REFERENCES
Coulter CL, Goldsmith PC, Mesiano S et al: Functional maturation of the primate fetal adrenal in vivo. II. Ontogeny of corticosteroid synthesis is dependent upon specific zonal expression of 3 beta-hydroxysteroid dehydrogenase/isomerase. Endocrinology 1996 Nov;137(11):4953–9 |
|
Sadovsky Y, Crawford PA: Developmental and physiologic roles of the nuclear receptor steroidogenic factor-1 in the reproductive system. J Soc Gynecol Investig 1998 Jan-Feb;5(1):6–12 |
|
Vachharajani A, Bethin K, Mouillet JF et al: The rare occurrence of absent adrenals in a term infant: a case report and review of the literature. Am J Perinatol 2006 Feb;23(2):111–4 |
|
Luo X, Ikeda Y, Parker KL: A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell 1994 May 20;77(4):481–90 |
|
Reutens AT, Achermann JC, Ito M et al: Clinical and functional effects of mutations in the DAX-1 gene in patients with adrenal hypoplasia congenita. J Clin Endocrinol Metab 1999 Feb;84(2):504–11 |
|
Kawabe K, Shikayama T, Tsuboi H et al: Dax-1 as one of the target genes of Ad4BP/SF-1. Mol Endocrinol 1999 Aug;13(8):1267–84. |
|
Hammer GD, Parker KL, Schimmer BP: Minireview: transcriptional regulation of adrenocortical development. Endocrinology 2005 Mar;146(3):1018–24 |
|
Kelch RP, Virdis R, Rapaport R et al: Congenital adrenal hypoplasia. Pediatr Adolesc Endocrinol 1984;13:156–161 |
|
Muscatelli F, Strom TM, Walker AP et al. Mutations in the DAX-1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. Nature 1994;372:672–676 |
|
Tait SAS, Schulster D, Okamoto M et al: Production of steroids by in vitro superfusion of endocrine tissue, part II. Steroid output from whole, capsular and decapsulated adrenals of normal intact, hypophysectomized and hypophysectomized-nephrectomized rats as a function of time of superfusion. Endocrinology 86: 360, 1970 |
|
Neher R: Aldosterone: Chemical aspects and related enzymology. J Endocrinol 81: 25P, 1979 |
|
Plump AS, Erickson SK, Weng W, Partin JS, Breslow JL, Williams DL. Apolipoprotein A-1 is Required for Cholesteryl Ester Accumulation in Steroidogenic Cells and for Normal Adrenal Steroid Production. J Clin Invest 97: 2660–2671, 1996 |
|
Borkowski AJ, Levin S, Delcroix C et al: Blood cholesterol and hydrocortisone production in man: quantitative aspects ofthe utilization of circulating cholesterol by the adrenals at rest and under adrenocorticotropin stimulation. J Clin Invest 1967 May;46(5):797–811 |
|
Vale W, Spiess J, Rivier C et al: Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta;-endorphin. Science 213: 1394, 1981 |
|
Shibahara S, Morimoto Y, Furutani Y et al: Isolation and sequence analysis of the human corticotropin-releasing factor precursor gene. EMBO J 2: 775, 1983 |
|
Swanson LW, Sawchenko PE, Rivier J et al: Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: An immunohistochemical study. Neuroendocrinology 36: 165, 1983 |
|
Mouri T, Suda T, Sasano N et al: Immunocytochemical identification of CRF in the human hypothalamus. Tohoku J Exp Med 142: 423, 1984 |
|
Bresson JL, Clavequin MC, Fellmann D, Bugnon C: Anatomical and ontogenetic studies of the human paraventriculo-infundibular corticoliberin system. Neurosciences 14: 1077, 1985 |
|
Gibbs DM, Vale W: Presence of corticotropin releasing factor like immunoreactivity in hypophyseal portal blood. Endocrinology 111: 1418, 1982 |
|
Chen R, Lewis KA, Perrin MH et al: Expression cloning of a human corticotropin-releasing-factor receptor. Proc Natl Acad Sci USA 90: 8967, 1993 |
|
Liu JH, Muse K, Contreras P et al: Augmentation of ACTH-releasing activity of synthetic corticotropin releasing factor (CRF) by vasopressin in women. J Clin Endocrinol Metab 57: 1087, 1983 |
|
Sugimoto T, Saito M, Mochzuki S, et al. Molecular Cloning and Functional Expression of a cDNA encoding the human V1b vasopressin receptor. J Biol Chem 269: 27088, 1994 |
|
Celio MR, Pasi A, Burgisser E et al: ‘Proopiomelanocortin fragments’ in normal adult pituitary: Distribution and ultrastructural characterization of immunoreactive cells. Acta Endocrinol 95: 27, 1980 |
|
Mains RE, Eipper BA, Ling N: Common precursor to corticotropins and endorphins. Proc Natl Acad Sci USA 74: 3014, 1977 |
|
Krieger DT, Liotta AS, Brownstein MJ et al: ACTH, &b.beta;-lipotropin, and related peptides in brain, pituitary, and blood. Recent Prog Horm Res 36: 277, 1980 |
|
Tanaka K, Nicholson WE, Orth DN: Diurnal rhythm and disappearance half time of endogenous plasma immunoreactive &b.beta;-MSH (LPH) and ACTH in man. J Clin Endocrinol Metab 46: 883, 1978 |
|
Krieger DT, Allen W, Rizzo P et al: Characterization of the normal temporal pattern of plasma corticosteroid levels. J Clin Endocrinol Metab 32: 266, 1971 |
|
Weitzman ED, Fukushima D, Nogeire C et al: Twenty-four hour pattern of the episodic secretion of cortisol in normal subjects. J Clin Endocrinol Metab 33: 14, 1971 |
|
Krieger DT: Serotonin regulation of ACTH secretion. Ann N Y Acad Sci 297: 527, 1977 |
|
Petraglia F, Facchinetti F, Martignoni E et al: Serotoninergic agonists increase plasma levels of &b.beta;-endorphin and &b.beta;-lipotropin in humans. J Clin Endocrinol Metab 59: 1138, 1984 |
|
Grossman A, Gaillard RC, McCartney P et al: Opiate modulation of the pituitary-adrenal axis: Effects of stress and circadian rhythm. Clin Endocrinol 17: 279, 1982 |
|
Allolio B, Deuss U, Kaulen D et al: FK 33–824: A metenkephalin analog, blocks corticotropin-releasing hormone-induced adrenocorticotropin secretion in normal subjects but not in patients with Cushing's disease. J Clin Endocrinol Metab 63: 1427, 1986 |
|
Ishizuka B, Quigley ME, Yen SSC: Pituitary hormone release in response to food ingestion: Evidence for neuroendocrine signals from gut to brain. J Clin Endocrinol Metab 57: 1111, 1983 |
|
Keller-Wood ME, Dallman MF: Corticosteroid inhibition of ACTH secretion. Endocr Rev 5: 1, 1984 |
|
Fehm HL, Voigt KH, Kummer G et al: Differential and integral corticosteroid feedback effects on ACTH secretion in hypoadrenocorticism. J Clin Invest 63: 247, 1979 |
|
Fraser R, Brown JJ, Lever AF et al: Control of aldosterone secretion. Clin Sci 56: 389, 1979 |
|
Sladek CD, Joynt RJ: Role of angiotensin in the osmotic control of vasopressin release by the organ-cultured rat hypothalamoneurohypophyseal system. Endocrinology 106: 173, 1980 |
|
Ganong WF: Sympathetic effects on renin secretion: Mechanism and physiological role. Adv Exp Med Biol 17: 17, 1972 |
|
Laragh JH: Atrial natriuretic hormone, the renin-aldosterone axis, and blood pressure-electrolyte homeostasis. N Engl J Med 313: 1330, 1985 |
|
Aguilera G, Marusic ET: Role of the renin-angiotensin system in the biosynthesis of aldosterone. Endocrinology 89: 1524, 1971 |
|
Dluhy RG, Greenfield M, Williams GH: Effect of simultaneous potassium and saline loading on plasma aldosterone levels. J Clin Endocrinol Metab 45: 141, 1977 |
|
Nicholls MG, Espiner EA, Donald RA: Plasma aldosterone response to low dose ACTH stimulation. J Clin Endocrinol Metab 41: 186, 1975 |
|
Matsuoka H, Mulrow PJ, Franco-Saenz R, Li CH: Effects of beta-lipotropin and beta lipotropin derived peptides an aldosterone production in the rat adrenal gland. J Clin Invest 68: 752, 1981 |
|
Gullner HG, Gill JR Jr: Beta endorphin selectivity stimulates aldosterone secretion in hypophysectomized, nephrectomized dogs. J Clin Invest 71: 124, 1983 |
|
Lis M, Hamet P, Gutkowska J et al: Effect of N-terminal portion of pro-opiomelanocortin on aldosterone release by human adrenal adenoma in vitro. J Clin Endocrinol Metab 52: 1053, 1981 |
|
Romero JC, Dunlap CL, Strong CG: The effect of indomethacin and other anti-inflammatory drugs on the renin-angiotensin system. J Clin Invest 83: 282, 1976 |
|
Cain MD, Walters WA, Catt KJ: Effects of oral contraceptive therapy on the renin-angiotensin system. J Clin Endocrinol Metab 33: 671, 1971 |
|
Derkx FHM, Stuenkel C, Schalekamp MPA et al: Immunoreactive renin, prorenin, and enzymatically active renin in plasma during pregnancy and in women taking oral contraceptives. J Clin Endocrinol Metab 63: 1008, 1986 |
|
Baxter JD, Forsham PH: Tissue effects of glucocorticoids. Am J Med 53: 573, 1972 |
|
Kopin IJ: Catecholamines, adrenal hormones, and stress. In Krieger DT, Hughes JC (eds): Neuroendocrinology, p 159. Sunderland, MA, Sinauer Associates, 1980 |
|
Walters MR: Steroid hormone receptors and the nucleus. Endocr Rev 6: 512, 1985 |
|
Otsuki M, Dakoda M, Baba S: Influence of glucocorticoids on TRF-induced TSH response in man. J Clin Endocrinol 36: 95, 1973 |
|
Copinschi G, L'Hermite M, Leclercq R et al: Effects of glucocorticoids on pituitary hormonal responses to hypoglycemia: Inhibition of prolactin release. J Clin Endocrinol Metab 40: 442, 1975 |
|
Dubey AK, Plant TM: A suppression of gonadotropin secretion by cortisol in castrated male rhesus monkeys (Macaca mulatta) mediated by the interruption of hypothalamic gonadotropin-releasing hormone release. Biol Reprod 33: 423, 1985 |
|
Haynes RC, Murad F: Adrenocorticotropic hormone: adrenocortical steroids and their synthetic analogs: inhibitors of adrenocortical steroid biosynthesis. In Gilman AG, Goodman LS, Rall TW (eds): Goodman and Gilman's The Pharmacologic Basis of Therapeutics, 7th ed, p 1459. New York, Macmillan, 1985 |
|
Vandewalle A, Farman N, Bencsath P, Vonvalet JP: Aldosterone binding along the rabbit nephron: An autoradiographic study on isolated tubules. Am J Physiol 240: F172, 1981 |
|
Stone DK, Seldin DW, Kokko JP, Jacobsen HR: Mineralocorticoid modulation of rabbit medullary collecting duct acidification: A sodium independent effect. J Clin Invest 72: 77, 1983 |
|
Allen GG, Barrett LJ: Effect of aldosterone on the transepithelial potential difference on the rat distal tubule. Kidney Int 19: 678, 1981 |
|
Mugais SK, Chekel MA, Jones WJ et al: Regulation of renal Na+-K+-ATPase in the rat: Role of the natural mineralo- and glucocorticoid hormone. J Clin Invest 73: 13, 1984 |
|
Fejes-Toth G, Naray-Fejes, Toth A: Effect of aldosterone on urinary kallikrein and prostaglandin synthesis in the rat. J Physiol 354: 79, 1984 |
|
Nasjletti A, Malik KU: The renal kallikrein-kinin and prostaglandin systems interaction. Ann Rev Physiol 43: 597, 1981 |
|
Law PY, Edelman IS: Induction of citrate synthesis by aldosterone in the rat kidney. J Membr Biol 41: 41, 1978 |
|
Goodman DBP, Wong M, Rasmussen H: Aldosterone-induced membrane phospholipid fatty acid metabolism in the toad bladder. Biochim Biophys Acta 421: 210, 1976 |
|
Wrong O, Metcalf-Gibson A: The electrolyte content of faeces. Proc R Soc Med 58: 1007, 1965 |
|
Weiner JS, Hellmann K: The sweat glands. Biol Rev 35: 141, 1960 |
|
Schneyer LH, Young JA, Schneyer CA: Salivary secretion of electrolytes. Physiol Rev 52: 720, 1972 |
|
Maroulis GB: Evaluation of hirsutism and hyperandrogenemia. In Wallach EE, Kempers RD (eds): Modern Trends in Infertility and Conception Control, Vol 2, p 32. Philadelphia, Harper & Row, 1982 |
|
Rosenfeld RS, Rosenberg BJ, Fukushima DK et al: 24 hour secretory pattern of dehydroisoandrosterone and dehydroisoandrosterone sulfate. J Clin Endocrinol Metab 40: 850, 1975 |
|
Parker LN, Lifrak ET, Odell WD: A 60,000 molecular weight human pituitary glycopeptide stimulates adrenal androgen secretion. Endocrinology 113: 2092, 1983 |
|
Schiebinger RJ, Chrousos GP, Cutler BG et al: The effect of serum prolactin on plasma adrenal androgens and the production and metabolic clearance rate of dehydroepiandrosterone sulfate in normal and hyperprolactinemic subjects. J Clin Endocrinol Metab 62: 202, 1986 |
|
Yuen BH, Kelch RP, Jaffe RB: Adrenal contribution to plasma oestrogens in adrenal disorders. Acta Endocrinol 76: 117, 1974 |
|
Orentreich N, Brind JL, Rizer RL et al: Age changes and sex differences in serum dehydroepiandrosterone sulfate concentrations throughout adulthood. J Clin Endocrinol Metab 59: 551, 1984 |
|
Vermeulen A: The hormonal activity of the postmenopausal ovary. J Clin Endocrinol Metab 42: 247, 1976 |
|
Nimrod A, Ryan KJ: Aromatization of androgens by human abdominal and breast fat tissue. J Clin Endocrinol Metab 40: 367, 1975 |
|
Hammond GL: Molecular properties of corticosteroid binding globulin and the sex-steroid binding proteins. Endocr Rev 11: 85, 1990 |
|
Mendel CM: The free hormone hypothesis: A physiologically based mathematical model. Endocr Rev 10: 232, 1989 |
|
Rosner W: The functions of corticosteroid-binding globulin and sex hormone-binding globulin: Recent advances. Endocr Rev 11: 80, 1990 |
|
Hyrb DJ, Khan MS, Romas NA et al: Specific binding of human corticosteroid-binding globulin to cell membranes. Proc Natl Acad Sci USA 83: 3253, 1986 |
|
Seralini GE, Berube D, Gagne R, Hammond GL: The human corticosteroid binding globulin gene is located on chromosome 14q31-q32. 1 near two other serine protease inhibitor genes. Hum Genet 86: 73, 1990 |
|
Rosner W: Plasma steroid binding proteins. Endocrinol Metab Clin North Am 20: 697, 1991 |
|
Peterson RE: Metabolism of adrenal cortical steroids. In Christy NP (ed): The Human Adrenal Cortex, p 87. New York, Harper & Row, 1971 |
|
Findling JW, Raff H: Newer Diagnostic Techniques and Problems in Cushing's disease. Endocr Metab Clinics 28: 191, 1999. |
|
Carpenter PC: Diagnostic evaluation of Cushing's syndrome. Endocrinol Metab Clin North Am 17: 445, 1988 |
|
Kaye TB, Crapo L: The Cushing syndrome: An update on diagnostic tests. Ann Intern Med 112: 434, 1990 |
|
Baulieu E-E, Corpechot C, Dray F et al: An adrenal-secreted androgen: Dehydroisoandrosterone sulfate. Its metabolism and a tentative generalization on the metabolism of other steroid conjugates in man. Recent Prog Horm Res 21: 411, 1965 |
|
Conrad SH, Lindberg MC, Herrman WL: Significance of plasma dehydroisoandrosterone and androsterone sulfates in the diagnosis of virilizing disorders. Am J Obstet Gynecol 91: 449, 1965 |
|
Yamaji T, Ibayashi H: Plasma dehydroepiandrosterone sulfate in normal and pathological conditions. J Clin Endocrinol 29: 273, 1969 |
|
Rosenfeld RS, Hellman L, Gallagher TF: Metabolism and interconversion of dehydroisoandrosterone and dehydroisoandrosterone sulfate. J Clin Endocrinol Metab 35: 187, 1972 |
|
Kirschner MA, Sinhamahapatra S, Zucker IR et al: The production, origin and role of dehydroepiandrosterone and delta 5-androstenediol as androgen prehormones in hirsute women. J Clin Endocrinol Metab 37: 183, 1973 |
|
Rosenfeld RS, Rosenberg BJ, Fukushima DK: 24-hour secretory pattern of dehydroisoandrosterone and dehydroisoandrosterone sulfate. J Clin Endocrinol Metab 40: 850, 1975 |
|
Korth-Schutz S, Levine LS, New MI: Dehydroepinandrosterone sulfate (DS) levels: A rapid test for abnormal adrenal androgen secretion. J Clin Endocrinol Metab 42: 1005, 1976 |
|
Maroulis GB, Manlimos FS, Abraham GE: Comparison between urinary 17-ketosteroids and serum androgens in hirsute patients. Obstet Gynecol 49: 454, 1977 |
|
Lobo RA, Paul WL, Goebelsmann U: Dehydroepiandrosterone sulfate as an indicator of adrenal androgen function. Obstet Gynecol 57: 69, 1981 |
|
Vermeulen A, Verdonck L: Studies on the binding of testosterone to human plasma. Steroids 11: 609, 1968 |
|
Forest MG, Rivarola MA, Migeon CJ: Percentage binding of testosterone, androstenedione and dehydroisoandrosterone in human plasma. Steroids 12: 323, 1968 |
|
Kappas A, Pearson OH, West CD, Gallagher TF: A study of idiopathic hirsutism: A transitional adrenal abnormality. J Clin Endocrinol 16: 517, 1956 |
|
Lobo RA, Paul W, Goebelsmann U: Serum levels of DHEAS in gynecologic endocrinopathy and infertility. Obstet Gynecol 57: 607, 1981 |
|
Hoffman DI, Klove K, Lobo RA: The prevalence and significance of elevated dehydroepiandrosterone sulfate levels in anovulatory women. Fertil Steril 42: 76, 1984 |
|
Steinberger E, Smith KD, Rodriguez-Rigau LJ: Testosterone, dehydroepiandrosterone, and dehydroepiandrosterone sulfate in hyperandrogenic women. J Clin Endocrinol Metab 59: 471, 1984 |
|
Madden JD, Milewich L, Parker CR Jr et al:The effect of an oral contraceptive treatment on the serum concentration of dehydroisoandrosterone sulfate. Am J Obstet Gynecol 132: 380, 1978 |
|
Carr BR, Parker CR Jr, Madden JD et al: Plasma levels of adrenocorticotropin and cortisol in women receiving oral contraceptive steroid treatment. J Clin Endocrinol Metab 49: 346, 1979 |
|
Wild RA, Umstot ES, Andersen RN, Givens JR: Adrenal function in hirsutism, part II. Effect of an oral contraceptive. J Clin Endocrinol Metab 54: 676, 1982 |
|
Howlett TA, Rees LH, Besser GM: Cushing's syndrome. Clin Endocrinol Metab 14: 911, 1985 |
|
Horrocks PM, London DR: Diagnostic value of 9 am plasma adrenocorticotrophic hormone concentrations in Cushing's disease. Br Med J 285: 1302, 1982 |
|
Wand GS, Ney RL: Disorders of the hypothalamic-pituitary-adrenal axis. Clin Endocrinol Metab 14: 33, 1985 |
|
Nelson JC, Tindall DJ: A comparison of the adrenal responses to hypoglycemia, metyrapone and ACTH. Indian J Med Sci 275: 165, 1978 |
|
Rose LL, Williams GH, Jagger PL et al: The 48 hour adrenocorticotrophin infusion test for adrenocortical insufficiency. Ann Intern Med 73: 49, 1970 |
|
Liddle GW: Tests of pituitary-adrenal suppressibility in the diagnosis of Cushing's syndrome. J Clin Endocrinol Metab 20: 1539, 1960 |
|
Steeten DHP, Anderson GH, Dalakos TG et al: Normal and abnormal function of the hypothalamic-pituitary-adrenocortical system in man. Endocr Rev 5: 371, 1984 |
|
Spark RF: Simplified assessment of pituitary-adrenal reserve: Measurement of serum 11-deoxycortisol and cortisol after metyrapone. Ann Intern Med 75: 717, 1971 |
|
Burke CW: Adrenocortical insufficiency. Clin Endocrinol Metab 14: 947, 1985 |
|
Nieman LK, Loriaux DL: Corticotropin-releasing hormone: Clinical applications. Ann Rev Med 40: 331, 1989 |
|
Muller OA, Stalla GK, von Werder K: CRH in Cushing's syndrome. Horm Metab Res [Suppl] 16: 51, 1987 |
|
Chrousos GP, Schulte HM, Oldfield EH et al: The corticotropin-releasing factor stimulation test. N Engl J Med 310: 622, 1984 |
|
Zacharieva S, Matrosov P, Stoeva I, Kirilov G: Corticotropin-releasing factor (CRF) stimulation test in normal subjects and patients with Cushing's syndrome. Exp Clin Endocrinol 93: 19, 1989 |
|
Findling JW, Raff H; Clinical Review: Cushing's Syndrome: Important Issues in Diagnosis and Management. J Clin Endocrinol Metab 91: 3746-3753, 2006. |
|
Gold PW, Loriaux DL, Roy A et al: Responses to corticotropin-releasing hormone in the hypercortisolism of depression and Cushing's disease. N Engl J Med 314: 1329, 1986 |
|
Gold PW, Gwirtsman H, Avgerinos PC et al: Abnormal hypothalamic-pituitary-adrenal function in anorexia nervosa. N Engl J Med 314: 1355, 1986 |
|
Schulte HM, Chrousos GP, Avgerinos PC et al: The corticotropin-releasing factor stimulation test: A possible aid in the evaluation of patients with adrenal insufficiency. J Clin Endocrinol Metab 58: 1064, 1984 |