
Placental Physiology
Authors
INTRODUCTION
The placenta is a unique fetal organ that performs a number of physiologic functions. Paramount is the placenta's interrelationship between the mother and fetus in the delivery of oxygen and nutrients and in the removal of waste. The health and growth of the fetus are dependent on this complex interaction. The growing fetus requires nutrients as fuel for generating energy as well as building blocks for growth. A shortage of nutrients may restrict growth and may lead to fetal compromise. An excess supply may be equally detrimental. We now realize that the effects of this process extend well beyond the time in utero, and influence growth in adolescence and adulthood. This chapter focuses on how the placenta performs this delicate balance. Important basic anatomic and transport mechanisms are presented, followed by current theories on transport of respiratory gases, macronutrients and micronutrients, and waste products. Interlaced are clinical scenarios that may be the result of abnormalities in these mechanisms. For an in-depth review of other placental functions, such as endocrine or immunologic, the reader is referred to excellent review articles.1,2
Our current understanding of placental function comes from several models. First, simple comparisons have been made between maternal and newborn concentrations of serum components. With periumbilical cord sampling, comparisons now include maternal-fetal values. Second, investigators have used in vitro models, such as perfused human placentae or microvesicles, directly measuring transfer of molecules and performing microscopic analysis. Finally, investigators have developed whole animal models to monitor transport in vivo. Each of these models has its limitations, but taken collectively, they aid our understanding of placental transfer mechanisms.
PLACENTAL STRUCTURE
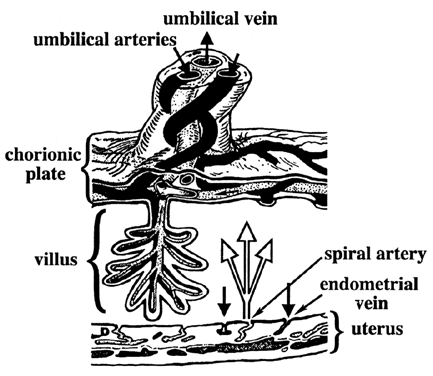
At term, the human placenta is a villus, hemomonochorial structure. That is, maternal blood (heme) is in direct contact with the fetal villus. The villus is composed of a single layer of trophoblast (monochorial) separating maternal blood from fetal vessels. The transfer of gases and nutrients can be divided into three stages: (1) delivery via maternal blood, (2) transfer across trophoblast tissue, and (3) uptake by fetal circulation (Fig. 1). The term placenta receives approximately 70% of uterine blood flow.3 Maternal blood enters the intervillous space via the spiral arteries and bathes the outer lining of the villi, the syncytiotrophoblast. The syncytiotrophoblast layer is a polarized, multinucleated epithelium lacking lateral cell membranes. Therefore, there is no “paracellular” or “between cell” transport pathway. Fetal blood enters arteries on the basal surface of the placenta, which branch into villus capillaries. Once transfer has taken place, nutrient-rich blood returns to the fetus via the umbilical vein.
|
The simplest pathway across a membrane is diffusion: movement of particles from areas of high to areas of low concentration. A substance can diffuse by the following means:
Simple diffusion: Particles pass (1) directly across the lipid membrane, or (2) through channel proteins
Facilitated diffusion: Particles diffuse by becoming attached to a carrier protein (Fig. 2).
|
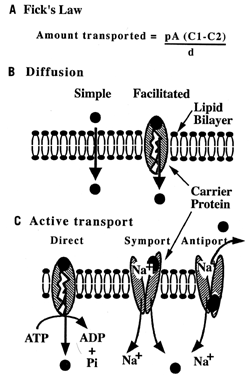
Small, neutrally charged particles, such as oxygen and carbon dioxide, pass directly through the lipid bilayer. Fick's first law of diffusion can be used to calculate the net amount of molecules transferred, or net flux. Charged or large particles cannot pass through the lipid bilayer easily; therefore diffusion either does not occur or progresses at a slow rate. Proteins embedded in the lipid bilayer can increase the rate of diffusion by creating an aqueous channel for these particles to travel, or by shuttling molecules across the bilayer. Hormones often regulate whether channels are “open” or “closed.” In carrier-mediated diffusion, both the numbers of carriers and the concentration difference of the solute may limit rate of diffusion across the membrane.
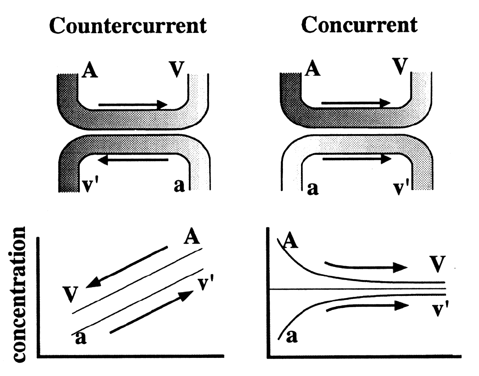
Many factors influence simple diffusion by affecting variables in Fick's law (Table 1).4,5,6 Some of these factors, such as protein binding and placental blood flow, influence the relative concentration gradient of a particle (C1 - C2). Noncovalently bound molecules are in a constant flux, binding and dissociating with the carrier. Only unbound molecules are free to diffuse across membranes. The greater the affinity a serum carrier protein has with a molecule, the less time it will spend “free” in solution, thereby limiting its ability to diffuse. A second factor is the relationship between direction of maternal and fetal blood flow (Fig. 3). Several species (e.g., guinea pig, rabbit) have maternal-fetal blood flow traveling in opposite directions, or countercurrent. This orientation of blood flow allows fetal venous blood to equilibrate to maternal artery concentrations of nutrients. This is a highly efficient means for transporting substances. In concurrent exchange, blood flow travels in the same direction. Therefore, blood returning to the fetus equilibrates to the maternal venous concentration, limiting concentration gradient. The human placenta acts as a concurrent exchanger.7 Maternal and fetal blood flow also affect diffusion. Several authors have reviewed factors involved in the regulation of blood flow.8,9
TABLE 1. Placental Characteristics Affecting Transport
| Trimester | |
Factor | First | Third |
Surface area (m2) | 1.5 | 12–14 |
Villus diameter (μm) | 170 | 40 |
Syncytiotrophoblast thickness (μm) | 10 | 1.7 |
Number of microvilli × 106/cm2) | 600 | 1200 |
Uteroplacental blood flow (mL/min) | 50 | 600 |
|
Cells require many substances that are in relatively low concentrations in the blood. Because diffusion is bidirectional, the concentration of a substance inside the cell is limited by the substance's extracellular concentration. Particles found in greater concentration in the cell would diffuse into the extracellular space. Cells may concentrate diffused substances by “trapping” the particle. For example, glucose is transported by facilitated diffusion. By phosphorylating glucose and forming a highly charged and nonpermeable molecule, the cell traps the glucose inside.
Cells can also concentrate substances by actively transporting particles across the membrane (see Fig. 2). Active transport is similar to facilitated diffusion in that it involves the formation of a protein-substrate complex. The difference, however, is that cellular energy is used, directly or indirectly, to pump the substance against its concentration gradient. When a protein pump hydrolyzes adenosine triphosphate (ATP) for energy, it is called direct active transport. A ubiquitous active transport protein is the sodium:potassium (Na+:K+) pump. By moving sodium out and potassium into the cell, an electrochemical gradient across the membrane is formed. The cell may use the energy “stored” in this electrochemical gradient to secondarily pump molecules against a concentration gradient. For example, cells concentrate amino acids by linking the movement of sodium (from high to low concentrations) to transport of an amino acid (from low to high). Sodium and a molecule can move in the same direction (symport) or in the opposite direction (antiport).
Another transport mechanism is receptor-mediated endocytosis. The cell membrane contains receptor proteins that bind specific ligands, such as low-density lipoprotein, a lipid carrier complex. Receptor-ligand complexes can migrate in the fluid lipid layer and cluster in groups. Endocytosis, or internalization of these complexes, occurs at specialized regions called pits. Some pits are “coated” by protein (clathrin) on the cytoplasmic side. Once internalized, the coat is removed, and the remaining endosome is degraded or transported to the opposing cell surface.
RESPIRATORY GASES
In accordance with Fick's law, gases cross plasma membranes by simple diffusion. The rate of diffusion of a particular gas depends on its lipid solubility and size. This rate is defined as a constant, “D.” Each gas has a specific D that is proportional to its solubility, and inversely proportional to the square root of its molecular weight. Because of their solubility differences, carbon dioxide diffuses across lipid membranes 20 times faster than oxygen (even though the square roots of their molecular weights are similar).10 Although Fick's law describes the rate of transfer, there are several factors that affect the total amount of oxygen transferred to the fetus.
Diffusion of oxygen occurs in several steps. Under normal conditions, 100 mL of maternal arterial blood contains 0.3 mL of dissolved oxygen and 20 mL of oxygen bound to hemoglobin. Dissolved and hemoglobin-bound oxygen is in constant equilibrium. Only dissolved oxygen is free to diffuse across the syncytiotrophoblast. Most of the oxygen transferred to the fetus must first dissociate from hemoglobin and travel across the erythrocyte membrane (Fig. 4).
|
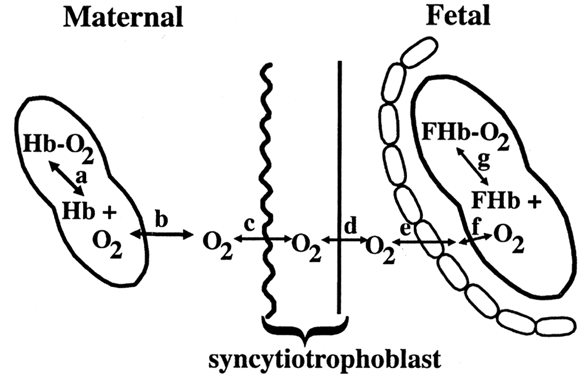
Critical to fetal oxygen delivery and uptake are the characteristics of maternal and fetal hemoglobin. Both maternal (HbA) and fetal (HbF) hemoglobin have four binding sites for oxygen. The number of sites occupied is the percent saturation. The partial pressure of oxygen (PO2) measures the small amount of dissolved oxygen. The relationship between partial pressure of oxygen and the degree of hemoglobin saturation is known as the oxygen dissociation curve (Fig. 5).11 The shape of the curve reflects many properties of oxygen transport:
|
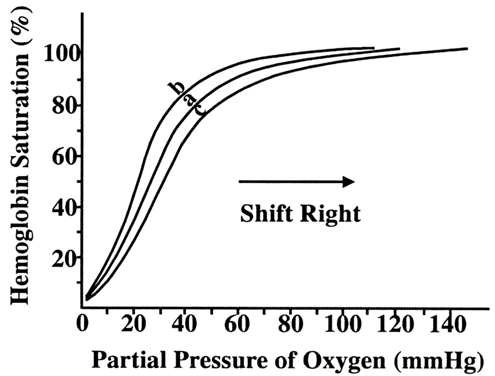
- Under normal conditions, there is nearly 100% saturation of hemoglobin in the maternal artery.
- The steep portion of the curve represents unloading of oxygen in peripheral tissues, such as the placenta.
- Fetal hemoglobin has a greater affinity for oxygen at all partial pressures.
Various factors affect the position on both the maternal and fetal oxygen dissociation curves. For example, the curve shifts to the right (affinity reduced) with an increase in temperature, hydrogen ion concentration, PCO2, and 2,3-diphosphoglycerate concentration in erythrocytes.12
Combining these principles, we can describe the steps of oxygen transport to the human fetus. Under normal conditions, the PO2 in maternal arterial blood is the same as in the alveolar sac. As the PO2 increases, hemoglobin becomes more saturated. This reaction is relatively fast (0.2 seconds). Total oxygen content in the maternal arterial system is 20.3 mL/100 mL (20 mL bound to hemoglobin and 0.3 mL dissolved in blood). As blood enters the villus space, oxygen, carbon dioxide, and hydrogen ions diffuse, equilibrating concentrations in maternal and fetal venous systems. The net effect is that maternal blood loses oxygen and gains carbon dioxide and hydrogen ions. These changes, plus the increase of temperature from fetal metabolism, decrease maternal hemoglobin affinity for oxygen. The PO2 equilibrates between maternal and fetal venous blood because the placenta is a countercurrent exchanger. Because of greater affinity for oxygen, fetal hemoglobin is more saturated than maternal hemoglobin at the same PO2.
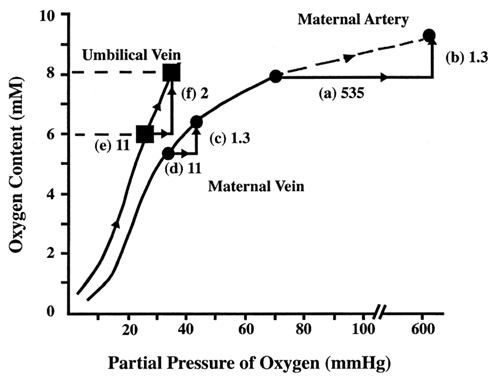
Fetal blood has a greater oxygen-carrying capacity than maternal blood. Not only does fetal hemoglobin have greater affinity for oxygen, but fetal hemoglobin concentrations are greater than the maternal hemoglobin concentrations. As a result, relatively small changes in fetal PO2 can greatly increase the fetal venous oxygen content. This can be dramatized in a clinical scenario: what happens to fetal oxygen measurements when the mother breathes 100% oxygen (Fig. 6)?13 In this example, maternal arterial PO2 is 65 mmHg, and the total amount of oxygen is 7.5 millimolar (mM) at room air. Breathing 100% oxygen increases the amount of dissolved oxygen (PO2) from 65 to 600 mmHg (step a). Because most hemoglobin is already saturated at room air, there is little change in bound O2. Total maternal oxygen content has a modest increase of 1.3 mM (step b). As the blood enters the villus space, oxygen, carbon dioxide, and hydrogen ions equilibrate between maternal and fetal venous circulations. Because changes in maternal arterial PO2 have no effect on the rate of oxygen consumption, maternal venous oxygen content also increases by 1.3 mM (step c). The flux of carbon dioxide and hydrogen ions into the villus space decreases maternal hemoglobin's affinity for oxygen. The extra 1.3 mM of oxygen increases the amount of unbound, dissolved oxygen by 11 mmHg (step d). Higher maternal venous PO2 results in a greater concentration gradient, and more oxygen diffuses across into umbilical venous blood. The PO2 in the fetal vein also increases 11 mmHg (step e). Fetal hemoglobin saturates at the higher PO2, and thereby fetal venous oxygen content increases (step f). In summary, administration of 100% oxygen to the mother slightly increases venous PO2, but significantly increases fetal oxygen content.
|
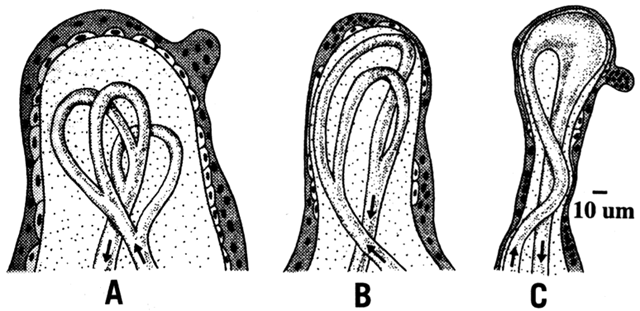
Thus far, this chapter has concentrated on the differences in maternal and fetal concentrations of oxygen. Other components of Fick's law are the area and thickness of the membrane, the placenta villus structure. The diffusing capacity of the placenta increases with gestational age as a result of growth and differentiation. As villi mature, their surface area increases and thickness decreases (Fig. 7).14 The mature villi are more vascular, and the vessels are closer to the syncytiotrophoblast layer. From 10 to 40 weeks of gestation, there is a 30-fold increase in diffusing capacity, the greatest change occurring after 20 weeks.15 The thinning of the villus stroma and trophoblast layer accounts for most of the increase.
|
Blood carries carbon dioxide in three forms: dissolved, as bicarbonate ion, and in combination with proteins as carbarno compounds. The erythrocyte enzyme carbonic anhydrase catalyzes the hydration of carbon dioxide to carbonic acid. The hydrogen ion formed reduces hemoglobin and increases hemoglobin affinity with carbon dioxide (Haldane effect). Carbon dioxide is very lipid soluble and is rapidly transferred across the placenta. The concentration gradient is due to a high fetal production of carbon dioxide as a byproduct of oxidation, and a low maternal PCO2 due to increased minute ventilation during pregnancy. A low fetal PCO2 occurs only when maternal carbon dioxide concentration decreases (maternal and fetal respiratory alkalosis). Increased fetal PCO2 may occur with poor gas exchange between fetal and maternal blood (e.g., abruption or severe placental infarcts) or when the PCO2 in the maternal blood greatly increases (e.g., respiratory failure).16
MACRONUTRIENTS
Carbohydrates
Glucose is the principle substrate for energy metabolism. Glucose transporters are located on the maternal (apical) and fetal (basal) surfaces of trophoblast.17,18 These transporters belong to a class of glucose carrier proteins called GLUT. All animal tissues contain GLUT proteins that allow facilitated diffusion of glucose. The other class of glucose carrier proteins comprises the SGLT proteins. These proteins actively transport glucose in the gastrointestinal tract and renal tubules, but have not been identified in the placenta. Like respiratory gases, placental transport of glucose relies on a concentration gradient. Glucose transport can be bidirectional, but under most conditions, flows from higher maternal concentration to lower fetal concentration.19 This process occurs in two steps. The first gradient drives glucose across the apical surface, from maternal blood to the intracellular compartment of the syncytiotrophoblast. The second gradient drives glucose across the basal surface, between syncytiotrophoblast and fetal blood. Most of the gradient is due to the high glucose consumption of the placenta, which may account for as much as 80% of the glucose uptake. Placental glucose consumption declines when fetal serum glucose concentrations are low, allowing a greater proportion of glucose to enter the fetal circulation (Fig. 8).20
|
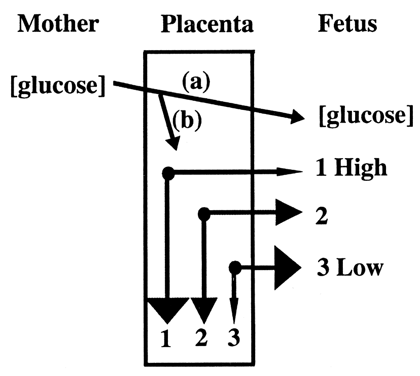
Fetal glucose comes almost exclusively from placental transport. Most fetuses, including the human fetus, require a glucose concentration of 4 to 8 mg/kg/min.21 The fetal brain, erythrocytes, and adrenal medulla rely entirely on glucose as a metabolic fuel. There is a greater demand for nutrients as the fetus grows. In the sheep, glucose utilization increases more than 10 fold in mid gestation.22 This demand is met by an increase in placental surface area and a greater diffusion gradient. The greater surface area of the placenta increases the capacity fivefold (density of glucose transporter remains constant).23 The remainder of glucose demand is met by the growth of insulin-sensitive fetal tissue. Insulin drives glucose into muscle, liver, and adipose tissue, thus lowering the fetal serum concentration. The lower serum concentration has two effects: (1) the gradient between fetal and maternal blood increases; and (2) the placenta utilization of glucose decreases.19
The placenta consumes much of the glucose transported, metabolizing as much as 50% to 70% to lactate.24 Trophoblasts use a small portion of the lactate, but export the majority into maternal and fetal circulations. Early in pregnancy, the fetus cannot utilize much of the lactate, and most lactate diffuses into the maternal circulation. Late in pregnancy, more fetal tissues take up lactate, which lowers fetal serum concentrations. This causes lactate to diffuse into the fetal circulation. Lactate is believed to be an important metabolic fuel, especially to the heart, as well as a substrate for fetal growth.
When maternal glucose concentrations are abnormal, there can be a marked effect on the fetus. Because of facilitated diffusion, any elevation in maternal glucose concentration results in an elevation in fetal serum concentration. This may invoke an insulin response as early as 14 to 16 weeks' gestation.25 The net effect is to drive glucose into cells, which in turn lowers fetal serum concentration. The magnitude of this response depends on the amount of insulin-sensitive tissue present. If there is still an excess, glucose diffuses back into the trophoblast. This increases placental metabolism, to as high as 80% of the transported glucose, and potentially decreases the fetal serum level by 60%.20 The placental response may have a greater effect but a slower response than the fetal secretion of insulin. Sustained, mild increases in glucose levels may therefore have limited fetal effect. However, if sharp fluctuations in glucose occur, or if a marked increase in glucose concentration occurs, insulin drives excess nutrients into fetal tissue. An opposite response occurs with maternal hypoglycemia: fetal insulin secretion drops, as well as placental utilization. This conserves the glucose supply for nervous tissue, adrenals, and erythrocytes at the expense of fetal growth.
Amino Acids
The placenta actively transports all amino acids, with fetal concentrations exceeding maternal levels. Transporters are not specific, and are grouped by the types of amino acids transferred and whether there is a cotransport of sodium (Na+).26 The sodium-dependent systems are as follows: “A,” preferentially transporting polar or neutral side chains (glycine and alanine); “ASC,” transferring alanine, serine, and cysteine; and XAG, selectively permeable to acidic amino acids (glutamate and aspartate). The following are the sodium-independent pathways: “L,” open to neutral, branched, and aromatic acids (leucine, phenylalanine); and y+, transporting basic amino acids (lysine, ornithine, and arginine). Several other amino acid transporters have been identified in the placenta that do not fit into this simple classification.27
A dynamic interaction exists between placenta and fetal liver, producing an amino acid pool for growth and metabolism. Both supply, modify, and metabolize amino acids in the fetal circulation. For example, the placenta actively takes up acidic amino acids (glutamate and aspartate) from maternal blood, but transfers little to the fetus. In addition, certain branched amino acids (leucine, isoleucine, and valine) are metabolized to form glutamate. Trophoblast metabolizes as much as 80% of the glutamate.28 There are two important consequences: (1) production of nicotinamide adenine dinucleotide phosphate (NADPH), important in fatty acid and steroid synthesis; and (2) reduction in glutamate concentration, a potential neurotoxin. In animals, the placenta converts almost all of the transferred maternal serine to glycine.29 This reaction can be linked to generation of tetrahydrofolate, an important cofactor in metabolism. The net pool of amino acids transported to fetal blood consists of maternally and placentally derived amino acids and their metabolites.
As blood enters the fetal liver via the umbilical vein, the hepatocytes may take up and modify amino acids. The liver often performs reciprocal functions to the placenta. For instance, it converts acidic and branched amino acids into corresponding keto acid with release of ammonia (e.g., glutamine back to its keto acid, glutamate).28 Hepatocytes have the ability to produce amino acids de novo from ammonia.30 The total amino acid pool greatly exceeds fetal requirements for protein synthesis. Fetal tissue utilizes some of the excess as another source of energy, producing urea and ammonia and dumping these molecules into the circulation. As fetal blood returns to the placenta, there is active transport of amino acids. Because the urea cycle is negligible in the placenta, there is constant production of ammonia. This dynamic amino acid pool and ammonia supply are sources of nitrogen for building blocks and metabolism.
Unlike the respiratory gases and glucose, fetal amino acid concentrations remain relatively stable over a large range of maternal values.31 Whether disruption of this process produces a fetal effect is unclear. Lower fetal serum concentrations and decreased placental transport of amino acids (specifically, branched amino acids) have been observed in growth-restricted fetuses. In vitro, compounds associated with intrauterine growth restriction (e.g., nicotine, ethanol, cocaine) decrease amino acid transport. Limiting supplies of oxygen or glucose to the placenta decreases amino acid transport.
Lipids
Lipids play a major role in fetal growth and development. Lipids are a basic constituent of plasma membranes, act as fuel for oxidative metabolism, and are precursors for compounds such as prostaglandin. Unfortunately, we have limited knowledge on lipid transport in the human placenta. Although lipids play similar roles in other animals, there are striking differences between animal models and humans. For instance, the sheep fetus has only 3% body fat at birth, compared to 18% for the human fetus. This difference is probably due to differences in lipid transport.19 Some conclusions have been drawn from a variety of animal models: (1) the relative composition of lipids in the fetal serum is similar to that in adults; (2) within a given species, fatter babies develop from mothers with higher plasma concentrations of lipids; and (3) between species, there is a relationship between lipid transport and fetal fat content.32 Thus, it is likely that fetal lipid content is dependent on the transplacental concentration gradient.
The human fetus has minimal carnitine and enzymes for lipid synthesis.33 As a result, the fetus depends on the placenta for most lipid requirements, for the partial breakdown of fatty acids to medium and short chains, and as a source of ketones. Lipids originate from the fatty acids in the maternal blood. Once transported into the trophoblast, breakdown occurs inside the mitochondria. Short- and medium-chain fatty acids diffuse into the mitochondria without difficulty. Long-chain fatty acids must be bound to carnitine, a derivative of lysine and methoin. Oxidation then continues within the matrix space of the mitochondria. A byproduct of lipid metabolism is the production of ketone bodies. For synthesis, reversing oxidation in the mitochondria can produce long-chain fatty acids from small and medium chains. More commonly, microsomes produce fatty acids de novo from acetyl coenzyme A.34
In maternal serum, the majority of lipids do not circulate in a free form. Free fatty acids are bound to albumin or are part of the chylomicron, whereas cholesterol, triglyceride, and phospholipids are transported as lipoprotein complexes.34 A membrane-bound enzyme, lipoprotein lipase, acts on chylomicrons and lipoprotein complexes in maternal blood to liberate fatty acids. Some lipids may diffuse directly across the membrane, whereas specific carrier proteins transport fatty acids by facilitated diffusion.35 The trophoblast combines transported lipids with lipids produced de novo. From the common pool, the trophoblast may oxidize lipids for cellular energy or transport fatty acids directly into the fetal serum. More important, the placenta produces medium- and short-chain fatty acids and ketones and transports them to the fetus.
Clinically, abnormal lipid transport may lead to abnormal fat content in the fetus. Two points are important: (1) fetal and maternal concentrations of lipids are similar; and (2) fatty acids in the fetal adipose tissue come mainly from placental sources. Therefore, high maternal lipid concentrations result in higher fat accumulation in adipose. This may be seen in pregnant women with diabetes or hyperlipidemia.
Water and Electrolytes
Besides transport of nutrients, cells must maintain a specific ion composition in the cytosol. The relative amounts of ions determine cell turgor pressure, pH, and cellular metabolism. In all cells, intracellular fluid contains a lower concentration of sodium and a higher concentration of potassium. Trophoblasts must maintain this relationship and allow for transport of the same water and ions to the fetus. The exact mechanism is not well understood. Comparisons of fetal and maternal values of selected ions are given in Table 2.36 Although the placenta contains a Na+:K+ pump at the basal (fetal) surface, net transfer of Na+ and Cl- is probably due to simple diffusion. Therefore, maternal and fetal concentrations are similar. Increases or decreases of sodium or chloride on one side are reflected by a proportional change on the other. In contrast, fetal K+ concentration is tightly regulated, and is constant over a range of maternal levels.37
TABLE 2. Maternal and Fetal Serum Concentrations of Selected Ions
Ion | Maternal Plasma (mM) | Fetal Plasma (mM) |
Sodium (Na+) | 138 ± 2 | 139 ± 4 |
Potassium (K-) | 4.6 ± 0.5 | 6.4 ± 0.2 |
Calcium (Ca2+) | 2.23 ± 0.12 | 2.81 ± 0.17 |
Chloride(Cl-) | 107 ± 2 | 108 ± 2 |
Phosphate (PO4-2) | 0.46 ± 0.20 | 0.62 ± 0.10 |
(Shennan DB, Boyd CAR: Ion transport by the placenta: A review of membrane transport systems. Biochem Biophys Acta 906:437, 1987)
Water easily moves across placental tissue by osmosis, causing a high turnover rate. At 14 weeks' gestation, the rate is 100 mL/hour, increasing to a maximum of 3500 mL/hour at 35 weeks.38 In early pregnancy, osmosis occurs between three compartments: maternal blood, fetal blood, and amniotic fluid. As the fetal skin becomes more keratinized, less water is transferred from fetus to amniotic fluid by osmosis, resulting in 95% of the maternal and fetal water exchange occurring across the placenta.38 Although large amounts of water move across the placenta, the net transfer is relatively balanced because the solute concentrations on either side are similar. Accumulation of water into the fetal compartments is 20 to 30 mL/day.39 Net transfer changes if the amount or concentration of solutes changes. Infusion of hypotonic dextrose to mothers decreases solute concentration, increasing net flow of water to the fetus. Hypertonic solutions, such as mannitol, increase solute concentration; water moves from fetus to mother, decreasing the net flow.40
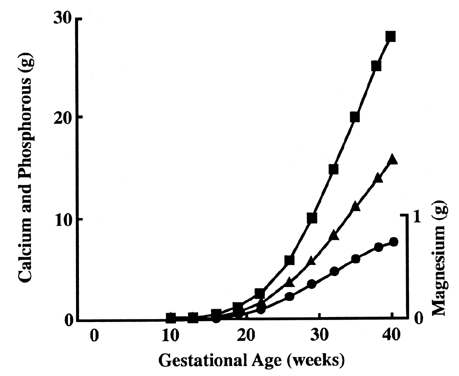
Serum calcium exists in three forms: bound to protein (40%), as ion complexes (10%), and free or ionized (50%). The placenta concentrates calcium on the fetal side while maintaining a low intracellular level necessary for cellular function. The exact mechanism is still unclear. Calcium concentration is 10,000 times lower in intracellular fluid than in extracellular fluid.41 Calcium channels are present at the maternal (apical) surface, allowing ionized calcium to flow in by simple diffusion.42 Intracellular free calcium is sequestered to maintain a low intracellular level. This sequestration of calcium may be in organelles, such as the Golgi apparatus or endoplasmic reticulum, or bound to intracellular protein, such as calcium-binding protein. Then trophoblasts actively pump calcium into the fetal circulation via Ca-ATPase located on the basal surface.43 Calcium demand increases greatly during the latter part of the pregnancy as bone mineralization occurs (Fig. 9).44 It is not known how placenta transport changes to match demand. Levels of calcium-binding protein increase dramatically in some animals, but not in humans.45,46 Levels of Ca-ATPase do not increase in animal or human placentas.45 Regulation may occur via hormonal control. Severe maternal hypocalcemia (less than 1.0 mmol), untreated hypoparathyroidism, and vitamin D deficiencies have caused cases of neonatal rickets and hypocalcemia.47,48,49 In addition, the fetus may modify calcium transport via hormones. The fetal parathyroid secretes parathyroid hormone and a parathyroid-related protein.50 The fetal kidney and placenta can produce vitamin D.51 Both maternal and fetal hormones modify calcium transport in vitro, but what roles they play in vivo still remain a mystery. Even less is known about phosphate and magnesium transfer. Both minerals are concentrated on the fetal side, and therefore active transport is likely to occur.44
|
MICRONUTRIENTS
Trace Metals
Iron transport across the placenta is similar to that for other tissues. Iron is carried in maternal blood bound to transferrin (Tf). The syncytiotrophoblast contains Tf receptors on its apical cell surface, which binds transferrin. The binding is pH dependent, having a greater affinity at a pH of 7.4 than at 5.5.52 After binding, the Tf—Tf-receptor complex is internalized, forming a vesicle. By activating hydrogen pumps, the vesicle drops the internal pH.53 The lower pH allows dissociation of iron from maternal transferrin (MTf), and causes conformation changes in MTf and its receptor, which protect them from proteases. Once unbound, iron is actively pumped out into the cytosol, where it can bind to intracellular ferritin. The receptor and MTf return to the apical surface. The Tf receptor releases the empty transferrin into the maternal circulation.53
Ferritin inside the trophoblast assimilates iron into fetal transferrin (FTf). The FTf-iron complex is carried to the basal surface, and iron is released into the fetal circulation.54 It is unclear how the trophoblasts maintain unidirectional iron transport, but it is believed that maternal and fetal transferrin represent two different pools.54 The amount of iron transported across the placenta depends on the number of transferrin receptors on the apical (maternal) surface and the concentration of ferritin inside the syncytiotrophoblast. Likewise, the number of fetal transferrin receptors affects fetal transfer.55 Upregulation occurs with both receptors when fetal stores of iron are low, and downregulation occurs when stores are high. Therefore the fetal need directly affects transport.
Placental transport of other trace metals, such as copper, zinc, and selenium, are much less understood. The body requires copper and zinc in trace amounts. Deficiencies or excesses can be detrimental to cells. The body stores excess amounts in the liver, bound to chelating proteins such as metallothionein or related proteins. Copper circulates bound to ceruloplasmin, albumin, and amino acids, specially histidine.56 Trophoblasts may reduce copper from bound Cu2+ to soluble Cu+ before transporting it via a membrane carrier or channel.57 Copper bound to albumin or histidine is more likely to be reduced and exchanged, whereas copper is tightly bound to ceruloplasmin. Reduction may occur by an enzymatic reaction such as NADH reductase, or by reducing agents such as vitamin C. Trophoblasts contain metallothionein, which may be used in sequestration and transport to the basal side.
Vitamins
Vitamins can be divided into two groups based on their solubility. Fat-soluble vitamins A, D, E, and K refer to groups of compounds that do not dissolve in water, but are able to diffuse across lipid layers of cell membranes. They are usually bound to carriers in the blood and must dissociate before diffusion can occur. Cells utilize these vitamins in differentiation and growth. The body stores fat-soluble vitamins; therefore deficiencies are rarely seen, but there is a potential for toxicity. The water-soluble vitamins, B complex and vitamin C, easily dissolve in blood, but poorly diffuse across cell membranes. Active transport usually occurs, forming a concentration gradient between fetus and mother of between 2:1 and 5:1.58 Water-soluble vitamins function as cofactors for enzymatic reactions. Because of high turnover and lack of storage, there is a greater potential for deficiencies. Table 3 summarizes proposed methods of transport as well as some biological functions of vitamins.
TABLE 3. Summary of Placental Transport and Fetal Uses of Vitamins
Common |
|
|
|
|
Name | Chemical Name | Biological Activity | Pregnant RDA* | Placenta Transport |
Fat Soluble |
|
|
|
|
Vitamin A | Retinol | Vision, growth differentiation | 800 RE (0%) | Receptor-mediated diffusion |
Vitamin D | Ergocalciferol, cholecalciferol | Calcium hemostasis | 10 μg (100%) | Simple diffusion |
Vitamin E | Tocopherols | Antioxidant | 10 mg (25%) | Simple diffusion |
Vitamin K | Phylloquinones, menaquinones | Blood clotting | 65 μg (0%) | Simple diffusion |
Water Soluble |
|
|
|
|
Vitamin B1 | Thiamine | Carbohydrate metabolism | 0.8 mg/kcal (36%) | Active transport |
Vitamin B2 | Riboflavin | Oxidation-reduction reactions | 1.6 mg (23%) | Active transport |
Vitamin B6 | Pyridoxine, pyridoxal, pyridoxamine | Amino acid, phospholipid, and glycogen metabolism | 2.2 mg (38%) | Diffusion, post-transport modification |
Niacin | Nicotinic acid | Oxidation-reduction | 17 mg (13%) | Active transport |
Folate | Pteroylglutamic | Nucleic acid and amino acid biosynthesis | 400 μg (122%) | Carrier-mediated diffusion |
Vitamin B12 | Cyanocobalamin | Amino acid and branched-chain ketoacid metabolism | 2.2 μg (12%) | Active transport, receptor-mediated endocytosis |
Biotin | — | Carboxylation | — | Simple diffusion |
Vitamin B5 | Pantothenic acid | Coenzyme A | — | Active transport |
Vitamin C | Ascorbic acid | Connective tissue formation, neurotransmitter? | 70 mg (17%) | Passive diffusion, carrier-mediated diffusion |
* The numbers in parentheses are the percent increase from nonpregnant. RE, retinol equivalent.
(Food and Nutrition Board, National Academy of Sciences: Nutrition During Pregnancy. Washington, DC, National Academy Press, 1990)
VITAMIN A.
Vitamin A is composed of a group of biological active substances chemically related to retinol. The term “retinoids” refers to vitamin A and a variety of chemically related natural and synthetic compounds. The natural sources of vitamin A include vegetables, which contain provitamins (carotenoids); and animal tissues, which have retinol esters. When ingested, the gut breaks down the precursors forming retinol and transports it to the liver in chylomicrons.59 The liver stores most retinol as fatty acids, such as retinyl palmitate. When needed, hepatocytes convert stored vitamin A to retinol; bind it to a small protein, retinol-binding protein (RBP); and then release the retinol-RBP complex into the blood stream. The retinol-RBP complex attaches to prealbumin, thereby preventing removal by glomerular filtration.60 Small amounts of retinol, which is very lipid soluble and easily diffuses across membranes, may exist unbound. At the maternal side of the placenta, the complex dissociates from albumin, binds to a cell receptor for RBP, and is internalized.
Our understanding of the trafficking of retinol inside trophoblasts is limited. Trophoblasts can transfer retinol to the fetal circulation or metabolize retinol to active or inactive forms. Albumin and lipoproteins in fetal blood pick up any transported retinol and carry it to the liver. Because the fetal liver has a limited ability to store vitamin A until late in gestation, increasing maternal intake has little effect on fetal stores.61 The placenta regulates the amount transported, and fetal concentrations remain uniform over a wide range of maternal dietary intake.
The process of placental transfer and the effect of vitamin A on the developing fetus have received much interest. In the embryo, vitamin A is important in cellular differentiation. Cells oxidize retinol to retinoic acid, which then binds to retinoic acid receptors (RARs). These complexes can modify gene expression by binding to DNA, producing actions similar to that of thyroid hormone.62 In animals, vitamin A deficiency has been linked to an increased rate of miscarriage, poor fetal and placental growth, and fetal death.63 In excess amounts, vitamin A may affect migration of neural crest cells, leading to defects in the central nervous system, limbs, cardiovascular system, and craniofacial formation.64,65 Clinically, deficient or excess states are rarely seen.66 Two noted exceptions are the synthetic retinoids, etretinate (Tegison) and isotretinoin (Accutane). Although both have prolonged half-lives, metabolites of these drugs may ultimately cause toxic effects. Etretinate is very lipid soluble, and it persists in the maternal system for months after delivery. There are case reports of fetal defects seen months to 1 year after discontinuation of the drug.67,68 Isotretinoin is water soluble with a half-life of its metabolites of up to 50 hours.69 It is likely to be cleared from the maternal system within 10 days of discontinuation.
VITAMIN D.
There are two dietary sources of vitamin D, ergocalciferol (plants) and cholecalciferol (animal). In addition, the body can produce vitamin D by the interaction of ultraviolet light with 7-dehydrocholesterol, an intermediate of cholesterol synthesis. All compounds undergo modifications. The liver hydroxylates at the 25th carbon, producing 25 hydroxy-cholecalciferol (25-OH D). The kidney hydroxylates at the first carbon, forming 1,25 (OH)2 D. The latter step is the regulatory step under the influence of parathyroid hormone. Both metabolites are bound to a carrier, vitamin D3—binding protein.70
For placental transfer, vitamin D dissociates from the binding protein and crosses by simple diffusion.71 Vitamin D3-binding protein has a greater affinity for 25-OH D than for 1,25(OH2)D. Because it is bound tighter, less 25-OH D is free to diffuse. This accounts for the 10-fold greater flux of the 1,25 OH D form. The placenta does have the ability to hydroxylate at the 25 position.71 As mentioned earlier, this may allow trophoblasts to modify calcium uptake. Intake of vitamin D at levels 600 times the recommended dose have been reported.72 Fetal concentrations increase in proportion to maternal concentration, but no toxic effects are seen.
VITAMIN E.
The vitamin E group is composed of tocol and tocotrienol derivatives. The naturally occurring form, α-tocopherol, is carried in blood by low-density lipoproteins and is an important antioxidant. Tissue concentrations are related to lipid content, with adipose, liver, and muscle representing major storage depots. Placental transfer occurs by diffusion, but fetal transfer is regulated.73 In animals, large doses increase maternal and placental concentrations, but minimal changes are seen in the fetus. There is little information on high doses in human pregnancy, but no adverse effects have been noted.
VITAMIN K.
Vitamin K is a group of fat-soluble substances important in the synthesis of coagulation factors. There are two naturally occurring forms: vitamin K1 (phylloquinone) is found in a variety of green plants, and vitamin K2 (menaquinones) is produced by intestinal bacteria. Vitamin K1 is actively transported across the gut, whereas menaquinones pass by diffusion.74 Both are carried to the liver via the lymphatic system. The liver can store a 30-day supply. The liver reduces both compounds to produce cofactors for clotting factor synthesis. Although there is no recommended daily allowance for vitamin K, most of the body's needs are supplied by intestinal bacteria.
The fetus and newborn depend on transplacental passage and fetal storage of vitamin K until normal intestinal flora are established. Vitamin K is transported by simple diffusion, but requires a large concentration gradient. A gradient of 30:1 has been reported in several studies.75 Because of slow transplacental passage and lack of the ability to store large amounts, some infants have a relative deficiency of vitamin K at birth. Vitamin K deficiency has been directly linked to intracranial hemorrhage (ICH) in the newborn.76 Three time intervals for ICH have been described: early, within the first 24 hours of life; classic, between the second and fifth day of life; and late, after 5 days.77 The first occurs because of low fetal concentrations; the latter two are related to lack of significant stores of vitamin K. Several studies have examined the value of prenatal vitamin K supplementation to prevent ICH, but have not shown a greater benefit than vitamin K administration to the newborn at the time of delivery.78
Maternal deficiency is rare, but may occur when medications destroy the intestinal bacteria or increase the rate of degradation of vitamin K. The latter mechanism may occur with anticonvulsants, such as phenytoin and barbital derivatives, which induce liver enzymes.79 Lower maternal concentrations mean a lower gradient for diffusion and lower fetal concentrations. In addition, anticonvulsants can cross the placenta and induce the fetal liver to degrade vitamin K.80 Several reports have linked maternal anticonvulsant therapy to abnormal clotting function in newborns as well as an increased rate of ICH. There is still much debate whether prenatal vitamin K administration is beneficial in these women.
VITAMIN C.
The active form of vitamin C is ascorbic acid, a six-carbon molecule resembling glucose. Humans do not have the ability to synthesize vitamin C, as do some animals, and therefore must depend on dietary sources to meet all requirements. Ascorbic acid does not easily diffuse across cell membranes, but can be oxidized to dehydroascorbic acid in the stomach.81 The latter is uncharged, and readily passes through cell membranes. In the human placenta, dehydroascorbic acid diffuses passively across the syncytiotrophoblast, where it is converted to ascorbic acid. Because of its poor permeability, ascorbic acid is “trapped” on the fetal side.82 The placenta can also transport ascorbic acid by a sodium-dependent mechanism, possibly using the same transporter as for glucose.83 At high concentrations, the fetus increases the rate of ascorbic acid catabolism.84 In women taking more than 400 mg/day, this increased rate of breakdown has been linked to rare cases of neonatal scurvy.85
FOLIC ACID.
Folic acid acts as a coenzyme for nucleotide synthesis as well as methionine, an important precursor for a variety of methylation reactions. In addition to its well-publicized link to neural tube defects, folic acid deficiency has been associated with pregnancy loss, low birth weight, and delayed maturation of the nervous system.86 In serum, folic acid is found as methyltetrahydrofolate. There are specific folate-binding proteins in serum, but most folic acid is carried nonspecifically by albumin. Although the function of these serum folate-binding proteins is not known, there is increased binding in pregnancy. The placenta has membrane-bound folate-binding proteins, which are thought to be involved in receptor-mediated endocytosis.87
VITAMIN B12.
Vitamin B12, or cobalamins, are organo-metallo complexes consisting of a cobalt-containing corrin ring and a nucleotide base. Although B12 is transported in the serum by three cobalamin-binding proteins, only transcobalamin II is useful for placental transport.88 The placenta highly concentrates vitamin B12 by receptor-mediated endocytosis and then transports it to the fetal serum down a concentration gradient.89 The final fetal concentration is twice the maternal concentration. Fetal accumulation is 0.1 to 0.2 μg/day, and the newborn usually has 25 μg in stores.90
Animal products are virtually the only dietary sources of vitamin B12. Under normal intake, the adult liver stores up to 2 mg, representing a 20-year supply. Deficiencies are rarely seen, except in strict vegetarians or in patients with malabsorption syndromes. There is evidence that current intake may be very important for placental transport.91 Most of the vitamin B12 in serum is carried on transcobalamin I, and is not available for placental transport. Transcobalamin II transports newly absorbed vitamin B12. In women who were vegans for 30 months, maternal serum and milk concentrations were diminished.
SUMMARY
Many factors are involved in the delivery of oxygen and nutrients to the fetus. Maternal delivery of products is limited by concentrations of nutrients, rate of uterine blood flow, and the countercurrent orientation of maternal and fetal blood flow. Transfer across the placenta occurs by several mechanisms, including diffusion, active transport, and receptor-mediated endocytosis. Finally, the fetus and placenta interact to continually modify transport of nutrients. As we learn more about the natural state, and what dysfunctions lead to fetal compromise, we may understand how we can modify the process to improve fetal outcome. Administration of oxygen to the mother to increase fetal oxygen content or dietary restrictions for diabetic mothers to limit abnormal fetal growth are examples already in use.
REFERENCES
Pepe GJ, Albrecht ED: Actions of placental and fetal adrenal steroid hormones in primate pregnancy. Endocrinol Rev 16: 608, 1995 |
|
Saji F, Koyama M, Matsuzaki N: Current topic: Human placental Fc receptors. Placenta 15: 453, 1994 |
|
Assali NS: Dynamics of the uteroplacental circulation in health and disease. Am J Perinatol 6: 105, 1989 |
|
Aherne W, Dunhill MS: Quantitative aspects of placental structure. J Pathol Bacteriol 91: 123, 1966 |
|
Boyd JD, Hamilton WH: Development and structure of the human placenta from the end of the third month of gestation. J Obstet Gynaecol Br Commonw 74: 161, 1967 |
|
Martin CB: The anatomy and circulation of the placenta. In Barnes AC (ed): Intrauterine Development, p 35. Philadelphia, Lea & Febiger, 1968 |
|
Wilkening RB, Meschia G: Comparative physiology of placental oxygen transport. Placenta 13: 1, 1992 |
|
Firth JA, Leach L: Not trophoblast alone: A review of the contribution of the fetal microvasculature to transplacental exchange. Placenta 17: 89, 1996 |
|
Myatt L: Control of vascular resistance in the human placenta. Placenta 13: 329, 1992 |
|
Lee G, De J, Dubois AB: Pulmonary capillary blood flow in man. J Clin Invest 34: 1380, 1955 |
|
Hellegers AE, Schruefer JJ: Nomograms and empirical equations relating oxygen tension, percentage saturation, and pH in maternal and fetal blood. Am J Obstet Gynecol 81: 377, 1961 |
|
Guyton AC, Hall JE: Transport of oxygen and carbon dioxide in the blood and body fluids. In Textbook of Medical Physiology, 9th ed, p 518. Philadelphia, WB Saunders, 1996 |
|
Meschia G: How oxygen is transferred across the placenta. Contemp Obstet Gynecol 14: 151, 1979 |
|
Jackson MR, Mayhew TM, Boyd PA: Quantitative description of elaboration and maturation of villi from 10 weeks of gestation to term. Placenta 13: 357, 1993 |
|
Mayhew TM, Jackson MR, Boyd PA: Changes in oxygen diffusive conductances of human placentae during gestation (10–41 weeks) are commensurate with the gain in fetal weight. Placenta 14: 51, 1993 |
|
Hollingsworth HM, Irwin RS: Acute respiratory failure in pregnancy (pulmonary disease in pregnancy). Clin Chest Med 13: 723, 1992 |
|
Smith CH, Moe AJ, Ganapathy V: Nutrient transport pathways across the epithelium of the placenta. Annu Rev Nutr 12: 183, 1992 |
|
Barros LF, Yudilevich DL, Jarvis SM et al: Quantitation and immunolocalization of glucose transporters in human placenta. Placenta 16: 19, 1995 |
|
Hay WW: Metabolic interrelationships of placenta and fetus. Placenta 16: 19, 1995 |
|
Hay WW: Energy and substrate requirements of the placenta and fetus. Proc Nutr Soc 50: 321, 1991 |
|
Aldoretta PW, Hay WW Jr: Metabolic substrates for fetal energy metabolism and growth. Clin Perinatol 22: 15, 1995 |
|
Hay WW Jr, Sparks JW, Quissell B et al: Simultaneous measurement of umbilical uptake, fetal utilization rate, and fetal turnover of glucose. Am J Physiol 240: E662, 1981 |
|
Molina RD, Meschia G, Battaglia FC et al: Gestational maturation of placental glucose transfer capacity in the sheep. Am J Physiol 261: R697, 1991 |
|
Simmons MA, Battaglia FC, Meschia G: Placental transfer of glucose. J Dev Physiol 1: 227, 1979 |
|
Ktorza A, Bihoreau MT, Nurjhan N et al: Insulin and glucagon during the perinatal period: Secretion and metabolic effects on the liver. Biol Neonate 48: 204, 1985 |
|
Christensen HN, Albrilton LM, Kakuda DK, MacLeod CL: Gene product designations for amino acid transporters. J Exp Biol 196: 51, 1994 |
|
Moe AJ: Placental amino acid transport. Am J Physiol 268: C1321, 1995 |
|
Vaughn PR, Lobo C, Battaglia FC et al: Glutamine-glutamate exchange between placenta and fetal liver. Am J Physiol 268: E705, 1995 |
|
Moores RR Jr, Carter BS, Meschia G et al: Placental and fetal serine fluxes at midgestation in the fetal lamb. Am J Physiol 267: E150, 1994 |
|
Holzman IR, Lemons JA, Meschia G et al: Ammonia production by the pregnant uterus. Proc Soc Exp Biol Med 156: 27, 1977 |
|
Domenech M, Gruppuso PA, Nishino VT et al: Preserved fetal plasma amino acid concentrations in the presence of maternal hypoaminoacidemia. Pediatr Res 20: 1071, 1986 |
|
Page K: Transport and metabolism of lipids. In Physiology of the Human Placentae, p 71. London, UCL Press, 1993 |
|
Liechty EA: Fat metabolism and requirements. In Pollin RA, Fox WW (ed): Fetal and Neonatal Physiology, p 237. Philadelphia, WB Saunders, 1996 |
|
Guyton AC, Hall JE: Lipid metabolism. In Textbook of Medical Physiology, 9th ed, pp 865–866. Philadelphia, WB Saunders, 1996 |
|
Campbell FM, Gordon MJ, Duha-Roy AK: Plasma membrane fatty acid-binding protein (FABP pm) of the sheep placenta. Biochim Biophys Acta 1214: 187, 1994 |
|
Shennan DB, Boyd CAR: Ion transport by the placenta: A review of membrane transport systems. Biochim Bio Physica Acta 906: 437, 1987 |
|
Stewart EL, Welt LG: Protection of the fetus in experimental potassium depletion. Am J Physiol 200: 824, 1961 |
|
Hutchinson DL, Gray MJ, Plertl AA et al: The role of the fetus in water exchange of the amniotic fluid of normal and hydramniotic patients. J Clin Invest 38: 971, 1959 |
|
Hytton F: Water transfer. In Chamberlain GVP, Wilkinson AW (eds): Placental Transfer, p 90. Tunbridge Wells, Pitman Medical, 1979 |
|
Battaglia F, Prystowsky H, Smisson C et al: Fetal blood studies: XIII. The effect of administration of fluids intravenously to mothers upon the concentrations of water and electrolytes in plasma of the human fetus. Pediatrics 25: 2, 1960 |
|
Husain SM, Mughal MZ: Mineral transport across the placenta. Arch Dis Child 67: 874, 1992 |
|
Stulc J, Stulcova B: Transport of calcium by the placenta of the rat. J Physiol 371: 1, 1986 |
|
Borke JL, Curide A, Verna AK et al: Calcium pump epitopes in placental trophoblast basal plasma membranes. Am J Physiol 257: C341, 1989 |
|
Widdowson EM: Changes in body composition during growth. In Davis JA, Dobbing J (eds): Scientific Foundations of Paediatrics, p 330. London, William Heinemann Medical Books, 1981 |
|
Glazier JD, Atkinson DE, Thornburg KL et al: Gestational changes in Ca2+ transport across rat placenta and mRNA for calbindin 9K and Ca(2+ )-ATPase. Am J Physiol 263 (4 Pt 2):R930, 1992 |
|
Tuan RS: Calcium-binding protein of the human placenta: Characterization, immunohistochemical localization and functional involvement in CA++ transport. Biochem J 227: 317, 1985 |
|
Maxwell JP: Further studies of adult rickets (osteo malacia) and fetal rickets. Proc R Soc Med 28: 265, 1934 |
|
Jacobson BB, Tersley E, Lund B, Sorensen OH: Neonatal hypocalcaemia associated with maternal hyperparathyroidism. Arch Dis Child 53: 308, 1978 |
|
Park W, Paust H, Kaufman HJ et al: Osteomalacia of the mother: Rickets of the newborn. Eur J Ped 146: 292, 1987 |
|
Shaw AJ, Mughal MZ, Maresh MJA, Sibley CP: Effects of two synthetic parathyroid hormone—related protein fragments on maternal fetal transfer of calcium and magnesium and release of cyclic AMP by the in situ perfused rat placenta. J Endocrinol 129: 399, 1991 |
|
Bruns ME, Bruns DE: Vitamin D metabolism and function during pregnancy and the neonatal period. Am Clin Lab Sci 13: 521, 1983 |
|
Paterson S, Armstrong N, Iacopetta BJ et al: Intra vesicular pH and iron uptake by immature erythroid cells. J Cell Physiol 120: 225, 1984 |
|
Harris ED: New insights into placenta iron transport. Nutr Rev 50: 329, 1992 |
|
Cerneus DP, Van der Ende A: Apical and basolateral transferrin receptors in polarized Be Wo cells recycle through separate endosomes. J Cell Biol 114: 1149, 1991 |
|
Bierings MB, Bert MRM, Van Eijk HG et al: Transferrin receptor expression and the regulation of placental iron intake. Mol Cell Biochem 100: 31, 1991 |
|
Mcardle HJ: The transport of iron and copper across the cell membrane: Different mechanisms for different metals? Proc Nutr Soc 51: 199, 1992 |
|
Percival SS, Harris ED: Ascorbate enhances copper transport from ceruloplasmin into human K 562 cells. J Nutr 119: 779, 1989 |
|
Zempleni J, Link G, Kobler W: The transport of thiamine, riboflavin and pyridoxal 5-phosphate by human placenta. Int J Vitam Nutr Res 62: 165, 1992 |
|
Ross AC: Overview of retinoid metabolism. J Nutr 123: 346, 1992 |
|
Noy N, Slasberg E, Scarlatta S: Interactions of retinol with binding proteins: Studies with retinol binding protein and with transthyretin. Biochemistry 31: 1118, 1992 |
|
Shena JP, Chytil F, Jhaveri A, Stalman MT: Plasma vitamin A and retinol binding protein in premature and term neonates. J Pediatr 99: 302, 1981 |
|
Zang X-K, Hoffman B, Tran PBV et al: Retinoid × receptor is an auxiliary protein for thyroid hormone and retinoic acid receptors. Nature 355: 441, 1992 |
|
Takahashi VI, Smith JE, Winick M et al: Vitamin A deficiency and fetal growth and development in the rat. J Nutr 105: 1299, 1975 |
|
DiGiacomo RF, Deeb BJ, Anderson RJ: Hypervitaminosis A and reproductive disorders in rabbits. Lab Anim Sci 42: 250, 1992 |
|
Rosa FW, Wilk AL, Kelsey FO: Teratogen update: Vitamin A congeners. Teratology 33: 355, 1986 |
|
Rothman KJ, Moore LL, Singer MR et al: Teratogenicity of high vitamin A intake. N Engl J Med 333: 1369, 1995 |
|
Grote W, Harms D, Janig U et al: Letter to the Editor: Malformation of fetus conceived 4 months after termination of maternal etretinate treatment. Lancet 1: 1276, 1985 |
|
Greaves MW: Embryopathy in infant conceived one year after termination of maternal etretinate: A reappraisal. Lancet 2: 1254, 1988 |
|
Anonymous: Recommendations for isotretinoin use in women of childbearing potential. Teratology 44:1, 1991 |
|
DeLuca HF, Krisinger J, Darwish H: The vitamin D system. Kidney Int 38: 52, 1990 |
|
Ron M, Levitz M, Chuba J, Dancis J: Transfer of 25-hydroxyvitamin D3 and 1,25-dihydroxyvitamin D3 across the perfused human placenta. Am J Obstet Gynecol 148: 370, 1984 |
|
O'Brien J, Roserwasser S, Feingold M et al: Prenatal exposure to milk with excessive D supplementation. Teratology 47: 387, 1993 |
|
Njeru CA, McDowell LR, Wilkinson NS et al: Pre and postpartum supplemental DL-alpha-tocopherol acetate effects on placental and mammary vitamin E transfer in sheep. J Anim Sci 72: 1636, 1994 |
|
Mosher DF: Disorders of blood coagulation. In Bennett JC, Plum F (eds): Cecil Textbook of Medicine, 20th ed, p 999. Philadelphia, WB Saunders, 1996 |
|
Sheaer MJ, Crampton OE, McCarthy PT et al: Vitamin K in plasma: Relationship to vitamin K status, age, pregnancy, diet and disease. Haemostasis 16 (suppl 5): 83, 1986 |
|
Clarkson PM, James AG: Parental vitamin K1: The effective prophylaxis against hemorrhagic disease for all newborn infants. NZ Med J 103: 95, 1990 |
|
Lane PA, Hathway WE: Vitamin K in infancy. J Pediatr 106: 351, 1985 |
|
Thorp JA, Gaston L, Caspers DR et al: Current concepts and controversies in the use of vitamin K. Drugs 49: 376, 1995 |
|
Moslet U, Hansen ES: A review of vitamin K, epilepsy and pregnancy. Acta Neurol Scand 85: 39, 1992 |
|
Cornelissen M, Steegers-Theunissen R, Kollee L et al: Increased incidence of neonatal vitamin K deficiency resulting from maternal anticonvulsant therapy. Am J Obstet Gynecol 168: 923, 1993 |
|
Rose RC: Intestinal absorption of water-soluble vitamins. Proc Soc Exp Biol Med 212: 191, 1996 |
|
Rybakowski C, Mohar B, Wohlers S et al: The transport of vitamin C in the isolated human near-term placenta. Eur J Obstet Gynecol Reprod Biol 62: 107, 1995 |
|
Rybakowski C, Leichtweiss HP, Schroder H: Do dehydroascorbic acid and D-glucose make use of the same transfer system in the human placenta? Placenta 12: 431, 1991 |
|
Norkus EP, Rosso P: Effects of maternal intake of ascorbic acid on the postnatal metabolism of this vitamin in the guinea pig. J Nutr 111: 624, 1981 |
|
Cochrane W: Overnutrition in prenatal and neonatal life: A problem? Can Med Assoc J 93: 893, 1965 |
|
Dansky LV, Rosenblatt DA, Andermann E: Mechanism of teratogenesis: Folic acid and antiepileptic therapy. Neurology 42 (suppl 5): 32, 1992 |
|
Selhub J: Folate binding proteins: Mechanism for placental and intestinal uptake. In Allen L, King J, Lonnerdal B (eds): Nutrient Regulation During Pregnancy, Lactation and Infant Growth, p 141. New York, Plenum Press, 1994 |
|
Fernandes-Costa F, Metz J: Transplacental transport in the rabbit of vitamin B12 bound to human transcobalamin I, II and III. Br J Haematol 43: 625, 1979 |
|
Moe A, Plas DR, Powell KA, Smith CH: Riboflavin uptake in microvillous and basal membrane vesicles isolated from full-term human placentas. Placenta 15: 137, 1994 |
|
Allen LH: Vitamin B12 metabolism and status during pregnancy, lactation, and infancy. In Allen L, King J, Lonnerdal B (eds): Nutrient Regulation During Pregnancy, Lactation and Infant Growth, p 173. New York, Plenum Press, 1994 |
|
Specker BL, Black A, Allen L et al: Vitamin B-12: Low milk concentrations are related to low serum concentrations in vegetarian women and to methylmalonic aciduria in their infants. Am J Clin Nutr 52: 1073, 1990 |